


Abstract
Biased G protein–coupled receptor agonists engender a restricted repertoire of downstream events from their cognate receptors, permitting them to produce mixed agonist-antagonist effects in vivo. While this opens the possibility of novel therapeutics, it complicates rational drug design, since the in vivo response to a biased agonist cannot be reliably predicted from its in cellula efficacy. We have employed novel informatic approaches to characterize the in vivo transcriptomic signature of the arrestin pathway-selective parathyroid hormone analog [d-Trp12, Tyr34]bovine PTH(7-34) in six different murine tissues after chronic drug exposure. We find that [d-Trp12, Tyr34]bovine PTH(7-34) elicits a distinctive arrestin-signaling focused transcriptomic response that is more coherently regulated across tissues than that of the pluripotent agonist, human PTH(1-34). This arrestin-focused network is closely associated with transcriptional control of cell growth and development. Our demonstration of a conserved arrestin-dependent transcriptomic signature suggests a framework within which the in vivo outcomes of arrestin-biased signaling may be generalized.
Introduction
The last decade of basic research in G protein–coupled receptor (GPCR) biology has produced two concepts that together may facilitate development of novel GPCR-based therapeutics, i.e., pluridimensional efficacy and ligand bias. The former refers to the discovery that GPCRs signal via multiple G protein and non–G protein effectors (Galandrin and Bouvier, 2006). The best-studied non–G protein effectors are the arrestins, which promote homologous desensitization of G protein–mediated signaling while simultaneously acting as ligand-regulated scaffolds that recruit additional signaling effectors (Luttrell et al., 1999). GPCR-arrestin interactions promote the assembly of multiprotein signalsomes that enable GPCRs to engage protein and lipid kinases, phosphatases, ubiquitin ligases, regulators of small G proteins, and other novel effectors (Maudsley et al., 2005; Luttrell and Gesty-Palmer, 2010; Shenoy and Lefkowitz, 2011).
Traditional models of ligand efficacy treat GPCRs as existing in spontaneous equilibrium between unitary on and off states, the relative proportion of which is changed upon ligand binding. However, these models cannot account for the phenomena of reversal of potency or efficacy, which require the existence of multiple active conformations that couple the receptor to downstream effectors with different efficiencies (Kenakin, 1995; Christopoulos and Kenakin, 2002). The concept of functional selectivity acknowledges that chemically distinct ligands may affect the conformational equilibrium of the receptor in ways that differ from the native ligand, causing the receptor to couple to only a subset of its potential effectors (Wei et al., 2003; Kenakin, 2007; Kenakin and Miller, 2010; Luttrell and Kenakin, 2011; Maudsley et al., 2012). In vivo, such biased ligands produce mixed effects, directly activating some downstream pathways while antagonizing the effects of circulating endogenous ligands on others.
We have previously reported that [d-Trp12,Tyr34]bovine PTH(7-34) behaves as an arrestin pathway-selective biased agonist of the type 1 parathyroid hormone receptor (PTH1R). In cellula, bovine PTH(7-34) [bPTH(7-34)] exhibits true reversal of efficacy, acting as an inverse agonist for PTH1R-Gs coupling and an agonist for arrestin-dependent signaling, e.g., ERK1/2 activation, anti-apoptotic signaling, and cell migration (Gesty-Palmer et al., 2006, 2009; Appleton et al., 2013). In vivo, intermittent injection of bPTH(7-34) increases trabecular bone mass, osteoblast number, and mineral apposition rate without stimulating osteoclast proliferation and bone resorption like its conventional agonist counterpart, human PTH(1-34) [hPTH(1-34)] (Gesty-Palmer et al., 2009). Surprisingly, functional transcriptomic analysis of bone from mice treated with bPTH(7-34) or hPTH(1-34) indicates that the two ligands increase bone mass in vivo through largely distinct transcriptomic mechanisms (Gesty-Palmer et al., 2013).
The promise of biased agonism lies in its potential to qualitatively change GPCR signaling (Whalen et al., 2011; Maudsley et al., 2012). While the example of bPTH(7-34) provides proof-of-principle that biased agonists can produce effects in vivo that cannot be achieved using conventional agonists or antagonists, it also underscores the following problem: because biased agonists activate GPCRs in unnatural ways, the in vivo response of a biased agonist cannot necessarily be predicted from either the in cellula efficacy profile or knowledge of the actions of the native ligand (Luttrell, 2014). This is particularly true of arrestin pathway-selective agonists since the biologically relevant GPCR signals transmitted via arrestins remain poorly characterized (Luttrell and Gesty-Palmer, 2010). Here, we have employed informatic approaches to compare the in vivo transcriptomic signatures of bPTH(7-34) and hPTH(1-34) in six different murine tissues after chronic drug exposure with the goal of delineating a conserved arrestin-biased PTH1R signaling repertoire in vivo that might be generalizable to other systems.
Materials and Methods
Animals and Drug Treatment.
The derivation of arrestin3 null mice has been previously described (Bohn et al., 1999). Mice were maintained under standard nonbarrier conditions, fed rodent chow (LabDiet, PMI Nutrition International, St. Louis, MO), and had access to water ad libitum. Human 40 µg/kg per day PTH(1-34), 40 µg/kg per day bovine [d-Trp12, Tyr34]PTH(7-34), or phosphate-buffered saline vehicle was administered to 11-week-old male wild-type (WT) C57BL/6J or congenic arrestin3 null mice via Alzet osmotic minipumps (Model 1004; Durect Corp., Cupertino, CA) for 28 days. Minipumps were implanted subcutaneously in the upper back of anesthetized mice. The pump releases 0.25 μl/h in vehicle of 1 mM acetic acid in sterile phosphate-buffered saline. At the end of the infusion period, animals were sacrificed and the target tissues (calvarial bone, heart, lung, liver, kidney, and aorta) were harvested and stored at −80°C until mRNA isolation. Animal protocols were approved by the institutional animal care and use committee at Duke University School of Medicine and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Quantitative Computed Tomography.
High-resolution quantitative computed tomography (µCT40; Scanco Medical, Basserdorf, Switzerland) was used to evaluate trabecular bone volume fraction and microarchitecture in the distal femur. Samples were scanned at 45 keV with conebeam mode and a slice increment of 6 µm. Images from each group were generated at 275 threshold. The three-dimensional volume of interest structure was constructed and analyzed with the internal software of the µCT system.
Urine Biochemistry and Bone Turnover Markers.
Urine was collected for 24 hours from mice housed in metabolic cages (Hatteras Instruments, Hatteras, NC). Dexoypyridinoline excretion was quantitated using the Pyrilinks-D assay kit (Metra Biosystems Inc., Mountain View, CA). Urine creatinine concentration was measured by the Jaffé alkaline picrate method using a kit from Exocell (Philadelphia, PA). Urine ionized calcium measurements were obtained using a micro/mono-calcium ion electrode (Lazar Research Laboratories, Los Angeles, CA).
RNA Extraction and Oligonucleotide Microarray Hybridization.
RNA isolation from three individual animals in each experimental group was carried out using a Qiagen RNeasy Mini Kit (Qiagen, Inc., Valencia, CA), as described previously (Gesty-Palmer et al., 2013). RNA conversion to cDNA and subsequent hybridization with Sentrix MouseRef-8 Expression BeadChips (Illumina, San Diego, CA) was performed as described previously (Chadwick et al., 2010; Martin et al., 2012a). Microarray data were analyzed using DIANE 6.0 (http://www.grc.nia.nih.gov/branches/rrb/dna/dna.htm), a spreadsheet-based microarray analysis program based on the SAS JMP7.0 system (http://www.sas.com/en_us/home.html). Raw microarray data were subjected to filtering and z normalization and tested for significant changes as described previously (Chadwick et al., 2010). Initial filtering identified genes with a z-ratio of ≥1.50, with the z-ratio being derived from the difference between the averages of the observed gene z-scores divided by the S.D. of all of the differences for that particular comparison. Genes were then refined by calculating the false discovery rate, which controls for the expected proportion of falsely rejected hypotheses, and including only those genes with false discovery rates <0.05. These data were further analyzed using analysis of variance with significance set at P < 0.05. This allowed us to identify transcripts that differed in their intensity across all of the animal replicates and the various experimental conditions of the mice employed in this study. We have deposited the raw data at GEO/ArrayExpress under Accession Number GSE64485, and we confirm all details are Minimum Information About a Microarray Experiment compliant.
Bioinformatic Analyses.
Visualization and quantitative comparison of holistic gene expression was performed using Omnimorph, a novel three-dimensional visualization application for high-dimensionality data sets (Maudsley et al., 2013). The analytical Omnimorph output is full-spectrum annotated (array z-scores: red, high positive; violet, high negative), and enables highly detailed holistic data set discrimination using center of mass and spherical deformation (spikiness) calculations based on transcriptomic z-score moduli (see Supplemental Material).
Array-derived gene lists were analyzed using multiple forms of functional annotational clustering. For each specific form of functional annotation we employed a cutoff of at least two genes (from the original filtered/analysis of variance gene set) needing to be present to fully populate a particular gene ontology (GO) term group, canonical signaling pathway, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, etc., with a probability (P) of enrichment value of ≤0.05. Parametric gene set enrichment analyses, such as GO or Molecular Signatures Database–PAGE, are primarily performed using raw data sets to test significance of enrichment at a group/collection level rather than at the individual gene significance level (Mootha et al., 2003). Ingenuity Pathway Analysis (http://www.ingenuity.com/) was employed for canonical signaling pathway analysis. For the GO term, KEGG pathway, and transcription factor, target enrichment was performed using WebGestalt (http://bioinfo.vanderbilt.edu/webgestalt/). BioCarta signaling pathway and Protein Information Resource keyword enrichment analysis was performed using NIH DAVID (http://david.abcc.ncifcrf.gov/). Functional protein/gene network interaction analysis was performed using STRING (http://string-db.org/). Where stated, a hybrid score process was employed to generate a single index for a specifically enriched category, e.g., GO terms or KEGG pathways. Hybrid scores are generated by the multiplication of the negative log10 of enrichment probability (P) with the enrichment factor (R).
Protein Expression Analysis.
Microarray results were validated by selective Western blots. Tissues excluding calvarial bone were homogenized using sonication followed by fractionation using a Qiagen Qproteome kit according to the manufacturer’s instructions (Qiagen, Inc., Valencia, CA). For all experiments, cytoplasmic fractions were used. Calvaria were pulverized on dry ice in a glass Dounce homogenizer and proteins were extracted for 30 minutes on wet ice in 200 μl of protein extraction buffer [2% sodium dodecyl sulfate; 2 M urea; 10 mM Tris-HCl (pH 6.8); 1 mM phenylmethlysulfonyl-fluoride]. Homogenates were clarified by microcentrifugation for 10 minutes at 15,000 rpm, and the buffer composition was adjusted to 10% glycerol, 10 mM dithiothreitol, and 0.0025% bromophenol blue for SDS-PAGE. Each tissue homogenate was loaded onto a BisTris 4–12% polyacrylamide gel (Invitrogen, Carlsbad, CA) before electrotransfer to a polyvinylidene difluoride membrane (Thermo Scientific, Rockford, IL). Proteins were identified using primary antisera at 1:1000 dilution, followed by species-specific alkaline phosphatase–conjugated secondary antibodies (Sigma-Aldrich, St Louis, MO) at 1:7000 dilution. Primary antibodies specific for 14-3-3 η (Ywhah), β-actin (Actb), glyceraldehyde 3-phosphate dehydrogenase (Gapdh), GRB2-associated binding protein 1 (Gab1), aspartyl-tRNA synthetase (Dars), and RAB1 member RAS oncogene family (Rab1), were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). TRAF family member–associated NF-κB activator (Tank), sirtuin 1 (Sirt1), antibodies were obtained from Cell Signaling Technology (Danvers, MA). Primary antibodies for insulin-induced gene 1 (Insig1) and dynactin 1 (Dctn1) were obtained from AbCam (Cambridge, MA). Primary antibodies for nuclear factor I/C (Nfic) and metaxin 1 (Mtx1) were obtained from Sigma-Aldrich. Primary antibodies for glycogenin (Gyg) were obtained from LifeSpan Biosciences, Inc. (Seattle, WA). Polyvinylidene difluoride–bound immune complexes were identified using enzyme-linked chemifluorescence and quantified using a Typhoon 9410 PhosphorImager (GE Healthcare, Piscataway, NJ). All antibodies used have been previously validated.
Statistical Analyses.
In each histogram, data represent the mean ± S.E. Statistical analyses (Student’s t test) were performed using GraphPad Prism (GraphPad Software, San Diego, CA). P ≤ 0.05 was considered statistically significant.
Results
bPTH(7-34) and hPTH(1-34) Produce Different Holistic Functional Signatures In Vivo
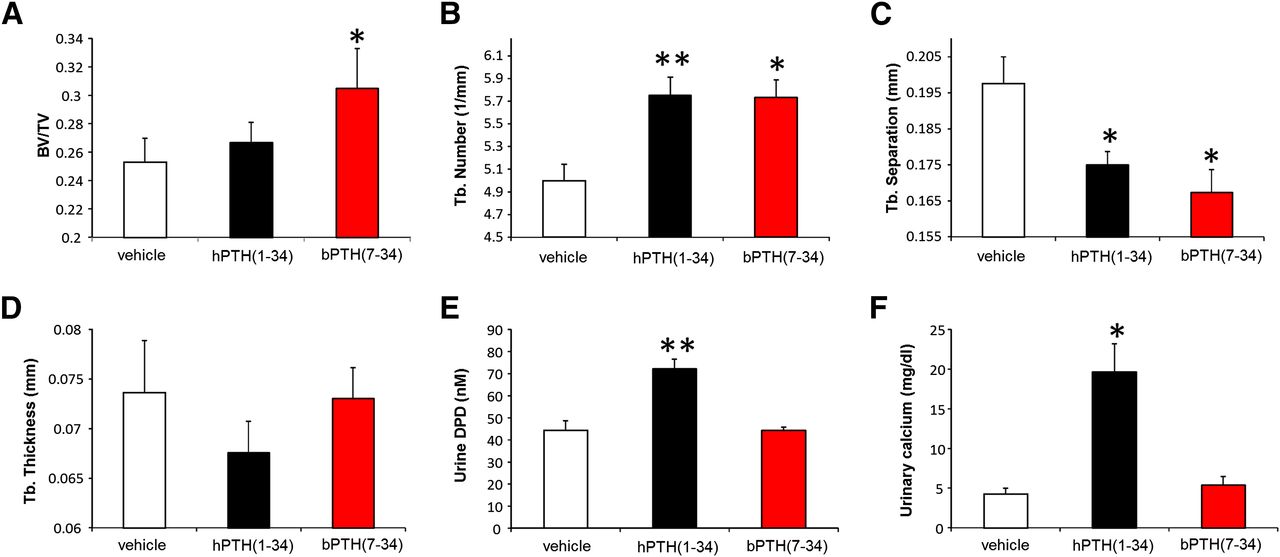
Male WT C57BL/6J mice were implanted with microinfusion pumps and dosed with hPTH(1-34) or bPTH(7-34) for 28 days. Fig. 1 depicts the distinct phenotypic responses of trabecular bone to treatment with the two ligands. Unlike the conventional agonist, hPTH(1-34), continuous exposure to hPTH(7-34) increased trabecular bone volume/total volume measured by quantitative computed tomography (Fig. 1A). While both agonists increased trabecular number and decreased trabecular separation, hPTH(1-34) provoked a loss of trabecular thickness that was not observed with bPTH(7-34) (Fig. 1, B–D). Reflective of the divergent effects of hPTH(1-34) and bPTH(7-34) on osteoclastic bone turnover (Gesty-Palmer et al., 2009), only hPTH(1-34) treatment was associated with increased urinary dexoypyridinoline and 24 hour urinary calcium excretion (Fig. 1, E and F).

Continuous infusion of bPTH(7-34) increases trabecular bone mass without stimulating bone resorption or causing increased urinary calcium excretion. Eleven-week-old male C57BL/6J mice were treated with phosphate-buffered saline vehicle, hPTH(1-34) (40μg/kg per day) or bPTH(7-34) (40μg/kg per day) via osmotic pump for 4 weeks. Quantitative computed tomography of distal femurs was performed to determine the effects of treatment on (A) trabecular bone (Tb) volume (BV/TV), (B) trabecular number, (C) trabecular separation, and (D) trabecular thickness. Twenty-four hour urine collections were analyzed for deoxypyrodinoline to assess bone turnover (E) and calcium excretion (F). Data represent the mean ± S.E.M. of measurements taken from at least seven mice. *P < 0.05 and **P < 0.01 compared with vehicle-treated mice. Significance was determined using one-way analysis of variance with Bonferonni multiple comparisons post-test. DPD, deoxypyridinoline.
Although highest in kidney and bone, the PTH1R is widely expressed, including aorta, adrenal gland, bladder, brain, cerebellum, breast, heart, ileum, liver, lung, skeletal muscle, ovary, placenta, skin, spleen, stomach, uterus, and testes (Ureña et al., 1993). To generate data sets representing the effects of long-term infusion of hPTH(1-34) or bPTH(7-34) across different cellular backgrounds, we performed quantitative transcriptomic analysis of six tissues (aorta, bone, heart, kidney, liver, and lung). Triplicate biologic replicates were analyzed for each ligand/tissue combination. Transcriptome data were assessed for quality control (labeling and hybridization), and then normalized and statistically filtered as described preeviously (Chadwick et al., 2010) [Supplemental Tables 1–6, hPTH(1-34); Supplemental Tables 7–12, bPTH(7-34)]. To generate a comprehensive appreciation of the systemic effects of these two compounds we first examined the data using a well characterized global data set analytical approach, principal component analysis. Applying principal component analysis to the transcriptomic data sets revealed a strong separation between different tissues, as would be expected (Supplemental Fig. 1, A–C). In contrast, minimal spatial separation between hPTH(1-34) and bPTH(7-34) was apparent (Supplemental Fig. 2, A–F).
Since principal component analysis lacked the sensitivity to identify global data set differences between vehicle and ligand-treated tissue samples, we next investigated the differential effects of bPTH(7-34) and hPTH(1-34) using topological data analysis (TDA). TDA facilitates the appreciation of high-dimensionality data sets in a more gestalt manner that is likely closer to the level of functional physiologic interactivity of transcripts/proteins (Carlsson, 2009; Lum et al., 2013). Recently, TDA has been successfully applied to investigate transcriptomic drug signatures (Martin et al., 2013), protein-protein interaction networks (Sardiu et al., 2014), and pharmacogenomic profiles (Nicolau et al., 2011). Using our newly developed three-dimensional visualization application, Omnimorph, we created exemplary highly complex data structures using the averaged microarray z-score ligand-response data sets from each tissue/drug combination, e.g., hPTH(1-34) and bPTH(7-34) in murine kidney (Fig. 2A). The upper panels in Fig. 2A represent a binary view of expression polarity, while the lower panels demonstrate strong polychromatic differences in Omnimorph expression polarities between hPTH(1-34) and bPTH(7-34). For differential topological analysis the extraction of distinct and divergent features from the structure is important. Thus, we assessed indices indicative of the overall data structure, i.e., the coordinates of its ultimate center of mass converted from spherical coordinates to Cartesian coordinates r, q, and h (see Supplemental Material), as well as surface features such as deformation variability, i.e., range and number of surface deformation magnitudes. The extraction of these two feature types gives both a holistic structural analysis that integrates data from all the respective data points linked together, and an appreciation of differential surface topologies representing smaller scale transcriptomic features (Watts and Strogatz, 1998; Martin et al., 2013). We found that Omnimorph structures created from hPTH(1-34) and bPTH(7-34) data sets possessed significant differences with respect to both their holistic global structure (i.e., their center of mass location; specifically, the Cartesian r transformation) and their degree of surface structure variability [i.e., bPTH(7-34) data sets generated structures with larger numbers of stronger surface deformations than hPTH(1-34)] (Fig. 2, B–D). Therefore, when using high-dimensionality TDA, the two ligands exhibit characteristic and significant structural differences in their holistic transcriptomic responses.
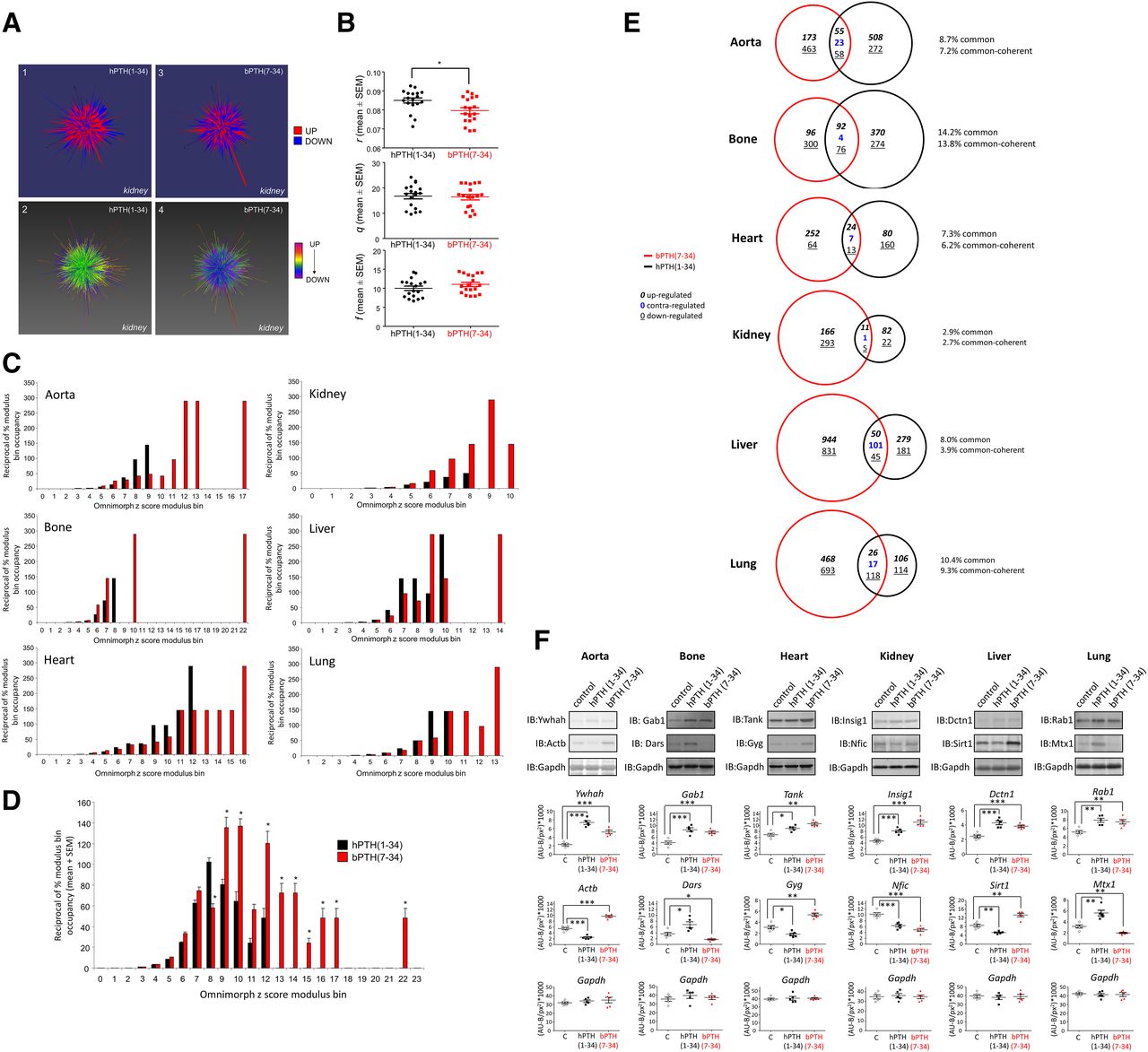
bPTH(7-34) generates a distinct transcriptomic response in multiple tissues compared to hPTH(1-34). (A) Omnimorph structures depicting a high-dimensionality z-score representation of the transcriptional responses to hPTH(1-34) (panels 1-2) or bPTH(7-34) (panels 3-4). Center of mass calculations from triplicate values for each Omnimorph from hPTH(1-34)– and bPTH(7-34)–treated mice were compared using the standard factors (r, q, and f) used to deconvolute spherical to Cartesian coordinates. (B) Cartesian deconvolution of Omnimorph center of mass spherical coordinates. A statistically significant difference between hPTH(1-34)– and bPTH(7-34)–treated tissues exists only with respect to r, the Euclidean distance from the origin to the center of mass, demonstrating that a global deformation of the Omnimorph is characteristic of the different ligands. Each data point represents the specific r, q, or f measurement from an individual Omnimorph generated from a single mouse subject [*P < 0.05 for bPTH(7-34) compared with hPTH(1-34)]. (C) Omnimorph spike deformation data, percentage of total transcript spikes in a specific modulus bin, for hPTH(1-34) (black bars) and bPTH(7-34) (red bars) transcriptomic data across multiple murine tissues. Omnimorph deformations are binned according to their z-score expression modulus. (D) Mean data for the six-tissue comparison of z-score modulus Omnimorph spike distribution between hPTH(1-34) and bPTH(7-34). Across multiple tissues bPTH(7-34)–associated Omnimorphs possess a distinct profile of spike deformations compared with hPTH(1-34). (E) Venn diagram analysis of the intersections of significantly-regulated transcripts for hPTH(1-34)–treated (black) and bPTH(7-34)–treated (red) murine tissues. The percentage of shared drug transcript identity ranged from only 2.9% (kidney) to 14.2% (bone). Coherent regulation of common transcripts refers to the preservation of the same expression polarity (up- or downregulation) induced by either ligand. Contra-regulation indicates divergent expression polarities, induced by PTH ligands, for common transcripts. (F) Western blot validation of protein expression levels correlating to significantly-modulated gene transcripts across six murine tissues (hPTH(1-34), black; bPTH(7-34), red. Protein expression values are indicated as Arbitrary fluorescent units; background/pixels2*1000 [(AU-B)/px2)*1000]).
Given that hPTH(1-34) and bPTH(7-34) would have opposite effects on heterotrimeric G protein signaling by the PTH1R in vivo (agonism versus antagonism), we hypothesized that the structural differences in their holistic transcriptomic responses observed using TDA were reflective of marked differences between the two ligands at the level of individual transcript regulation. Confirming this posit, we found at the transcript level that the commonality between significantly modulated hPTH(1-34) and bPTH(7-34) transcripts ranged between only 2.9 and 14.2% across the six different tissues (mean commonality 8.6%) (Fig. 2E). Considering only those transcripts that were regulated by both ligands and the same tissue, we nonetheless found substantial coherence in the polarity of change, i.e., upregulation versus downregulation compared with vehicle control. With the exception of liver (49% coherence), the majority of commonly regulated transcripts within a given tissue were coherently regulated (83–97% coherence), suggesting the two ligands possess shared as well as distinct transcriptomic effects. For transcriptomic validation using an orthogonal technology, we chose two significantly regulated transcripts from each tissue that were both readily identifiable using Western analysis and demonstrated a variety of expression polarity effects in response to the two ligands (Fig. 2F). A strong correlation between the protein expression data and the transcriptomic regulation was evident, suggesting that our transcriptomic data sets are closely linked to functional signaling alterations in the different tissues.
bPTH(7-34) Activates a Distinct and More Focused Set of Transcripts across Multiple Tissues than Does hPTH(1-34).
Our comparison of the regulated gene transcript identities in each target tissue demonstrated that hPTH(1-34) and bPTH(7-34) generated distinctly separate response profiles (Fig. 2, E and F). However, lack of concordance in individual regulated transcripts within a tissue may nonetheless represent substantial functional overlap if the two treatments are activating shared signaling pathways. To address this, we statistically clustered the significantly regulated hPTH(1-34) and bPTH(7-34) transcripts into canonical signaling pathways using Ingenuity Pathway Analysis (http://www.ingenuity.com/) [hPTH(1-34), Supplemental Tables 13–18; bPTH(7-34), Supplemental Tables 19–24]. After functional clustering, we did observe an increase in the percentage of pathway similarity across tissues, i.e., the levels of commonality between significantly modulated hPTH(1-34) and bPTH(7-34) signaling pathways ranged between 1.4% in kidney and 38.9% in bone (mean commonality, 19.9%) (Fig. 3, A–F). Considering only coherently modulated pathways, i.e., pathways with the same predicted direction of change, the degree of commonality ranged between 0.0% in kidney and 35.4% in bone (yellow shaded areas in Fig. 3, A–F).

hPTH(1-34) and bPTH(7-34) stimulate distinct signaling pathway profiles in multiple tissues. (A) Significantly populated canonical signaling pathways induced by bPTH(7-34) (red bars) or by hPTH(1-34) (black bars) in murine aorta. Each histogram bar represents the signaling pathway score (number of ligand upregulated pathway-populating transcripts minus the number of ligand downregulated pathway-populating transcripts. Signaling pathways within the yellow block represent the coherently regulated common pathways between the two ligands in the respective tissue. The associated Venn diagram indicates the functional signaling separation between bPTH(7-34) and hPTH(1-34) in the specific tissue experiment. (B) Bone, (C) heart, (D) kidney, (E) liver, and (F) lung all represent analogous data to (A).
We next investigated the potential nature of the signaling bias of bPTH(7-34) compared with hPTH(1-34) by clustering the signaling pathways that were uniquely regulated by each ligand in each tissue (Supplemental Tables 25–30). Tissue-by-tissue inspection of the signaling pathway clusters (Supplemental Fig. 3, A–F) demonstrated that, compared with hPTH(1-34), bPTH(7-34) signaling disproportionately stimulated signaling activity linked with cell cycle control, cell damage/repair, GPCR modulation, and tissue development. Therefore, in addition to the divergence of bPTH(7-34)P and hPTH(1-34) activity at the transcript level, a strong pathway divergence and bPTH(7-34) bias toward tissue development and cell cycle control signaling was apparent.
We next investigated the posit that a GPCR ligand with a focused signaling repertoire, i.e., bPTH(7-34), would demonstrate a more conserved transcriptomic profile between tissues than a more pluripotent ligand, i.e., hPTH(1-34) (Maudsley et al., 2012). We analyzed the multitissue distribution of significantly regulated gene transcripts and signaling pathways for hPTH(1-34) and bPTH(7-34) using VENNTURE, a 6-way Edwards Venn diagram application (Martin et al., 2012b). We found that at both the transcript and pathway levels the effects of bPTH(7-34) were more strongly conserved across multiple tissues than the actions of hPTH(1-34) (Fig. 4, A and B; Supplemental Fig. 4). Using a unified gene set normalization and scaling system for cross-tissue conservation (10× multiplication for each successive level of multitissue conservation), it was evident that bPTH(7-34) regulates transcriptional activity across diverse tissues in a more coherent manner than hPTH(1-34) (Fig. 4C). Analyzing cross-tissue signaling pathway annotation in a similar manner again demonstrated a more coherent signaling activity of bPTH(7-34) compared with hPTH(1-34) (Fig. 4D; Supplemental Fig. 4). While no common signaling pathways were significantly populated in all six tissues by hPTH(1-34), bPTH(7-34) significantly (P < 0.05) populated five common pathways across all six tissues, i.e., molecular mechanisms of cancer; IGF-1 signaling; PPARα/RXRα activation; IL-8 signaling; and prostate cancer signaling. Thus, bPTH(7-34) regulates more multitissue conserved transcripts in a coherent manner than hPTH(1-34), indicating a potentially more rigid and less tissue-sensitive signaling paradigm for bPTH(7-34).
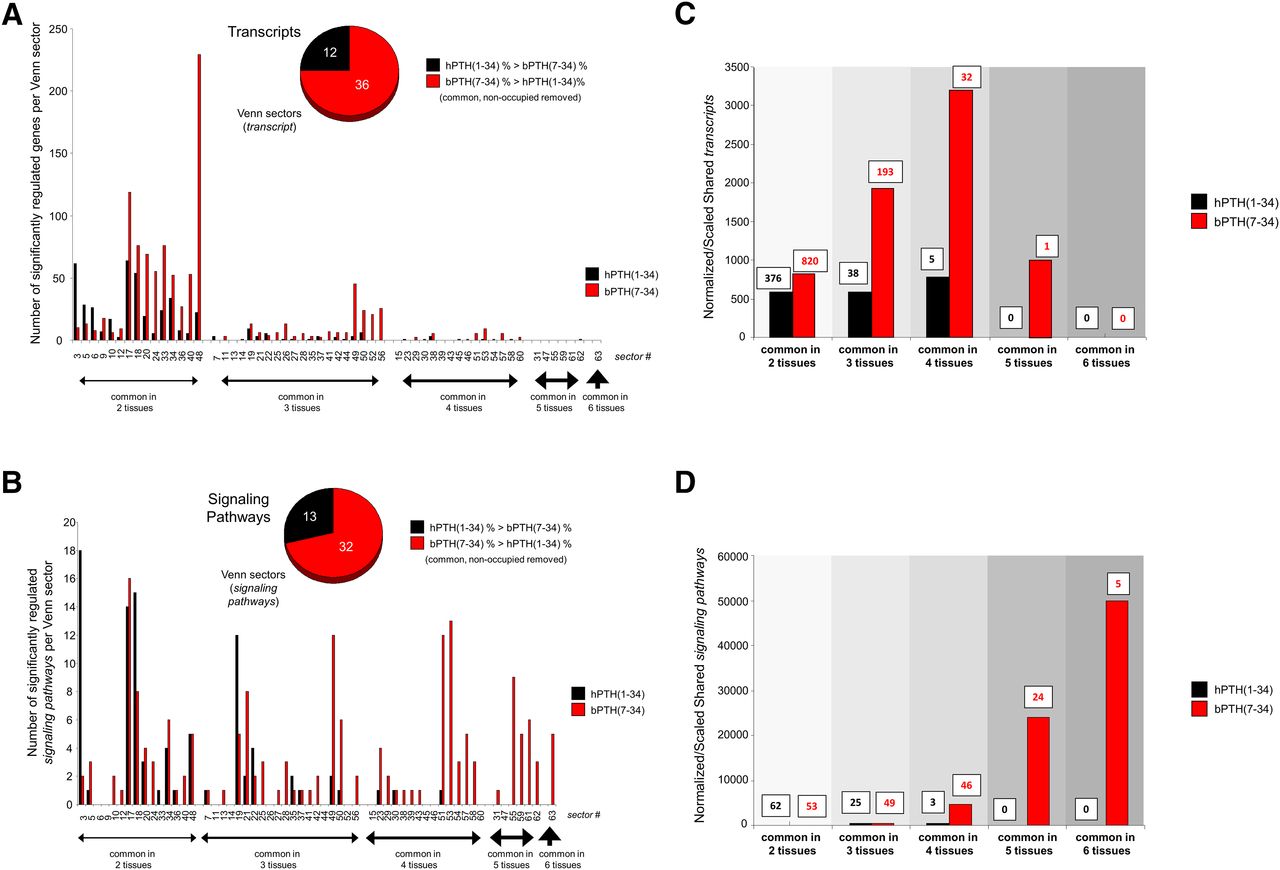
bPTH(7-34)–mediated transcriptomic activity is more conserved across multiple tissues compared with hPTH(1-34). (A) Edwards Venn distribution, excluding unoccupied and tissue-unique sectors, for hPTH(1-34) or bPTH(7-34) significantly regulated transcripts. The majority (75%) of multiple tissue-shared (>2) Venn sectors were more strongly populated (normalized to input data set size) with bPTH(7-34)–regulated transcripts compared with hPTH(1-34)–regulated transcripts. (B) Edwards Venn distribution (excluding unoccupied and tissue-unique sectors) for the signaling pathways significantly populated by the transcripts controlled by hPTH(1-34) or bPTH(7-34). bPTH(7-34) demonstrated a numerically greater population of each multitissue sector compared with hPTH(1-34), i.e., 71% of sectors had greater bPTH(7-34) population, and affected a considerably greater population of the most cross-tissue conserved Venn sectors, i.e., 3–6 tissues conserved. No common signaling pathways were significantly populated in all six tissues by hPTH(1-34); however, bPTH(7-34) activity significantly (P < 0.05) populated five common pathways across all six tissues. (C) Classification of the degree of cross-tissue commonality of transcript regulation for hPTH(1-34) or bPTH(7-34). The histogram displays a scaled (for ascending levels of tissue commonality) and normalized (for data set size) representation of the ability of each ligand to control the same transcripts over multiple tissues. The number of significantly regulated transcripts associated with each histogram bar are indicated above the bar [hPTH(1-34), black numbers; bPTH(7-34), red numbers]. (D) Classification of the degree of cross-tissue commonality of signaling pathway regulation for hPTH(1-34) or bPTH(7-34). The histogram displays a scaled and normalized representation of the ability of each ligand to control the same transcripts over multiple tissues. The number of significantly regulated transcripts associated with each histogram bar are indicated above the bar [hPTH(1-34), black numbers; bPTH(7-34), red numbers].
While signaling pathway analysis can describe potential functional effects of transcript groups, pathway annotation is incomplete and considerable overlap often exists at the populating transcript level. As a result, a few significantly regulated transcripts in an empirically derived data set may identify multiple pathways that are closely related functionally and do not represent discrete processes. This limitation did not apply to the five bPTH(7-34)–regulated pathways identified in all six tissues. For example, despite the similar functional activity suggested by the cross-tissue conserved prostate cancer signaling and molecular mechanisms of cancer pathways, of the total 138 empirically observed bPTH(7-34)–regulated transcripts that drove the selection of these two pathways, only 23 (16%) were transcripts that were common to both. Therefore, it is likely that the predicted signaling functions of bPTH(7-34) encompassed by these two terms are functionally distinct.
The Conserved Transcriptomic Signature of bPTH(7-34) Is Focused on Pathways Related to Transcriptional Control of Cell Growth and Development.
Having identified the most conserved hPTH(1-34)– and bPTH(7-34)–regulated signaling pathway clusters generated by considering each tissue separately, we reanalyzed the data using a complementary approach that pooled the array data from all tissues prior to analysis. To do this, we constructed two lists of similar proportional size representing the top 5% most cross-tissue conserved significantly regulated transcripts for each ligand. The top 5% lists comprised 130 out of 2590 total transcripts for hPTH(1-34) and 200 out of 4016 total transcripts for bPTH(7-34). Since no transcripts changed in all six tissues in response to either ligand (Fig. 4C), i.e., no individual transcript was coherently regulated, the top 5% lists were constructed without regard to the polarity of change. The top 5% most conserved transcripts for hPTH(1-34) and bPTH(7-34), respectively, along with the magnitude (z-ratio) and polarity (direction) of change in each tissue where a significant change was observed are included in Supplemental Tables 31 and 32.
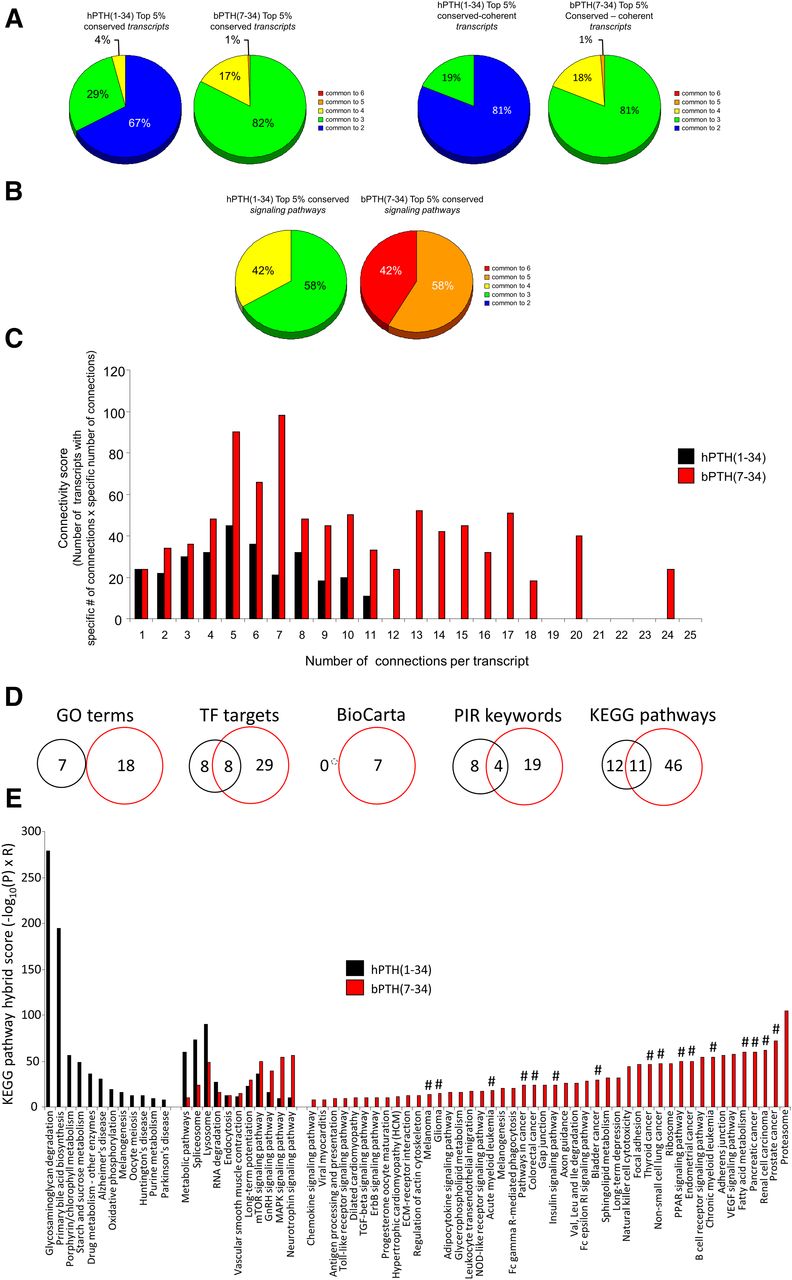
As seen in the tissue-by-tissue analysis, a considerably greater proportion of bPTH(7-34)–regulated transcripts in the top 5% data set were conserved across multiple tissues, compared with hPTH(1-34) (Fig. 5A). Identical results were obtained if the top 5% lists were narrowed to include only those transcripts that were coherently regulated in each tissue where a significant change was observed. The same bias for conserved bPTH(7-34)–mediated multitissue signaling was also present when examining the degree of cross-tissue conservation at the top 5% signaling pathway level (Fig. 5B). We next assessed whether the top 5% bPTH(7-34) multitissue regulated transcripts formed a more functionally interrelated group than the top 5% of hPTH(1-34) multitissue regulated transcripts. Using the STRING evidence-based network analysis (http://string-db.org/), we found that the bPTH(7-34) top 5% transcript data set possessed a considerably greater number of functionally connected factors (Fig. 5C).

The conserved multitissue bPTH(7-34)–regulated transcriptional response is distinct and more coherent than that of hPTH(1-34). (A) Percentage distribution across multiple tissues of the top 5% conserved transcripts for hPTH(1-34) and bPTH(7-34) (left panel) and the top 5% coherently regulated conserved transcripts for hPTH(1-34) and bPTH(7-34) (right panel). Among the top 5% of cross-tissue conserved transcripts, all 200 of those regulated by bPTH(7-34) were common to at least three different tissues, while only 42 out of 130 of those regulated by hPTH(1-34) were common to at least three tissues. Similarly, 100% of the bPTH(7-34) coherently regulated transcripts were common to at least three different tissues, while only 19% of hPTH(1-34) coherently regulated transcripts were shared across three different tissues. (B) Percentage distribution across multiple tissues of hPTH(1-34)– or bPTH(7-34)–regulated signaling pathways. All of the bPTH(7-34) top 5% pathways were common to at least five tissues, while no hPTH(1-34)–mediated pathways were common to more than four tissues. (C) The STRING-derived network (http://string-db.org/) connectivity score (i.e., the number of transcripts with a specific connection number score multiplied by the specific number of connections) histogram demonstrates that bPTH(7-34) generates a transcriptomic response comprising a greater number of more highly interrelated signaling factors than hPTH(1-34). (D) Strong functional distinction between conserved transcriptomic responses to hPTH(1-34) and bPTH(7-34). Using the top 5% most cross-tissue conserved transcripts controlled by each ligand, the resultant similarity between the GO term annotation, target transcription factor (TF) enrichment, BioCarta pathway population, Protein Information Resource (PIR) keyword analysis, and KEGG pathway enrichment, was calculated [hPTH(1-34), black circles: bPTH(7-34), red circles]. (E) KEGG pathway hybrid scores for the top 5% most conserved transcriptomes controlled by the two ligands demonstrate a strong functional divergence. The most strongly populated KEGG pathways controlled by bPTH(7-34) show a functional correlation to the most conserved canonical signaling pathways observed using a different mode of analysis. Hash mark symbols (#) demonstrate the functional convergence between bPTH(7-34)–generated KEGG pathways and previously identified bPTH(7-34) conserved canonical signaling pathways.
Given the greater connectivity between bPTH(7-34)–regulated transcripts, i.e., more functionally connected factors coherently regulated across more tissues, we hypothesized that the top 5% most cross-tissue conserved gene set for bPTH(7-34) would reflect the inherent functionality of the ligand in vivo. To investigate these functions, we employed multiple convergent informatic analyses (Supplemental Tables 33–37). Across multiple forms of data annotation we found that a distinct functional prediction of bPTH(7-34) versus hPTH(1-34) activity was clear (Fig. 5D). Focusing upon the divergence in KEGG pathway activation between the hPTH(1-34) and bPTH(7-34) top 5% conserved transcriptomes (Fig. 5E) we found that many of the significantly populated bPTH(7-34)–specific pathways were strongly associated with the previously identified 6-tissue–conserved canonical signaling pathways (Fig. 4B). Therefore, a consistent multitissue bPTH(7-34) signaling phenotype associated with alterations in cell growth (cancer-related pathways), PPAR activity, insulin system activity, and interleukin/cytokine signaling was revealed using two distinct and complementary methodologies.
bPTH(7-34) Signaling in a Native In Vivo Background Is Strongly Arrestin3 Dependent.
Compared with WT, adult arrestin3 knockout (Arrb2KO) null mice exhibit normal skeletal morphology and similar whole-body bone-mineral density (Gesty-Palmer et al., 2009). As might be expected given that ablation of arrestin3 should globally impair GPCR desensitization, circulating endogenous PTH levels are suppressed in the Arrb2KO background (Pi et al., 2005), and dynamic indices of bone turnover indicate a higher basal rate of both bone formation and bone resorption (Gesty-Palmer et al., 2009). The effects of Arrb2KO on basal bone transcription are included in Supplemental Table 38 (see also Gesty-Palmer et al., 2013). Although intermittently dosed hPTH(1-34) still increases bone mass in congenic arrestin3 null mice, the anabolic effects of bPTH(7-34) are lost (Gesty-Palmer et al., 2009). Whereas pathway analysis of transcriptomic data from calvaria of hPTH(1-34)–treated mice demonstrates that its anabolic effects are associated with changes in pathways classically associated with enhanced bone formation, including collagen synthesis and matrix mineralization, bPTH(7-34) primarily regulates pathways associated with cell cycle control, cell survival, and cytoskeletal rearrangement (Gesty-Palmer et al., 2013). Importantly, the reciprocal effects of arrestin3 deletion and bPTH(7-34) action can be demonstrated in cellula using primary calvarial osteoblasts, i.e., Arrb2KO osteoblasts proliferate faster and are more susceptible to apoptotic stimuli than WT osteoblasts, while bPTH(7-34) slows cell proliferation, promotes survival, and stimulates cell migration in WT, but not Arrb2KO, cells (Gesty-Palmer et al., 2013).
To test the in vivo arrestin dependence of bPTH(7-34) signaling in the present study where the ligand was dosed via continuous infusion, we compared the extent to which the observed transcriptomic responses to bPTH(7-34) and hPTH(1-34) were conserved between WT and Arrb2KO mice. We reproduced our WT infusion experiments for hPTH(1-34) (Supplemental Table 39) and bPTH(7-34) (Supplemental Table 40) in age- and gender-matched Arrb2KO mice. Using Omnimorph data topological analysis, we noted only minimal alterations in the holistic bone response to hPTH(1-34) resulting from deletion of arrestin3 (Fig. 6A, panels 1–4). In contrast, the bone transcriptomic activity of bPTH(7-34) was dramatically altered (Fig. 6A, panels 5–8). We had previously observed that WT bPTH(7-34) and hPTH(1-34) Omnimorphs differed significantly in the deconvolution factor r. Compared with the WT condition, r was more significantly altered in the Arrb2KO background for bPTH(7-34) (P = 0.036) when compared with hPTH(1-34) (Fig. 6A). No significant differences in q or f between bPTH(7-34) and hPTH(1-34) in the Arrb2KO background were observed.

bPTH(7-34) in vivo transcriptomic activity is strongly arrestin dependent. (A) Arrestin3 genomic ablation in bone significantly affects the Omnimorph center of mass radial distance from the origin (r) of the bPTH(7-34) data sets compared with hPTH(1-34). (B) Venn analysis of the effects of arrestin3 ablation upon bPTH(7-34) and hPTH(1-34) transcriptome regulation reveals that arrestin3 deletion disproportionately affects bPTH(7-34) signaling compared with hPTH(1-34). Only 1.9% of significantly regulated transcripts were conserved between WT and Arrb2KO mice for bPTH(7-34) compared with 7.9% conservancy for hPTH(1-34). Multiple forms of informatic and signaling pathway annotation to the bPTH(7-34) and hPTH(1-34) transcriptomes in WT and Arrb2KO bone were applied: GO term analysis, transcription factor (TF) target analysis, BioCarta pathway analysis, Protein Information Resource (PIR) keyword analysis, and KEGG pathway analysis. As with the individual transcript identities, in each of these higher-order informatic analyses it was demonstrated that the genetic ablation of arrestin3 more profoundly reduced bPTH(7-34) signaling conservation than hPTH(1-34). (C) Genomic ablation of arrestin3 in bone has minimal effect on the coherency of regulation of the transcriptional response to hPTH(1-34) (upper panel), but profoundly disrupts bPTH(7-34) (lower panel); 98.5% of the common (between WT and Arrb2KO) hPTH(1-34)–regulated transcripts exhibited a conserved direction of regulation between the two genomic backgrounds, while 73% of the bPTH(7-34) commonly regulated transcripts (WT and Arrb2KO) showed a reversal in expression polarity when arrestin3 was ablated. The hPTH(1-34) or bPTH(7-34) labeled histograms depict the polarity of transcripts commonly regulated by either ligand in WT or Arrb2KO bone. (D) The moduli of the z-ratio differences between transcript expression values in WT versus Arrb2KO bone for the WT/Arrb2KO-conserved bPTH(7-34) transcripts was significantly (***P < 0.001) greater than that for hPTH(1-34). (E) Percentages of bPTH(7-34)– or hPTH(1-34)–regulated transcripts in WT bone that are also differentially regulated between Arrb2KO bone and WT bone. (F) Disease and biofunction annotation using Ingenuity (http://www.ingenuity.com/) activation z-score analysis of the differential effects of bPTH(7-34) in WT bone (upper panel) and Arrb2KO bone (lower panel). In the WT bone bPTH(7-34) presents a predicted capacity to increase bone cell number, while in Arrb2KO bone bPTH(7-34) presents a predicted capacity to reduce bone cell number. In both cases the transcript interactions that mediate these predictions are indicated with the associated explanatory Ingenuity key. (G) Disease/biofunction activation z-score histogram for bPTH(7-34) extracted bone-related biofunctions from WT bone (red bars) and from Arrb2KO bone (red bars with blue border). Positive activation z-scores indicate predicted biofunction stimulation, negative z-scores predict biofunction inhibition.
We next inspected the specific differences in bPTH(7-34) and hPTH(1-34) transcriptome regulation in WT versus Arrb2KO bone (Fig. 6B). Comparing the percentage of the total WT ligand-regulated transcriptome that was preserved in the Arrb2KO background, we found a greater loss of transcriptional regulation for bPTH(7-34) than for hPTH(1-34). As with the individual transcript identities, we found that the genetic ablation of arrestin3 more profoundly reduced bPTH(7-34) signaling conservation, measured by multiple and distinct forms of data annotation, than hPTH(1-34) between WT and Arrb2KO mice.
Despite the differences between the individual transcripts significantly regulated by bPTH(7-34) and hPTH(1-34) in WT versus Arrb2KO bone, several were commonly regulated in both genetic backgrounds (Fig. 6C). The expression polarity of the 65 hPTH(1-34)–controlled transcripts that were common to WT and Arrb2KO bone was largely preserved, yet the 11 transcripts that were common between WT and Arrb2KO for bPTH(7-34) treatment were almost completely reversed in expression polarity (Fig. 6C). The degree of transcript expression divergence for WT versus Arrb2KO common genes was significantly greater for bPTH(7-34) compared with hPTH(1-34) (Fig. 6D). Thus, as previously observed (Gesty-Palmer et al., 2013), the transcriptomic actions of bPTH(7-34) were more consistently disrupted by genetic deletion of its putative effector than were the actions of the pluripotent ligand, hPTH(1-34).
We next compared the extent to which bPTH(7-34) treatment of WT bone affected transcripts whose expression was significantly different between WT and Arrb2KO bone. Of the 568 total transcripts affected by bPTH(7-34) in WT bone, 251 (44.2%) were also disrupted by Arrb2KO. In contrast, only 27.6% (226 out of 816) of transcripts regulated by hPTH(1-34), which retains G protein signaling capacity in the congenic arrestin3 null background, were affected by Arrb2KO. To examine the linkage between arrestin-regulated transcripts and bPTH(7-34) actions in bone, we compared the transcriptomic effects of bPTH(7-34) in the WT and Arrb2KO backgrounds. Deriving pathway activation z-scores using ingenuity disease and biofunction classification (http://www.ingenuity.com/), we found that bPTH(7-34) activity in bone was strongly affected by Arrb2KO (Fig. 6F; Supplemental Tables 41 and 42). In WT bone bPTH(7-34) was predicted to increase the quantity of bone cells, while in Arrb2KO bone this activity was largely reversed with respect to the biofunction activation z-score (Fig. 6F). Identical disease and biofunction inspection of the differences between hPTH(1-34) activity in WT versus Arrb2KO bone failed to demonstrate any clear disruption of bone-related signaling (Supplemental Tables 43 and 44). Using the activation z-scores from all the bone-related terms in the bPTH(7-34) disease/biofunction readouts from WT and Arrb2KO bone (Fig. 6G) it was clear that the positive effects of bPTH(7-34) on bone growth in WT were largely absent in the Arrb2KO context.
Discussion
Our analyses illustrate the stark contrast between signaling by a conventional pluripotent GPCR agonist and an arrestin pathway-selective agonist at the level of in vivo transcriptional regulation. Whereas hPTH(1-34) activates Gs, Gq/11, Gi/o, and arrestin signaling pathways in differing proportions depending on the tissue-specific complement of these effectors and the expression of modulators like the Na+/H+ exchanger regulatory factors 1 and 2 (Mahon et al., 2002, 2003), bPTH(7-34) possesses a narrower efficacy profile that in cellula appears restricted to arrestin signaling pathways that primarily affect cell proliferation, survival, and cytoskeletal rearrangement (Gesty-Palmer et al., 2006, 2009; Appleton et al., 2013). In addition, hPTH(1-34), but not bPTH(7-34), mimics the calcitropic effects of endogenous PTH, producing hypercalcemia, hypophosphatemia, and metabolic acidosis, which would change the extracellular milieu and potentially alter the transcriptional response not only to PTH but to other circulating factors. Thus, in vivo, the transcriptional response to conventional and biased PTH analogs reflects the complex interplay between the structure-dependent differences of the intrinsic efficacy of the ligand, cellular factors such as tissue-specific expression of downstream effectors, and systemic factors like hypercalcemia and hyperchloremic metabolic acidosis. Thus, it is not surprising that the transcriptomic signature of hPTH(1-34) is markedly different from that of bPTH(7-34), that the response to hPTH(1-34) differs between tissues, or that the transcriptomic response to bPTH(7-34) is more highly conserved.
When studied in cellula using homogenous cell populations in a controlled physiologic milieu, GPCR efficacy most closely reflects the distribution of active receptor states stabilized by the ligand. Indeed, most studies comparing conventional and arrestin-selective efficacy using short-term in cellula assays of receptor conformation (Liu et al., 2012), effector coupling (Saulière et al., 2012), second messenger generation (Gesty-Palmer et al., 2006, 2009; Saulière et al., 2012; Appleton et al., 2013), or protein phosphorylation (Kendall et al., 2011; Christensen et al., 2010), indicate that biased ligands regulate a subset of the pathways controlled by the native ligand (Luttrell, 2014). However, our examination of the transcriptomic responses produced by hPTH(1-34) and bPTH(7-34) in six different tissues in vivo reveals a striking degree of functional distinction between a conventional ligand and an arrestin-selective agonist targeting the same receptor. Whereas signaling by native hormones is balanced to meet the needs of the organism, biased agonists, by definition, alter the distribution of active receptor states. The resulting signals may not be qualitatively different, but they are unbalanced compared with the native ligand, which can lead to profound, and unpredictable, differences at the tissue level.
This unpredictability poses a challenge for drug design, in that the transcriptional effects of an arrestin-selective GPCR agonist in vivo cannot be reliably predicted from its measurable in cellula efficacy (Maudsley et al., 2012; Luttrell, 2014). One goal of this study was to test the hypothesis that examination of cross-tissue conserved bPTH(7-34) transcripts/pathways would yield insights into the physiologically relevant mechanisms of action of an arrestin-biased GPCR ligand in vivo. We therefore employed multiple convergent forms of informatic analysis to determine whether there is a conserved signature associated with the arrestin-selective agonism of the PTH1R that offers a framework for relating arrestin signaling to physiologic responses. We found that bPTH(7-34) generates a more focused and coherent transcriptomic response across multiple tissues compared with hPTH(1-34). Unlike the pluripotent ligand that is susceptible to pleiotropic cellular and systemic factors that produce tissue-specific responses, the bPTH(7-34) response, due to its limited signal transduction repertoire and lack of calcitropic effects, more likely reflects actions mediated directly via the PTH1R. The most conserved bPTH(7-34)–activated multitissue data set was strongly associated with signaling pathways and biologic processes related to cell cycle control, modulation of cell growth, somatic energy regulation, and interleukin/cytokine-mediated signaling. Importantly, these results are consistent with our independent functional genomic and biologic analysis of bPTH(7-34) actions in bone (Gesty-Palmer et al., 2009, 2013), and with emerging data in the cancer field suggesting that arrestins are critical regulators of tumor cell proliferation, survival, and metastasis (Buchanan et al., 2006; Dasgupta et al., 2006; Moussa et al., 2008; Chun et al., 2009; Lakshmikanthan et al., 2009; Li et al., 2009; Rosanò et al., 2009; Liu et al., 2011; Bonnans et al., 2012; Fereshteh, et al., 2012).
In conclusion, we have demonstrated that ligands that preferentially activate an arrestin-coupled form of a GPCR generate a highly characteristic transcriptomic phenotype that can be discerned even after chronic treatment. Our data and methodology therefore suggest the presence of a conserved generic arrestin response mechanism for GPCRs. Eventual identification and screening for this arrestin signature may facilitate the rational development of biased drugs that target arrestin signaling pathways.
Authorship Contributions
Participated in research design: Maudsley, Gesty-Palmer, Luttrell.
Conducted experiments: Maudsley, Gesty-Palmer.
Contributed new reagents or analytic tools: Martin, Maudsley.
Performed data analysis: Maudsley, Martin, Gesty-Palmer, Cheung, Johnson, Patel, Becker, Wood, Zhang, Lehrmann.
Wrote or contributed to the writing of the manuscript: Maudsley, Martin, Gesty-Palmer, Cheung, Johnson, Patel, Becker, Wood, Zhang, Lehrmann, Luttrell.
Footnotes
- Received August 5, 2014.
- Accepted January 30, 2015.
S.M., B.M., and D.G.-P. contributed equally to this work.
This research was supported by the Intramural Research Program of the National Institutes of Health National Institute on Aging; the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM095497]; the National Institutes of Health National Institute of Diabetes, Digestive and Kidney Diseases [Grant R01-DK055524]; the National Institutes of Health National Institute of Child Health and Human Development [Grant T32-HD043446]; the Arthritis Foundation; and the Research Service of the Charleston, SC Veterans Affairs Medical Center. The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- Arrb2KO
- arrestin3 knockout
- bPTH(7-34)
- bovine PTH(7-34)
- GO
- gene ontology
- GPCR
- G protein–coupled receptor
- hPTH(1-34)
- human PTH(1-34)
- KEGG
- Kyoto encyclopedia of genes and genomes
- TDA
- topological data analysis
- WT
- wild type
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}