


Visual Overview

Abstract
G protein–coupled receptor (GPCR) structural biology has progressed dramatically in the last decade. There are now over 120 GPCR crystal structures deposited in the Protein Data Bank of 32 different receptors from families scattered across the phylogenetic tree, including class B, C, and Frizzled GPCRs. These structures have been obtained in combination with a wide variety of ligands and captured in a range of conformational states. This surge in structural knowledge has enlightened research into the molecular recognition of biologically active molecules, the mechanisms of receptor activation, the dynamics of functional selectivity, and fueled structure-based drug design efforts for GPCRs. Here we summarize the innovations in both protein engineering/molecular biology and crystallography techniques that have led to these advances in GPCR structural biology and discuss how they may influence the resulting structural models. We also provide a brief molecular pharmacologist’s guide to GPCR X-ray crystallography, outlining some key aspects in the process of structure determination, with the goal to encourage noncrystallographers to interrogate structures at the molecular level. Finally, we show how chemogenomics approaches can be used to marry the wealth of existing receptor pharmacology data with the expanding repertoire of structures, providing a deeper understanding of the mechanistic details of GPCR function.
Introduction
The mechanisms by which drugs act on receptors involve a complex interplay of thermodynamic and kinetic parameters, dictated in large part by the structures of the molecules involved. Researchers studying molecular pharmacology use a wide range of techniques to elucidate the mechanisms of drug action. These include the measurement of direct interactions between ligands and receptors, such as labeled ligand-binding studies and surface plasmon resonance, functional assays to analyze signaling pathways, and monitoring conformational changes through fluorescence labeling of receptor subdomains. Decades of elegant pharmacology research have been heavily augmented in the last years by the increasing availability of crystallographic structural data, which have provided a slew of molecular models of protein-ligand complexes, often of therapeutic interest. G protein–coupled receptors (GPCRs) have exemplified this trend with a veritable explosion in the number of receptor structures in the last 8 years (Fig. 1, top panels), providing the opportunity to take an exquisite look into the atomic details of drug-binding and receptor-activation mechanisms and propelling the application of virtual screening and structure-based drug design to this family of receptors. Interestingly, many of these structures have been obtained in complex with pharmaceutically relevant drugs (Table 1).
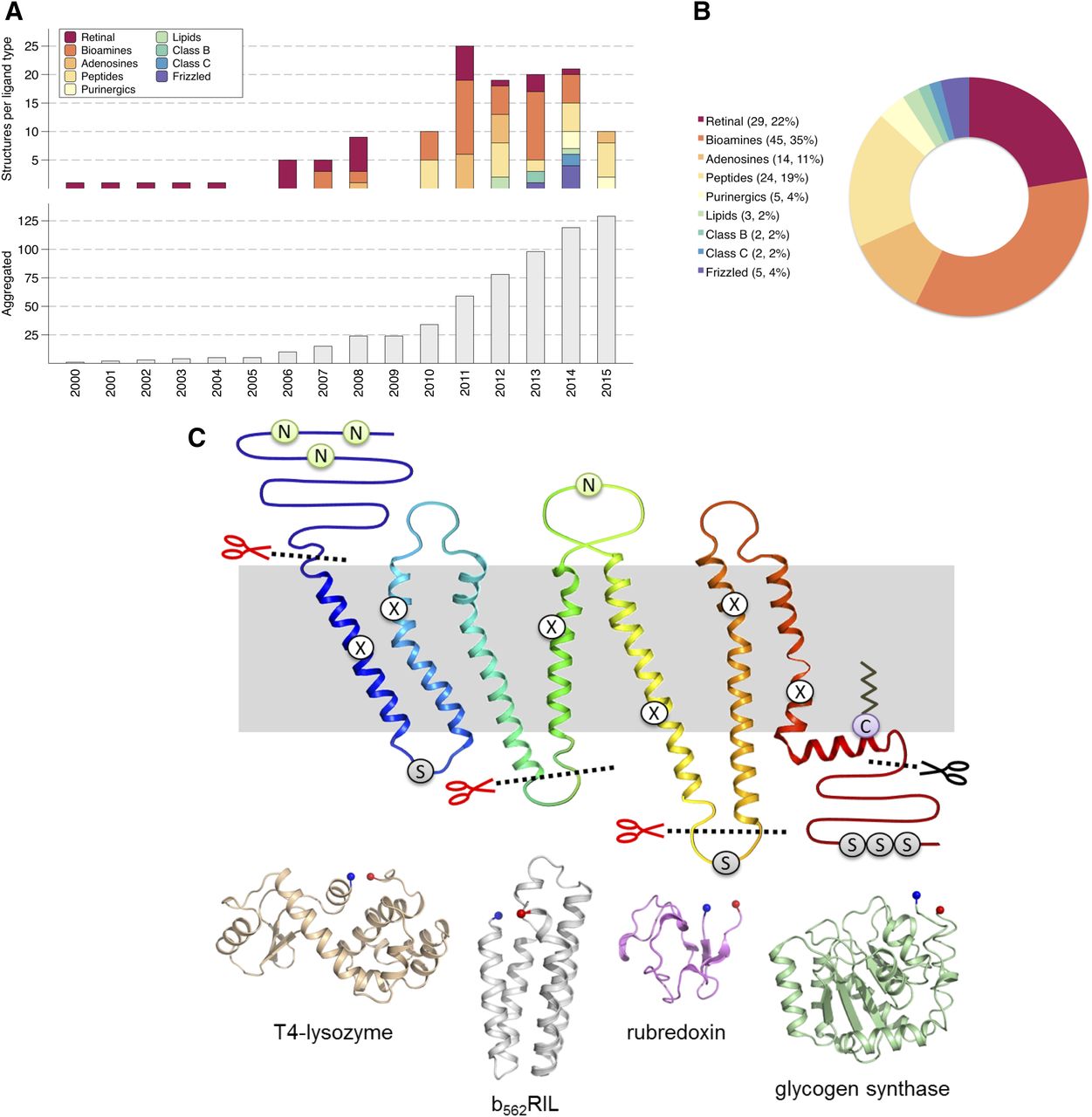
(A) Number of GPCR structures deposited in the Protein Data Bank per year (top) and total (bottom) as in July 2015. (B) Solved GPCR structures in each main class and ligand family. (C) The two-dimensional schematic diagram shows a summary of the main receptor modifications that enable structure determination of GPCRs. Sites of N-linked glycosylation are represented by the letter N; these are typically removed through mutagenesis or enzymatic digestion. Phosphorylation sites are indicated by the letter S, and palmitoylation sites are indicated by the letter C. Mutations that improve expression or stability are represented by the letter X, and have been found throughout the transmembrane domains and loops. Sites of receptor truncation are shown by dotted lines, and sites of protein fusion are indicated by red scissors. The fusion proteins used, either as an N-terminal tag, ICL2 or ICL3 fusion, are shown below with N and C termini represented as blue and red spheres, respectively (T4 lysozyme, PDB ID 3NY8; b562RIL, PDB ID 1M6T; rubredoxin, PDB ID 4MBS; catalytic domain of glycogen synthase, PDB ID 4S0V).
List of pharmaceutically relevant compounds cocrystallized with GPCRs
A prerequisite to attempt solving the structure of a protein by X-ray crystallography is the preparation of large quantities (milligrams) of purified, stable, and homogeneous protein. GPCRs have been historically hard targets for structural biology due to the difficulty to obtain samples that satisfy these ideal conditions (Kobilka, 2013). However, over the last 10 years the structural biology of GPCRs has advanced dramatically with the emergence of new technologies and techniques to tackle the problems of receptor instability and homogeneity. Perhaps the most important of such advances has been the use of protein engineering (e.g., mutagenesis, truncations, and the creation of chimeric constructs) and the generation of receptor-specific protein-binding partners for cocrystallization (such as conformational antibodies (Rasmussen et al., 2007; Hino et al., 2012), camelid antibody fragments (nanobodies) directed against the receptor (Rasmussen et al., 2011a) or the G protein to stabilize the active state ternary complex (Rasmussen et al., 2011b)). Furthermore, the discovery of new detergents for protein solubilization (Chae et al., 2010; Cho et al., 2015), the development of novel crystallization techniques such as lipidic cubic phase (LCP) crystallization (Caffrey, 2015), and key advances in several aspects of crystallography, such as microcrystallography (Moukhametzianov et al., 2008), X-ray free electron lasers (XFELs), and serial femtosecond crystallography (SFX) (Liu et al., 2013) have also contributed decisively to the field of GPCR structural biology.
It is important to realize that despite the outstanding value of three-dimensional (3D) GPCR structures for understanding the mechanisms of drug action at the molecular level, they must be viewed primarily as molecular models fitted to crystallographic data. The accuracy of these models, especially at the level of individual amino acid side chains, ligands, water molecules, and polar networks is completely dependent on the quality and completeness of the measured X-ray diffraction data sets. As a general rule, 3D structures should be backed up with pharmacologic data to verify the models and provide a broader context to the analysis of ligand-receptor interactions. Such approaches can provide new insights into the mechanisms of GPCR activation and form the basis for structure-based drug design (Congreve et al., 2014).
In this review, we provide an overview of the innovations in protein engineering and crystallography that have led to the recent successes in GPCR structural biology. We first outline the molecular biology techniques currently used to facilitate GPCR crystallography, discussing the rationale behind each type of receptor modification and how these can influence the information that can be obtained from the resulting structure. We then present an overview of the process of structural determination by X-ray crystallography, focusing on the concepts more relevant to molecular pharmacologists. The second section provides some guidance for assessing crystallographic information and interpreting the quality of structural models. Finally, we show an example of how structural and pharmacologic data can be combined using chemogenomics techniques to gain a deeper understanding of GPCR molecular pharmacology.
Molecular Biology Approaches to Facilitate GPCR Crystallography
As with soluble proteins in the early days of crystallography, the structures of the first GPCRs were solved thanks to the natural advantages that they offered to the crystallographer. Specifically, the first structures of a GPCR, bovine rhodopsin, were obtained through isolation of large quantities of the receptor from a natural source—retinal rod outer segments (Palczewski et al., 2000; Li et al., 2004). Later, crystallization of squid rhodopsin was achieved through a similar route (Murakami and Kouyama, 2008). In addition, rhodopsin is surprisingly stable when solubilized from the membrane using short-chain detergents, making it viable for vapor diffusion crystallization (Standfuss et al., 2007). Unfortunately, other GPCRs cannot be obtained as easily as rhodopsin and need to be overexpressed in recombinant systems, often leading to heterogeneity due to posttranslational modifications. Furthermore, unlike rhodopsin, most GPCRs are highly dynamic and unstable upon solubilization. Herein we describe the molecular biology approaches used by crystallographers to overcome these difficulties and optimize GPCR constructs for successful crystallization (Fig. 1, bottom panel).
Heterogeneity from Posttranslational Modifications.
The choice of expression system has important consequences for the characteristics of the expressed protein (Tate and Grisshammer, 1996), and expression of GPCRs using heterologous systems such as bacteria, yeast, insect or mammalian cells, or cell-free systems has had varying degrees of success (Milic and Veprintsev, 2015). The folding and maturation pathways of membrane proteins differ among these expression organisms. For instance, bacterial systems do not produce N-linked glycosylation, which could be the reason why functional expression of GPCRs in bacteria has had only limited success; N-linked glycosylation is cotranslational and, therefore, can be important for the expression, folding, and cell surface localization of GPCRs.
In mammalian cells, the glycosylation process occurs in the endoplasmic reticulum and the Golgi apparatus as the receptor is trafficked through the cell (Hossler et al., 2009). The glycosylation process is very complex, resulting in varying combinations of branched glycans attached to specific extracellular sites. Also, the more glycosylation sites a receptor has, the higher its potential heterogeneity. However, numerous studies have shown that simply eliminating receptor glycosylation either by mutagenesis or by chemical inhibition of the glycosylation machinery in eukaryotic expression systems results in poorly trafficked receptor and retardation in the endoplasmic reticulum and Golgi apparatus (Lanctot et al., 2006; Chen et al., 2010a; Norskov-Lauritsen et al., 2015). On the other hand, deglycosylation of the N terminus of GPCRs has not been shown to affect their pharmacology (Shimamura et al., 2011; Haga et al., 2012; Kruse et al., 2012).
The role of glycosylation on the extracellular loops of GPCRs may be more complicated. For instance, deglycosylation of extracellular loop 2 in protease-activated receptor type 1 (PAR1) has been shown to enhance the maximal signaling response (Soto and Trejo, 2010), possibly due to interactions between the sugar group and the ligand that influence the stability of the active receptor conformation. Unfortunately, complex sugar groups are usually not resolved in X-ray structures due to their flexibility, unless they form crystal contacts (Palczewski et al., 2000; Crispin et al., 2009).
Other posttranslational modifications, including phosphorylation and palmitoylation, are also sometimes removed for GPCR crystallography (Warne et al., 2008). Reversible phosphorylation occurs at the C terminus as well as the intracellular loops (ICLs) in response to receptor activation, and it is important for receptor desensitization and internalization (Tobin, 2008; Nobles et al., 2011). Phosphorylation is mediated through a number of different protein kinases, and different patterns of phosphorylation can occur in different tissue types. This leads to the possibility that phosphorylation influences the signaling capabilities of receptors in different tissues.
Palmitoylation occurs at the C terminus, typically at conserved cysteine residues present in helix 8, although some receptors are palmitoylated at multiple sites in the C terminus (Zuckerman et al., 2011). Palmitoylation is also reversible and serves to anchor the C terminus of the receptor to the lipid bilayer, effectively creating a fourth, and sometimes fifth, ICL. This structural rearrangement of the C terminus has been linked to biased signaling as well as to the dynamics of receptor phosphorylation and desensitization (Zuckerman et al., 2011). Removal of these modifications in most GPCR structures has usually been as a consequence of truncations to reduce flexibility of unstructured regions. Helix 8 is usually retained due to its role in GPCR signaling, although some receptors, such as metabotropic glutamate receptor 5 (mGlu5R) (Doré et al., 2014) and corticotropin-releasing factor receptor 1 (CRF1R) (Hollenstein et al., 2013), have been solved with helix 8 partially or completely removed.
Of the ∼120 GPCR crystal structures deposited in the Protein Data Bank (PDB) (Berman et al., 2000) (www.pdb.org), 54 correspond to unique sequences of 30 different nonrhodopsin receptors (e.g., the turkey β1 adrenergic receptor [β1AR] has been solved using three different constructs; see Supplemental Table 1). Of these sequences, 51 have some form of receptor truncation (Supplemental Table 1): 43 constructs have been crystallized with C-terminal truncations, 32 have been truncated at the N terminus, sometimes removing whole domains (Hollenstein et al., 2013; Siu et al., 2013; Doré et al., 2014; Wu et al., 2014), and 35 were crystallized with shortened ICLs (Warne et al., 2008; Zou et al., 2012; Egloff et al., 2014).
Mutagenesis and Conformational Stabilization.
Site-directed mutagenesis is an additional tool used to improve GPCR crystallizability. In addition to being used to remove sites of posttranslational modifications, mutagenesis is also used to enhance expression. For instance, mutations C1163.27L in the β1AR (Warne et al., 2008), E1223.41W in the β2AR (Hanson et al., 2008), I1353.29L in the κ-opioid receptor (Wu et al., 2012), and D2947.49N in the P2Y12 receptor (P2Y12R) (Zhang et al., 2014) resulted in phenotypes with an increased level of expression of functional receptor (Supplemental Table 1).
But perhaps the most creative use of site-directed mutagenesis in the field of GPCR crystallography has been its application to conformational stabilization. To crystallize GPCRs, the receptor molecules must first be extracted from the lipidic membrane using detergents. However, GPCRs are typically unstable in detergent solution. In addition, GPCRs are highly dynamic proteins, existing in a range of conformational states between inactive (R) and active forms (R*) (Lohse et al., 2014), which further hinders crystallization. The inactive conformation of the receptor is a low-energy state, and therefore a more stable form of the receptor. This is one of the reasons why the majority of GPCR structures have been solved in the inactive conformation in the presence of a stabilizing antagonist or inverse agonist (Ghosh et al., 2015). Some receptors are inherently more stable, and the formation of a ligand complex together with favorable binding kinetics is enough to stabilize the receptor for crystallization (Cherezov et al., 2007; Shimamura et al., 2011). In other cases (Wu et al., 2012), where the low stability of the receptor precluded purification of functional protein or sufficiently stabilizing ligands were unavailable, the receptor is stabilized by mutagenesis.
The first receptor to be stabilized using this approach was the turkey β1AR in the inactive conformation (Warne et al., 2008). The stabilized receptor contained six mutations and showed similar binding affinities to the wild-type receptor for antagonist ligands but a reduced affinity for agonist ligands, indicating that the receptor had been conformationally stabilized toward the R state. Comparison of the turkey β1AR structure to that of the homologous human β2AR inactive structures (solved without stabilizing mutations) (Cherezov et al., 2007; Rasmussen et al., 2007) showed few differences, even around the mutated positions (Warne et al., 2008).
Adenosine 2A receptor (A2AR) is another case of a GPCR stabilized using this approach, and it is the only example of a receptor solved using the same ligand with and without conformational stabilization by mutagenesis (Jaakola et al., 2008; Doré et al., 2011). Comparison of the structure around the stabilizing mutations did not show local perturbations (Doré et al., 2011), despite the receptors having different overall conformations, especially in the regions of transmembrane helices (TMs) 5 and 6. Consistent with the pharmacologic activity of the two constructs, the stabilized mutant is apparently locked into the inactive state, whereas the nonstabilized construct appears to be more conformationally flexible (Jaakola et al., 2008).
The molecular mechanisms by which certain point mutations result in thermostabilization are not yet fully understood (Tate and Schertler, 2009; Tate, 2012; Heydenreich et al., 2015). While such mutations are, to some extent, transferrable between close orthologs (Serrano-Vega and Tate, 2009), successful thermostabilizing mutations are difficult to predict, and computational methods are being developed to address this issue (Chen et al., 2012; Lee et al., 2014).
The structures mentioned here that are stabilized by mutagenesis correspond to inactive states. In an effort to crystallize an active conformation of the β2 adrenergic receptor (β2AR), a mutation was introduced into the ligand-binding site that enabled the covalent binding of a designed agonist, FAUC50 [(R)-5-(2-((4-(3-((2-aminoethyl)disulfanyl)propoxy)-3-methoxyphenethyl)amino)-1-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one] (Rosenbaum et al., 2011). However, the obtained structure retained an inactive conformation, indicating that agonist binding alone may not be sufficient for the stabilization of the active state. Indeed, when stabilization by mutagenesis was used to obtain the structures of agonist-bound A2AR (Lebon et al., 2011) and neurotensin 1 receptor (NTS1R) (White et al., 2012), this also resulted only in partially active states (Deupi, 2014). Remarkably, through the use of the large and highly potent agonist UK-432,097 [6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxytetrahydrofuran-2-yl)-N-(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide], the structure of A2AR (fused to T4 lysozyme [T4L]) was solved in an active-like conformation without the requirement of additional stabilization (Xu et al., 2011). However, despite these structures revealing the interactions involved in agonist binding, they are unlikely to represent the fully activated G protein–binding conformation comparable to the structure of active rhodopsin bound to the C-terminal G protein peptide (Choe et al., 2011; Deupi et al., 2012), or the structure of active β2AR bound to the Gαs protein (Rasmussen et al., 2011b).
Finally, site-directed mutagenesis for covalent trapping of ligand-receptor complexes has also been used in the elucidation of the structures of the murine μ-opioid receptor in complex with a morphinan antagonist (Manglik et al., 2012), and C-X-C chemokine receptor type 4 (CXCR4) in complex with a viral chemokine (Qin et al., 2015), among others (Weichert and Gmeiner, 2015).
The mutations we have discussed have no effect on the structure, but it has been observed that mutagenesis may sometimes have a dramatic effect. For instance, the structures of three similar constructs of CXCR4 with and without a T2406.36P mutation have shown that it caused the disruption of a short section of helix 6, effectively uncoupling ligand binding from receptor activation (Wu et al., 2010). To date, structures of 14 different receptors have been obtained using mutagenesis to stabilize a particular conformation of the receptor (Ghosh et al., 2015) (Supplemental Table 1).
Chimeric Constructs.
The most successful protein engineering technique to obtain crystalizable GPCRs has been the creation of chimeric constructs in which the receptors are genetically fused to a soluble protein. In such chimeric receptors, the fusion protein replaces an intracellular loop or is added as a tag to the N terminus (Wang et al., 2013b; Wu et al., 2014; Fenalti et al., 2015), providing a large and stable domain that favors the formation of crystal contacts. The fusion proteins themselves are highly crystalizable and feature N and C termini at the right distance for their insertion into GPCR loops without resulting in a significant distortion of the transmembrane bundle (Chun et al., 2012). These fusion proteins are typically T4L (Cherezov et al., 2007; Jaakola et al., 2008; Wu et al., 2010) or a thermostabilized apocytochrome (b562RIL) (Chun et al., 2012; Liu et al., 2012; Zhang et al., 2014), but more recently other proteins have also been used successfully as ICL3 fusions, such as the catalytic domain of Pyrococcus abyssi glycogen synthase in the orexin 2 receptor (OX2R) (Yin et al., 2015), or rubredoxin in C-C chemokine receptor type 5 (CCR5) (Tan et al., 2013) and the P2Y1 receptor (Zhang et al., 2015a) (Supplemental Table 1).
Such dramatic protein engineering is still required in many cases to obtain crystalizable GPCR constructs, which naturally raises questions about the reliability of the obtained structures. To address this concern, many efforts have been made to determine the effect of protein fusions on receptor activity. In β2AR and A2AR, T4L insertion into ICL3 does not appear to constrict the conformational changes associated with activation, as this fusion results in a higher affinity for agonists, a property associated with constitutive activity (Rosenbaum et al., 2007; Jaakola et al., 2008). Furthermore, although this fusion impedes coupling to the G protein, a fluorescence-based assay detects conformational changes in TM6 of β2AR that are consistent with agonist-induced movements upon activation (Rosenbaum et al., 2007).
It has been suggested that, in these cases, the fusion results in changes in the cytoplasmic side of TM6 that perturb an intramolecular ionic interaction (ionic lock) that stabilizes the inactive state of some class A GPCRs (Chien et al., 2010; Doré et al., 2011; Preininger et al., 2013). Supporting this idea, a structure of A2AR solved with the same ligand but without an ICL3 fusion did indeed show the presence of the ionic interaction (Doré et al., 2011). On the other hand, replacement of T4L by b562RIL in ICL3 produced a structure closer in conformation to the inactive state, although the ionic interaction was not fully formed (Liu et al., 2012). The insertion of b562RIL into ICL3 of the smoothened receptor has also been proposed as a reason for the lack of structural rearrangements at the cytoplasmic surface upon agonist binding (Wang et al., 2013b). Finally, comparison of the murine δ-opioid receptor structure solved with an ICL3 T4L fusion (Granier et al., 2012) and the human δ-opioid receptor with an N-terminal b562RIL fusion (Fenalti et al., 2014) shows a high degree of structural similarity, with the main deviations occurring proximal to the sites of fusion.
In summary, creation of fusion chimeras has proven a very successful strategy, greatly accelerating our knowledge of GPCR structure across a wide range of receptors. However, it is important to keep in mind that the use of this technique may introduce some artifacts in the obtained structures.
Cocrystallization Tools.
An additional strategy to facilitate structure determination of GPCRs is the use of crystallization chaperones to form stable complexes and/or trap the receptor in a given conformation. For instance, monoclonal antibody fragments (Fabs) have been used to determine the structure of β2AR (Rasmussen et al., 2007) and A2AR (Hino et al., 2012) in the presence of inverse agonists. Like in the fusion strategy, the Fabs create an extended hydrophilic surface area to mediate crystal contacts and reduce the flexibility at the receptor surface.
Fab5, directed against β2AR, binds to a structural epitope on ICL3 (one of the most structurally dynamic regions in many GPCRs) but does not affect the ligand-binding properties of the receptor (Day et al., 2007). However, the crystal structure of the β2AR-Fab5 complex bound to an inverse agonist showed an apparent intermediate conformational state that may have been influenced by Fab-mediated crystal packing constraints.
For A2AR, on the other hand, Fab2838 is conformationally selective for the antagonist-bound state, abrogating agonist binding while retaining wild-type antagonist pharmacology (Hino et al., 2012). Fab2838 binds to a similar pocket on the cytoplasmic side of the receptor to that used by the C-terminal α-helix of Gαs upon activation. However, Fab2838 binding results in an inactivated receptor by locking TM3, TM6, and TM7 together.
The development of G protein mimetics has enabled crystallographers to capture the active state of certain GPCRs. Specifically, nanobodies (Nbs), the recombinant antigen-binding domain of camelid heavy chain antibodies, are only a quarter of the size of conventional Fab fragments and very efficient at mimicking G proteins (Steyaert and Kobilka, 2011). For instance, immunization of a llama with purified agonist-bound β2AR generated a nanobody (Nb80) that recognized specifically the active state of the receptor. Interestingly, Nb80 shows similar attributes to Gαs with respect to its influence on agonist affinities and on the conformational changes stabilized in the receptor (Rasmussen et al., 2011a). Importantly, while the β2AR-T4L fusion construct was unable to activate G protein signaling, presumably due to steric clashes between T4L and the G protein (Rasmussen et al., 2007), Nb80 is small enough to stabilize the active state of the receptor even with T4L inserted into ICL3 (Rasmussen et al., 2011a). Further engineering of Nb80 resulted in the creation of a higher affinity nanobody (Nb6B9) that was used to solve structures of β2AR in the presence of a range of agonists, including some with low affinity (Ring et al., 2013). These structures revealed that different agonists can stabilize similar conformational changes during receptor activation using a different set of ligand-receptor interactions.
Nanobodies have also been used to crystallize the M2 muscarinic acetylcholine receptor in an active conformation with and without a positive allosteric modulator (Kruse et al., 2013a), and the constitutively active viral GPCR US28 in complex with the human chemokine domain fraktalkine (CX3CL1) receptor (Burg et al., 2015). But the most important application of nanobodies to GPCR structure determination has been their use in the elucidation of the complex between β2AR and the Gαs protein (Rasmussen et al., 2011b). This impressive feat, however, was only possible through the combined use of many of the techniques discussed here (Supplemental Table 1).
Crystallization Techniques.
GPCRs, as integral membrane proteins, must be extracted from the lipid bilayer using detergents before their purification. Outside the membrane environment GPCRs are typically unstable and tend to unfold unless they are stabilized by ligand binding, protein engineering, or mutagenesis, as previously discussed. Classic approaches to crystallography using the vapor diffusion technique are usually unsuitable for many GPCRs because they have a relatively small hydrophilic surface area and the long-chain detergents used for solubilization form large micelles that generally occlude the polar surfaces available to form crystal contacts.
The dichotomy of requiring short-chained detergents with small micelles to expose the receptor for crystallogenesis and the need to maintain folded functional receptor in such harsh conditions reduces the chances of success. Indeed, only four nonrhodopsin GPCRs have been solved using vapor diffusion crystallography, and these required either thermal stabilization (Warne et al., 2008; Doré et al., 2011; Egloff et al., 2014) or the use of cocrystallization tools such as Fabs (Hino et al., 2012) (Supplemental Table 1). However, whether this is because vapor diffusion crystallography has fallen out of fashion or if it is truly inhibitory is still debatable. New detergents such as neopentyl glycols (Chae et al., 2010), ganglio-tripod amphiphiles (Chae et al., 2014), and steroidal amphiphiles (Lee et al., 2013), some of which have the unique property of stabilizing nondenatured GPCRs when diluted below their critical micelle concentration, may aid further crystallization efforts.
Nevertheless, the majority of GPCR structures have been solved using LCP crystallization (for a comprehensive review, see Caffrey, 2015). In this method, the receptor is crystallized in a lipidic environment instead of from a detergent solution. The membrane-like environment of LCP is more stabilizing than detergents, so it is advantageous for crystallizing unstable receptors. However, due to the nature of the lipidic phase, crystal nucleation is slower compared with vapor diffusion, and crystals typically take longer to grow. Consequently LCP crystals tend to be small (10–30 μm) and pose challenges to their isolation and to obtain diffraction (Liu et al., 2014).
These drawbacks have been overcome by the development of microfocus beamlines and high-energy synchrotron sources, which have enabled data to be extracted from microcrystals. However, the radiation damage caused to such small crystals makes collection of high-resolution data difficult. The recent emergence of XFELs is allowing these challenges to be overcome. XFELs are capable of generating ultrafast pulses of X-rays at intensities several orders of magnitude above the brightest synchrotron sources (Chapman et al., 2011; Spence et al., 2012), enabling data collection from small protein crystals before the sample is damaged (and eventually destroyed) by the power of the beam. Hence, the crystals do not require cryoprotection, and data collection can be performed at room temperature.
When a continuous stream of crystals is supplied to the XFEL beam, a data set can be compiled from hundreds of thousands of diffraction images, which forms the basis of SFX. This method was used to determine the structure of serotonin 2B receptor (5-HT2BR) using crystals formed in LCP (Liu et al., 2013). Comparison of this structure to that of the same protein determined using traditional X-ray crystallography techniques (Wacker et al., 2013) showed a remarkable agreement, with only small differences in the loops, termini, and a few side chain rotamers.
As SFX allows diffraction data to be obtained at room temperature and in a lipidic environment, it can be argued that it provides a truer depiction of the native state of a receptor (Liu et al., 2013). The use of SFX in structure determination of GPCRs has since been validated in additional receptors, including smoothened (Weierstall et al., 2014), the δ-opioid receptor (Fenalti et al., 2015), and the AT1 angiotensin receptor (Zhang et al., 2015b). Further developments of this technique may enable the study of receptor kinetics within crystals and capture short-lived conformational states during activation (Barty et al., 2013; Kern et al., 2013).
Nonligand Molecules Present in Structures.
It is sometimes overlooked that the natural environment of the receptor in vivo, including the lipid bilayer and its components, affects GPCR activation and signaling. Lipids can affect the function of membrane proteins in a number of ways, either through direct interaction or by altering the physical properties of the membrane environment, such as bilayer thickness, curvature, or lateral pressure (Oates and Watts, 2011). In addition, many GPCRs have been shown to localize in certain regions of the cell membrane through association with lipid rafts, mediated by interactions with the palmitoylated cysteines of helix 8 (Chini and Parenti, 2004).
Lipid molecules and cholesterol have been observed in the crystal structures of many GPCRs (Cherezov et al., 2007; Jaakola et al., 2008; Manglik et al., 2012; Wu et al., 2014; Zhang et al., 2014). Due to the nature of protein crystals, the presence of a lipid molecule suggests a favorable interaction between the lipid and the receptor, although it is not clear whether these interactions are incidental associations or whether the interaction modulates receptor function.
In a structure of inactive β2AR, cholesterol was a prerequisite for crystallogenesis, and thus it was added in excess during crystallization (Cherezov et al., 2007). Accordingly, cholesterol was found to mediate parallel associations of receptors in the crystal lattice. A subsequent structure with a different crystal lattice showed the same cholesterol binding sites located in a shallow groove between TM1 and TM4, but in this case they did not participate in crystal contacts (Hanson et al., 2008). This observation led to the discovery of a putative cholesterol binding consensus motif present in almost half of all class A GPCRs (Hanson et al., 2008).
Other cholesterol binding sites suggested from modeling studies have been later observed in the structures of A2AR and μ-opioid receptor (Jaakola et al., 2008; Manglik et al., 2012; Cang et al., 2013). Interestingly, an additional cholesterol binding site was predicted in the β2AR at the top of TM1 and TM7 that could potentially influence ligand binding (Cang et al., 2013). Recently, a cholesterol molecule was found in the structure of P2Y12R in a similar position (Zhang et al., 2014).
Depletion of cholesterol from lipid bilayers has been shown to change the biased signaling properties of β2AR, from signaling through Gαs/Gαi to predominantly through Gαs (Xiang et al., 2002; Cherezov et al., 2007). However, it is uncertain whether this effect stems from a direct interaction between cholesterol and receptor, or from colocalization effects of cholesterol on the receptor with specific G proteins (Pontier et al., 2008). In addition, β2AR has been shown to form homodimers in vivo (Angers et al., 2000), and studies have shown that cholesterol may also affect the way β2AR molecules assemble into dimers, leading to the intriguing idea that cholesterol could modulate receptor function by changing the way GPCRs associate in the bilayer (Prasanna et al., 2014). However, if one entertains the idea of molecules such as cholesterol acting as modulators of receptor activity, one must also consider that absence of these molecules in receptor structures for which their natural environment is cholesterol-rich may result in an incomplete representation of the receptor.
GPCR X-Ray Structure Determination
The previous section outlined the recent techniques and approaches that have been used to obtain stable and purified GPCRs and grow crystals suitable to obtain X-ray diffraction data in synchrotrons or XFELs. In this section, we describe briefly how such diffraction data are translated into the final 3D structures that allow researchers to map the interactions between drugs and receptors. Comprehensive introductions to macromolecular crystallography for a general scientific audience can be found in excellent books on the subject by Rhodes (2006) and Rupp (2009).
X-Ray Crystallography.
In a nutshell, structural determination by X-ray crystallography involves measuring the directions and intensities of X-rays diffracted by the electron clouds of the molecules in the crystal, then using computer software to reconstruct a map of the electron density. In an iterative refinement process, the crystallographer builds a 3D model of the protein that fits the electron density while being consistent with the prior knowledge of general protein structure and on the protein that has been crystallized. It is important to keep in mind that the final 3D structure, which is all that most of noncrystallographers see, is a model representing the best fit of the protein atoms to the electron density map. Thus, to assess the quality of this model, it is useful for users of these models to be familiar with some basic concepts of structure determination by X-ray crystallography. This section is not meant to be a comprehensive guide to X-ray crystallography; rather, we provide just a brief conceptual overview, highlighting a few key points in the process along the way that are important for the critical use of crystallographic structure models.
Crystals and Diffraction
In a protein crystal, the molecules are arranged in an array of repeating elements called unit cells. The unit cells forming the crystal and the contents of the unit cells themselves are held together mainly by protein-protein contacts, but the molecules are loosely packed, and the solvent content in the crystal is very high (around 60% in GPCR crystal structures). The contacts mediating the crystal formation are generally weak and are not always biologically relevant. Protein-protein interactions in crystal structures should always be carefully evaluated with additional experimental data before drawing conclusions about their physiologic relevance, especially if the interface is small or nonconserved. As an example, in class A GPCR structures, contacts between transmembrane regions observed in the crystal structures so far may in some instances resemble the biologic interfaces that are hypothesized to be present in GPCR oligomers, but they are not definitive proof of physiologically relevant dimeric structures (Duarte et al., 2013).
When crystals are exposed to a beam of X-rays, the lattice-like array of unit cells causes the scattered radiation to be amplified into discrete spots (reflections) at specific angles relative to the incoming X-ray beam (Bragg and Bragg, 1913), resulting in a diffraction pattern measured by a detector (Fig. 2). The diffracted X-rays are waves and thus characterized by three parameters: amplitude, frequency, and phase. These parameters, together with the arrangement of the spots relative to each other on the detector, provide the details necessary to construct an “image” of the unit cell. The amplitude is recorded as intensity values on the detector pixels; the square root of the intensity value is simply the amplitude of the wave. The frequency of each reflection is related to the angle at which the reflection exits the crystal.

Main steps in the process of structure determination by X-ray crystallography.
Conceptually, the exact angle that the reflection exits the crystal is related to the resolution of the diffracted beam; higher angle reflections (recorded farther from the center of the detector) arise from diffraction of finer slices of the unit cell and thus bring higher resolution information toward the reconstruction of its contents. The phase of each reflection is, unfortunately, not recorded by the detector and therefore must be estimated in some way.
Finally, the spacing and relative arrangement of the reflections captured by the detector are related to the size and symmetry of the unit cell. The distance between reflections measured along diffraction axes are inversely proportional to the dimensions of the unit cell (e.g., closely spaced reflections measure out a large unit cell axis and vice versa). The symmetry of the molecular arrangements within the unit cell is also reflected in the diffraction data, which aid in the calculations as symmetry-related reflections can be averaged together to increase the signal-to-noise ratio of the data set.
Data Collection.
From a 3D crystal, reflections are recorded as the crystal is rotated to capture a complete data set. The recorded reflections on individual frames are then indexed with a coordinate value h,k,l (the so-called Miller index), the intensities are integrated, and finally the intensity values of the frames are scaled together, adjusted for systematic errors, and symmetry-related or multiply-measured spots are merged. The result is a table containing several thousand or more unique reflections, with intensity (I), standard deviation (σ), and Miller index (h,k,l).
As stated previously, each reflection is a discrete wave resulting from the periodic scattering of the contents of the unit cell. The sum of these individual waves adds together to produce a complex 3D form, which is the image of the electron density of the unit cell. In practice, reflections (amplitudes plus phase estimate) are combined together by Fourier synthesis (simulating the function of the lens in a microscope) to produce the image of the unit cell. To think about it another way, the electron density distribution of the unit cell can be imagined as a complex 3D waveform (high-density peaks where protein atoms are crystallized in place, low density where disordered solvent fills the spaces between), and the diffracted reflections are the individual waves that can be added back together to reconstruct the image of this density.
The quality of the crystal largely determines the quality of the diffraction data and, in turn, the accuracy of the structural model. Good-quality protein crystals that produce well-resolved high-resolution diffraction can be difficult to achieve, particularly for highly dynamic membrane proteins such as GPCRs. When crystals of such proteins can be coaxed into existence, they are often small, resulting in faint diffraction signals, and/or are insufficiently crystalline, resulting in poor diffraction. If there is too much variation between unit cells and the protein molecules are not well ordered, the diffraction breaks down, and the reflection data become smeared, weak, and ultimately absent. For instance, crystals may exhibit a large degree of “mosaicity” (i.e., are formed by a mosaic distribution of differently oriented blocks) or may be “twinned” (i.e., have crystalline blocks specifically oriented which give rise to overlapping diffraction). In such cases, the measured diffraction pattern is much more difficult to analyze. As such, recent technological advances in crystallography have allowed researchers to more easily overcome these hurdles.
As described in previous sections, extensive genetic modification of the receptor with thermostabilizing mutations, splicing in fusion proteins such as T4L, or by cocrystallization with Fab proteins or nanobodies, has allowed GPCRs to be crystallized with sufficient quality for diffraction studies. Furthermore, developments in synchrotron microcrystallography and XFEL instrumentation have made key contributions by providing very intense focused beams of radiation that allow diffraction data to be obtained from very small crystals.
Initial Phasing.
As mentioned previously, to reconstruct the electron density map of the unit cell from diffraction data, the phases of the diffracted X-rays are required. There are several techniques to obtain this information. In the first high-resolution X-ray crystal structure of a GPCR, bovine rhodopsin, phasing information was obtained by a technique called multiwavelength anomalous diffraction (Hendrickson et al., 1985) using mercury-soaked crystals (Palczewski et al., 2000). When heavy atoms such as mercury are incorporated into protein crystals, at certain wavelengths the diffraction pattern changes slightly but significantly in a very precise way. This change in diffraction can be exploited to extract a limited set of phase information, allowing the reconstruction of electron density.
With only two recent exceptions—the smoothened receptor (Wang et al., 2014) and the metabotropic glutamate receptor 1 (Wu et al., 2014), which were also solved by soaking crystals with heavy atom solutions and employing anomalous diffraction measurements—all the remaining GPCR crystal structures (over 120 to date) have been solved by a technique called molecular replacement (MR). With MR, basically, the phases are calculated from a known protein structure that is expected to be similar and are applied together with the intensities of the diffraction data to generate an initial electron density map. If the structure of the MR model is similar enough to the unknown structure, a reasonable electron density map is produced that can be further refined. With low-resolution and weak diffraction data, there is a significant risk of introducing “model bias” into the calculated maps, whereby the model phase information dominates the calculation, resulting in essentially an electron density map of the MR model only. A clue that the initial MR-derived map is of sufficient quality is to see if it contains new features not present in the MR model, such as truncated regions or side chains. With this method, the structure of the β2AR (Rosenbaum et al., 2007), the second GPCR structure to be solved, after rhodopsin, was obtained using rhodopsin as a phase template to reconstruct the electron density. In a similar fashion, the next structure, β1AR (Warne et al., 2008), was solved using β2AR as a template, and so on.
Model Building.
The final step corresponds to building a 3D molecular model that fits in the electron density (Fig. 2). As most GPCRs structures start from an MR-derived map, the MR model itself is generally a good starting point to begin building and refinement. High-sequence homology within GPCR transmembrane regions provides a convenient base upon which the structure of the crystallized receptor can be modeled. After a set of changes to the model are made, computer programs apply energy minimization algorithms to more finely fit the atomic coordinates and displacement factors (B-factors) to the electron density, restraining the model to known chemical properties of amino acids (e.g., length and angle of covalent bonds, van der Waals contact distances) and to expected protein structure properties (e.g., peptide backbone dihedral restraints, secondary structure hydrogen bond restraints).
The degree to which restraints are applied depends on the quality of the diffraction data. Lower resolution data demand stronger restraint whereas higher resolution data can be “freed” a bit more, letting the higher certainty of the electron density guide atom placement. Thus, low-resolution structures tend to have geometrical statistics with small variance (low root mean square deviation), closer to an average “ideal” value than higher-resolution structures.
Crystal Structure Quality Metrics.
When we visualize the structure of a protein solved by X-ray crystallography, we are looking at a molecular model that has been built to fit as well as possible into an electron density map, using advanced computational methods and, in many cases, some assumptions about missing data. To effectively use these models it is important to be familiar with some of the existing metrics available to validate their quality. Crystal structure validation is absolutely essential in the process of interpreting and adapting models for further research applications (Read et al., 2011). In this process, we want to understand the quality of the data that generated the model, the stereochemical quality of the model itself, and how well the model actually fits the data, both on a global basis and the local fit of residues and ligands.
Perhaps the most familiar metric to noncrystallographers is resolution, which, as we have stated, essentially reports the highest angle reflections recorded in the diffraction pattern. A reported resolution value of, for example, 2.5 Å, states that the diffraction data set contains reflections that arose from the scattering of unit cell contents in 2.5 Å increments. Importantly, resolution in this context does not refer specifically to the precision in the position of the atoms in the structure; rather, it can be thought of as a measure of the “fineness” of the data, which gets fed into the electron density calculation and should be regarded more as a metric of the data quality, not necessarily the model quality.
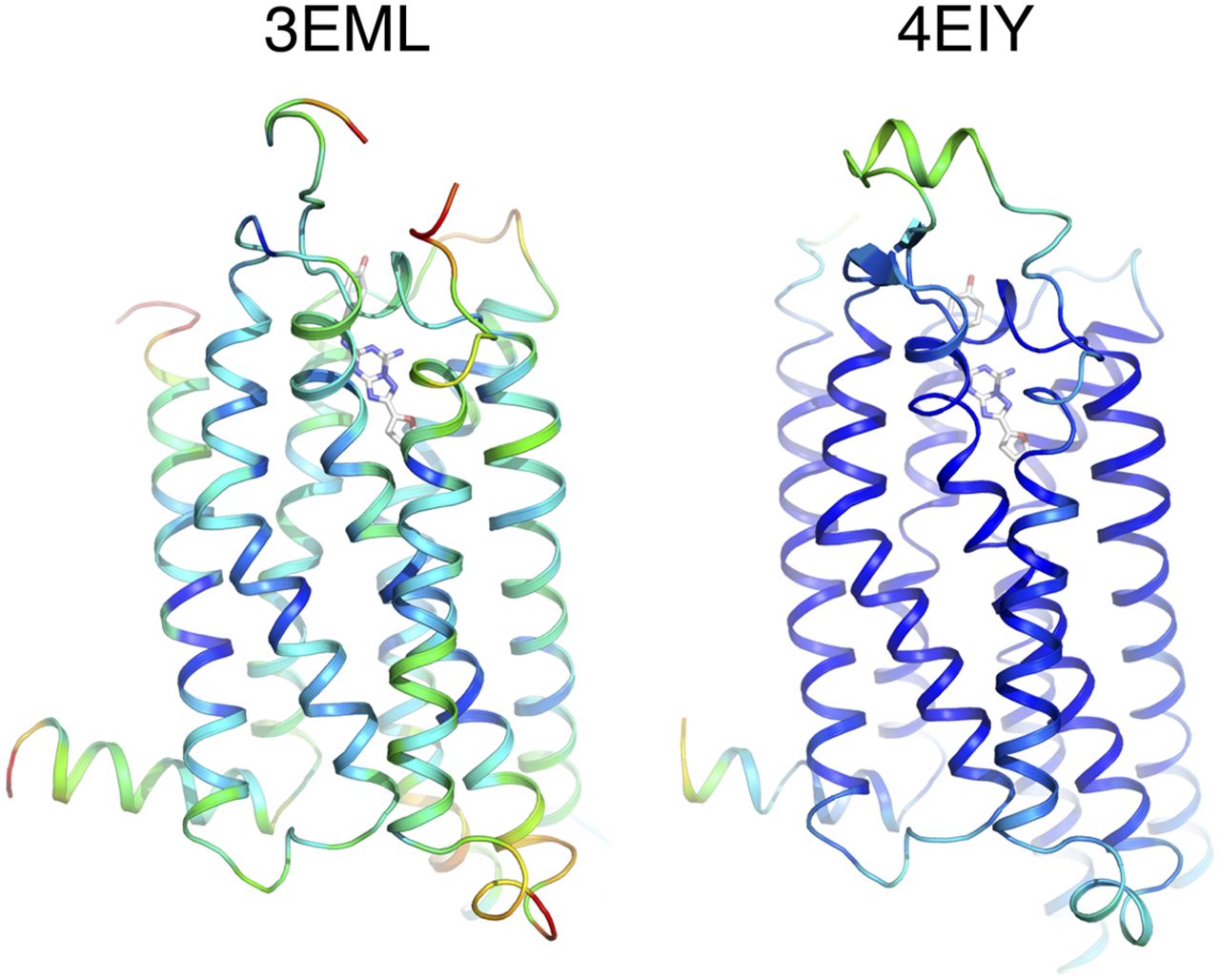
The precision in atom positions is reported as an estimated coordinate error value (ESD or ESU) and is typically between 1/5–1/10 of the resolution (Brunger, 1997); that is, a structure at a resolution of 3 Å provides a precision in the position of atoms within 0.6–0.3 Å, depending on the quality of the data. The average resolution of the solved GPCR structures is 3 Å, which, practically speaking, allows one to visualize in the electron density the basic contours of amino acid side chains and ligands. The highest resolution obtained for a GPCR is 1.8 Å (Liu et al., 2012), which allows significantly more detail to be modeled. This structure was able to include 57 ordered water molecules and a sodium ion inside the receptor, plus two cholesterol and 23 ordered lipid molecules (Fig. 3).

Electron densities in the binding site (top panels) of the A2A adenosine receptor in complex with ZM241385 [4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol] (bottom panels) solved at two different resolutions. Even at 2.6 Å resolution (left) side chains and ligand can be modeled reliably, but the electron density of water molecules at this contour level (2 σ) is too weak to be visible. On the other hand, at 1.8 Å resolution (right), the electron density of most water molecules in the binding site is clearly visible, and modeling of the side chains and ligand are unambiguous. The commands to produce this type of representation in PyMOL are provided in the Supplemental Material.
Another measure of crystal structure quality is the R-factor, which measures the agreement between the recorded diffraction data and the derived model built into the electron density. Thus, the R-factor quantifies how well the refined structure predicts the observed data. An R-factor of zero indicates perfect agreement, and an R-factor of approximately 0.54 indicates randomness, or essentially no agreement. Crystallographers realistically aim for R-factors of about 0.2 or less, if possible. In the available GPCR structures, this value ranges roughly from 0.2 to 0.4, with an average of 0.27.
R-factors are reported as two separate values: R-work and R-free. R-work is the R-factor calculated from all the data used in the refinement process. However, this statistic can become erroneously low if the model is “overfit” to the data. When refining against low-resolution data sets, if too few restraints are placed on the model, the refinement algorithms will, in a sense, take too many liberties in adjusting the coordinates and B-factors so that the calculated diffraction pattern matches the observed diffraction pattern. Thus, the R-work is not an independent and unbiased statistical indicator of model quality, and it is always in danger of being artificially low.
To guard against this, the R-free statistic was introduced. R-free is calculated from a subset of data that has been randomly selected at the very start of the process and withheld from refinement throughout the entire process. Therefore, in theory it should be “free” from model bias. If the refinement algorithms are performing properly and the model truly reflects the observed data, both R-work and R-free should be in agreement. If the model is overfit, R-work and R-free will diverge.
In practice, the R-work and R-free values may differ by about 5% points (e.g., 0.20 and 0.25, respectively) as it is nearly impossible to completely remove all bias from the procedure. Incidentally, however, if the values are too similar, this may indicate a bias in R-free, which can arise with careless application of molecular replacement phasing or if the R-free test reflections are correlated to R-work reflections by noncrystallographic symmetry elements.
While resolution, R-work, and R-free are global measures of the quality of the data and structure, temperature factors (also known as B-factors or atomic displacement factors) and occupancies are local descriptors at the atomic level. In essence, B-factors are a measure of how smeared out the electron density is for an atom, and they provide some insights into the local disorder of the molecules. Generally, loop regions or long amino acid side chains have a higher freedom of movement, which can be thought of as a certain “blurring” of the atom in space, which translates into a higher B-factor (Fig. 4, left panel).

Structures of the A2A adenosine receptor in complex with ZM241385 [4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol] colored by B-factor. The structure in the right was solved at a higher resolution and also presents a better order, which translates as a better definition (e.g., extracellular loop 2). The commands to produce this type of representation in PyMOL are provided in the Supplemental Material.
Although it is tempting to equate high B-factors with highly dynamic regions, this interpretation should be done with caution, as it is not always the case. High B-factors can arise from either dynamic disorder or static disorder. Because the electron density map is an average over all the unit cells contributing to the diffraction data, if atoms are locked into the crystal in slightly varying locations in each unit cell (static disorder), the electron density for that atom will be smeared out, and the B-factor will be high. Alternatively, if the atom is fluctuating within the unit cell (dynamic disorder), the electron density will be similarly smeared out, and the B-factor will also be high. The B-factor simply tells you how well the atom position is defined in the crystal.
Generally, the mean B-factor of a crystal structure is correlated with the resolution of the data set. Higher resolution diffraction arises from better ordered structures and a higher degree of crystallinity across all unit cells, which means that atom positions are less variable in the crystal and give rise to well defined electron density maps with low positional uncertainty and low B-factors.
For protein modeling and interpretation purposes, B-factors can be considered as a metric of uncertainty for the coordinates. High B-factors equate with high positional uncertainty. Thus, regions with high B-factors can be interpreted as being less well defined than regions with low B-factors. In molecular dynamics studies, for example, this can be related to the degree of fluctuation observed during a simulation run, especially for regions of the model where the crystal structure is not clearly constrained by crystal contacts or other artifacts of the crystal environment (e.g., bound buffer components). As such, a low root mean square fluctuation value in a molecular dynamics simulation might not always recapitulate a low B-factor observed in the crystal structure due to these environmental differences.
On the other hand, occupancies are an estimate of the fraction of the diffracting molecules in which the atom occupies the position specified in the model. For moderate- to low-resolution data sets, occupancy and B-factor are nearly impossible to refine independently. Thus, fractional occupancies are calculated and refined only for very specific cases where the reduced occupancy can be clearly supported by the electron density. This generally only occurs for highly electron-dense scatters (e.g., heavy atoms such as selenium or mercury, sometimes well ordered aromatic ligands), or for alternate conformations of amino acid side chains and loops. For instance, a side chain may exhibit two conformations, both supported by electron density. Two side chains can be built into the density, each with a fractional occupancy that adds to 1.0.
When evaluating a crystal structure, geometrical statistics should also be considered to understand how closely the model conforms to an accurate protein structure (Read et al., 2011). Covalent bond lengths and angles should be consistent with known chemical parameters, and van der Waals contacts should be within allowed distances, accounting for the placement of hydrogen atoms in the structure.
Of course, chirality of amino acids and ligands must be correct. The peptide backbone should generally contain planar peptide bonds, and the torsion angles φ and ψ should generally conform to expected values from updated Ramachandran plots. Side chain torsion angles χ should also be evaluated for outliers. A good quality average protein structure will not have outliers in any of these measurements, by definition.
It must be stressed, however, that quite often protein structures do, in fact, contain geometrical outlier values relative to the average structure as they may contain regions wound up energetically for some functional process. However, outliers in a good model must be supported by electron density. Going back to what was stated in the beginning of this section, this is why low-resolution structures should have better statistics than higher resolution structures. At low-resolution, stronger geometrical restraints are needed to prevent overfitting of the model to the map. At higher resolutions, the data are stronger to more confidently fit atoms to the electron density with less restraint.
Discerning users of protein structure models should always concern themselves with these measures. Although most of these values can be easily inspected in the corresponding entries in the PDB web site (e.g., the full validation report produced by the PDB) or in the PDB coordinate file itself (which is simply a text file that can be opened with any text editor), a better approach is to use an analysis program such as MolProbity (http://molprobity.biochem.duke.edu/) (Chen et al., 2010b). MolProbity can be run directly from a web interface and generates a thorough analysis of the protein structure. PDB files can be fetched directly from the PDB, or custom files can be uploaded. The program will add hydrogen atoms, identify whether the side chains of Asn, Gln, or His should be flipped based on hydrogen bonding patterns (a common mistake in protein models), and then perform an all-atom contact and geometry analysis.
Several clear tutorials are available at the Web site to assist the first-time user. A MolProbity analysis outputs a so-called multi-criterion chart which gives a residue-by-residue list of scores on several geometrical indicators of model quality, including all-atom contacts (clash score), Ramachandran score, Cβ deviations, side chain rotamer outliers, and general bond length and angle deviations. Although all these scores are important in evaluating a model, particular emphasis should be placed first on the all-atom contact clash score. This is a measure of the van der Waals overlaps, which must be minimized to the greatest extent possible in a well built protein structure no matter the resolution or quality of the data. The Ramachandran analysis provides a measure of the peptide backbone φ and ψ angles and how they relate to a benchmark population of high-resolution crystal structures. Most protein structures should not contain Ramachandran outliers; if they do, the presence of the outlier should be justified by strong electron density. Again, the lower the quality of the diffraction data (the harder the electron density maps are to interpret), the better the geometry statistics should be. Higher resolution data with strong electron density can provide evidence of deviations from an ideal protein structure, whereas poor data cannot justify outliers. MolProbity also outputs kinemage files and can be operated with the program Coot to provide a more graphic presentation of flagged outlier regions for closer inspection.
B-factors and occupancies can be visualized in a molecular graphics program (e.g., PyMOL; Schrödinger LLC, https://www.pymol.org/) by coloring each residue according to these values (Fig. 4). Coloring by B-factors will give an idea of which regions of the receptor are more disordered and probably have weaker electron density, while coloring by occupancy will highlight residues that were modeled with alternate conformations.
Analysis of Electron Density Maps.
For a more detailed analysis, electron density should be inspected. Maps calculated from deposited data sets can be obtained easily for most structures, in lieu of sophisticated crystallographic software packages, from the Electron Density Server (EDS, http://eds.bmc.uu.se/eds/) (Kleywegt et al., 2004). This Web service provides a summary analysis of the data and model, and can generate analytic plots for evaluating the electron density data. Particularly illustrative is the real-space R-factor plot, showing a residue-by-residue calculation of the fit of the model to the map. This can highlight potentially troubling areas of the model that warrant further inspection.
Of course, the best way to understand how the model fits the data is to look at the map itself. For this, the EDS can calculate so-called σ-A weighted maps, which are the most common type of maps crystallographers use for model building. Two useful flavors of these maps are the “standard” 2mFo-DFc map, and the “difference” mFo-DFc map. The standard 2mFo-DFc map shows, essentially, the experimental electron density map into which the crystallographer has built the model. Exploring this map is useful to verify the overall quality of the map, and how different regions of the model may be built into weaker or stronger density. The difference map mFo-DFc shows the residual electron density after subtracting the calculated model density from the experimental (observed) density. This is useful to highlight errors in the model. Substantial positive electron density peaks suggest an incomplete model (e.g., missing atoms) whereas negative electron density peaks show areas where the model is not supported by experimental density.
A useful derivative of the difference map is the “omit” map, whereby a small region of the model is purposely deleted, then the mFo-DFc map is recalculated (sometimes after performing a bit of simulating annealing dynamics to reduce model bias—this is then commonly referred to as an “SA-omit” map). Omit maps are routinely used to validate the placement of ligands in crystal structures. If the omitted region is highlighted by strong positive density peaks, this is good evidence that the model is correct. However, if the difference density for the omitted region is weak and uninterpretable, the model is probably wrong.
The recent versions of most popular molecular graphics programs (e.g., PyMOL, Schrödinger LLC; Coot, Emsley et al., 2010; or CCP4MG, McNicholas et al., 2011) provide well documented functionality to fetch, calculate, and display electron density maps directly from data deposited in the PDB or via EDS (Fig. 3). However, the omit maps must be recalculated using a crystallographic refinement program.
With a small amount of effort, the strengths and weaknesses of crystallographic data can be assessed to determine how to process the model for downstream applications. Overall quality factors such as resolution and R-free are important metrics to consider, but it is equally important, if not more so, to critically evaluate crystal structures at a finer level, down to the local environments of individual residues and ligands, in the context of the electron density maps. Not all parts of the model built by the crystallographer are equally supported by the diffraction data, thus interpretations from crystal structures require these density-driven inspections. This imperative is the central thesis of a recent review by Lamb et al. (2015). For flexible membrane proteins such as GPCRs, map and model validation is especially relevant as most cases have only moderate overall resolution and the electron density quality can vary widely among the transmembrane domains and the solvent-exposed loops and ligand binding sites.
Complementing GPCR Structural Chemogenomics with Molecular Pharmacology Data
Decades of intense research on GPCRs have produced vast amounts of molecular pharmacology data available, for instance, at the International Union of Basic and Clinical Pharmacology/British Pharmacological Society Guide to Pharmacology (Pawson et al., 2014). As discussed previously, the field of structural biology is catching up, and curated and up-to-date structural data can be obtained, for instance, at the G protein–coupled receptors database (http://www.gpcr.org/7tm/) (Isberg et al., 2014). The combination of molecular pharmacology and structural data provides a powerful lens to gain new insights into the mechanistic details of GPCR function. For instance, a systematic analysis of the functional data available for crystallized ligand-receptor complexes is crucial to elucidate the molecular determinants of ligand binding, including details of structure–activity relationships, which, in turn, can be used to extrapolate the available information to ligands and receptors that are related to known structures but have not yet been crystallized (Kooistra et al., 2013).
In this section we present an example of how structural data can complement chemogenomics studies. Supplemental Table 2 displays ligand and mutagenesis data for 27 crystallized GPCRs. Ligand data include chemical structure, name, and PDB identification number of cocrystallized ligands, plus the total number of small-molecule ligands (60 heavy atoms or fewer) with binding affinity (IC50/Ki) or functional potency (IC50/EC50) of at least 10 μM for each receptor, identified using the ChEMBLdb (Bento et al., 2014); below this number and in brackets, we specify how many of these ligands are similar (ECFP-4 Tanimoto similarity ≥0.4) to the cocrystallized compound. Mutagenesis data have been extracted from the G protein–coupled receptors database and recent literature, and include the number of ligands used in mutagenesis studies to assess ligand binding and activity (and how many of those are peptide ligands), the number of mutants and mutated positions in these studies, and the number of investigated combinations of mutants and ligands. Such mutagenesis data provide a quantitative measure of the amount of information available for each receptor that can be used in the process of drug discovery. The data in Supplemental Table 2 is summarized graphically in Fig. 5A.

(A) Graphical representation of the data in Supplemental Table 2 (see main text and the legend of Supplemental Table 2 for details). (B) Different ligand-binding modes in the P2Y1 receptor. (C) The ligands doxepin (H1R), (R)-3-quinuclidinylbenzilate (BZ) (M2R), and tiotropium (M3R) ligands share a similar butterfly shape.
About half of the analyzed GPCR structures have been cocrystallized with ligands that are chemically similar (red bar segments in Fig. 5A, top) to a substantial portion (≥10%) of the known ligands for that receptor (Fig. 5A, top, green background). Thus, for these receptors, the binding mode of a relatively large number of ligands can be confidently predicted using computational techniques such as molecular docking. Remarkably, for the P2Y1 and PAR1 receptors, the binding mode of about half the known ligands (191 of 131, and 236 of 574) can in principle be modeled in a relatively straightforward manner (although small differences in ligand may affect overall ligand binding mode).
Still, for most of these receptors the majority of compounds have different chemotypes than the cocrystallized ligands (Fig. 5A, top, blue bar segments). In these cases, binding modes can be inferred by analyzing mutagenesis data covering many different mutants/residue positions and different ligands (Fig. 5A, middle and bottom). For instance, in A2AR, β2AR, δ-opioid receptor, and CXCR4 there is a large amount of mutagenesis data (number of ligands used, number of unique mutants and mutated positions, and combinations of mutants and ligands; see Supplemental Table 2, mutagenesis data, for details) that can guide the prediction of binding modes of ligands that are not yet crystallized. Moreover, communitywide GPCR structure modeling assessments (GPCRDOCK) (Michino et al., 2009; Kufareva et al., 2011, 2014) to predict the coordinates of the GPCR-ligand crystal structures have indicated that the best A2AR (Costanzi et al., 2009), D3R (Obiol-Pardo et al., 2011), CXCR4 (Roumen et al., 2011; Bhattacharya et al., 2013), 5-HT1B (Rodriguez et al., 2014), and 5-HT2B models were constructed by the careful consideration of receptor mutation data. Conversely, PAR1, free fatty acid receptor 1 (FFA1), and P2Y12 receptors have relatively few mutation data available (Supplemental Table 2, mutation data), so additional structure–activity relationship data will be required to hypothesize binding modes for ligands that are dissimilar from the cocrystallized ligands. As an example, the P2Y1 crystal structures in complex with MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2ʹ-deoxyadenosine-3ʹ,5ʹ-bisphosphate] and 1-(2-[2-(tert-butyl)phenoxy]pyridin-3-yl)-3-[4-(trifluoromethoxy)phenyl]urea illustrate that ligands can target very different binding sites (Zhang et al., 2015a) (Fig. 5B).
At the opposite side of the spectrum, some GPCRs (κ-opioid receptor, muscarinic acetylcholine receptor M3 [M3R], dopamine 3 receptor [D3R], CRF1R, and histamine 1 receptor [H1R]) have been cocrystallized with ligands that cover 1% or less of the chemical space of known small drugs for each of these receptors. Clearly, cocrystallization of these receptors with chemically diverse ligands would greatly benefit the drug discovery efforts in these subfamilies.
Interestingly, there is a substantial amount of mutagenesis data available for these receptors (Supplemental Table 2, mutation data; and Fig. 5A, middle and bottom), which combined with structural information from related receptors, can facilitate the generation of reasonable docking models for other ligands. This is illustrated by successful crystal structure-based virtual screening studies in which novel potent D3R (Carlsson et al., 2011; Lane et al., 2013; Vass et al., 2014), M3R (Kruse et al., 2013b), and H1R (de Graaf et al., 2011) ligands were identified, in the case of H1R by so-called interaction fingerprint scoring to select molecules that make similar contacts with the receptor binding site as the cocrystallized ligand (de Graaf et al., 2011). It should furthermore be noted that in some receptors (Fig. 5A, top, starred), although there is a low number of known compounds similar to cocrystallized ligands, they nevertheless share some conserved substructures and/or a conserved shape/pharmacophore.
For instance, in the 5-HT1B receptor, the total number of known ligands similar to the cocrystallized (dihydro-)ergotamine (Wacker et al., 2013; Wang et al., 2013a) is negligible, but the tryptamine substructure is present in a large portion (24%) of known 5-HT1B ligands. Similarly, the phosphonic acid group of the cocrystallized ML056 [(R)-3-amino-(3-hexylphenylamino)-4-oxobutylphosphonic acid] (Hanson et al., 2012) is present in 11% of all sphingosine-1-phosphate receptor 1 (S1P1) ligands. Also, the CP-376395 [N-(1-ethylpropyl)-3,6-dimethyl-2-(2,4,6-trimethylphenoxy)-4-pyridinamine hydrochloride] cocrystallized antagonist (Hollenstein et al., 2013) shares perpendicularly oriented N-heterocyclic and hydrophobic aromatic rings with most CRF1R ligands, which can in principle facilitate modeling studies of other CRF1R ligands to the CRF1R binding site. Finally, the cocrystallized doxepin (H1R), (R)-3-quinuclidinylbenzilate (muscarinic acetylcholine receptor M2 [M2R]), and tiotropium (M3R) ligands share an amine, with two aromatic rings oriented in a butterfly shape (Fig. 5C), with many other H1R, M2R, and M3R ligands (Kooistra et al., 2013).
The large number of mutation data available for many of these receptors furthermore facilitates experimentally guided modeling of other ligand-binding modes (Fig. 5A, middle and bottom; Supplemental Table 2).
As a final note of caution, structural and chemogenomics data should only be combined when they refer to similar ligand-binding modes. For instance, the majority of metabotropic glutamate receptor 1 (mGlu1R) and metabotropic glutamate receptor 5 (mGlu5R) ligands extracted from ChEMBLdb target the (orthosteric) extracellular Venus Fly Trap domains of class C GPCRs, while the cocrystallized 4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazole-2-yl)-N-methylbenzamide (Wu et al., 2014) and mavoglurant (Doré et al., 2014) ligands target the (allosteric) transmembrane domain.
Conclusion
The rapid emergence of structural data for GPCRs is significantly advancing our ability to generate accurate models of ligand-receptor complexes of unknown structure, interpret ligand binding structure–activity relationships, and extrapolate these relationships to related systems. Many GPCRs have been crystallized in complex with clinically relevant drugs or close analogs of therapeutic compounds, providing a framework to understand the molecular basis for their pharmacologic activities. This, in turn, is fueling renewed efforts toward structure-based drug design and an expanding search for drugs that act through poorly understood mechanisms, such as allosteric modulation and biased signaling. Furthermore, the proliferation of structures generates new starting points for molecular dynamics simulations, providing insights into the dynamics of ligand binding and receptor activation.
These breakthroughs in GPCR crystallography have required the synergistic combination of numerous technical innovations in protein engineering, detergent chemistry, crystallization, and X-ray sources, breaking the intractability of this receptor family for structural studies. However, the techniques used to enable GPCR structure determination must not be overlooked when using the structures for further research, as many GPCR structures have been heavily modified and are far from the wild-type protein. We must also remember that deposited coordinates in a PDB file are the crystallographers’ best interpretation of a data set; the truest picture of a crystal structure only emerges when the model is validated and viewed in the context of electron density maps. With this, the structural information from crystallography and other methods, such as NMR and electron microscopy, combined with the decades of functional data on ligands, mutants, and signaling complexes is bringing forth the next chapter in molecular pharmacology.
Authorship Contributions
Participated in research design: Piscitelli, Kean, de Graaf, Deupi.
Performed data analysis: Piscitelli, Kean, de Graaf, Deupi.
Wrote or contributed to the writing of the manuscript: Piscitelli, Kean, de Graaf, Deupi.
Footnotes
- Received April 27, 2015.
- Accepted July 7, 2015.
C.L.P. and J.K. contributed equally to this work.
This work was supported by the Swiss National Science Foundation [Grant 146520] and by COST Action GLISTEN [CM1207]; J.K. is an employee of Heptares Therapeutics Ltd, which is a wholly owned subsidiary of the Sosei Group Corporation.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CP-376395
- N-(1-ethylpropyl)-3,6-dimethyl-2-(2,4,6-trimethylphenoxy)-4-pyridinamine hydrochloride
- 3D
- three-dimensional
- EDS
- Electron Density Server
- Fab
- monoclonal antibody fragments
- FAUC50
- (R)-5-(2-((4-(3-((2-aminoethyl)disulfanyl)propoxy)-3-methoxyphenethyl)amino)-1-hydroxyethyl)-8-hydroxyquinolin-2(1H)-one
- GPCR
- G protein–coupled receptor
- ICL
- intracellular loop
- LCP
- lipidic cubic phase
- ML056
- (R)-3-amino-(3-hexylphenylamino)-4-oxobutylphosphonic acid
- MR
- molecular replacement
- MRS2500
- 2-iodo-N6-methyl-(N)-methanocarba-2ʹ-deoxyadenosine-3ʹ,5ʹ-bisphosphate
- Nb
- nanobody
- PDB
- Protein Data Bank
- SFX
- serial femtosecond crystallography
- TM
- transmembrane helices
- UK-432,097
- 6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxytetrahydrofuran-2-yl)-N-(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide
- XFEL
- X-ray free electron laser
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}