


Visual Overview

Abstract
Recent reports regarding the significance of chemokine receptors in disease have put a spotlight on atypical chemokine receptor 3 (ACKR3). This atypical chemokine receptor is overexpressed in numerous cancer types and has been involved in the modulation of tumor cell proliferation and migration, tumor angiogenesis, or resistance to drugs, thus contributing to cancer progression and metastasis occurrence. Here, we focus on the clinical significance and potential mechanisms underlying the pathologic role of ACKR3 in breast, lung, and brain cancer and discuss its possible relevance as a prognostic factor and potential therapeutic target in these contexts.
Introduction
ACKR3 (atypical chemokine receptor 3, also known as CXCR7) is a seven-transmembrane domain atypical chemokine receptor that belongs to class A of G protein–coupled receptors. Both C-X-C motif chemokine 12 (CXCL12) and C-X-C motif chemokine 11 (CXCL11) bind to the receptor. CXCL12 also binds to CXCR4 (C-X-C chemokine receptor type 4), and likewise, CXCL11 binds to CXCR3, another chemokine receptor of this family. Since the first description of ACKR3 as a coreceptor for human immunodeficiency virus entry (Shimizu et al., 2000), it has become clear that this receptor has a prominent role in many pathologic contexts. ACKR3 expression was found to be upregulated in a variety of pathologic conditions related to inflammation, infection, or ischemia. For instance, ACKR3 expression is increased in inflammatory bowel disease, encephalitis, rheumatoid arthritis, acute renal failure, Epstein-Barr virus type I infection, or after permanent middle cerebral artery occlusion (Sánchez-Martín et al., 2013). Importantly, altered ACKR3 expression patterns have also been detected in numerous cancer types, such as prostate, kidney, liver, cervix, brain, lung, and breast cancer [reviewed by Sánchez-Martín et al. (2013) and Freitas et al. (2014)]. In this review, we focus on the evidence linking altered ACKR3 expression with the progression of breast, lung, and glioma tumors and discuss the underlying molecular mechanisms.
ACKR3 in Breast Cancer.
Breast cancer is the most prevalent cancer among women worldwide (Siegel et al., 2016). Several studies describe an association between the CXCL12-ACKR3 axis and disease progression and poor overall survival of breast cancer patients. As a result, interest in this receptor is markedly increasing, as it may be a potential therapeutic target. Previous reports indicated that ACKR3 expression in breast cancer patient samples (in situ and invasive lobular carcinomas) is much higher when compared with adjacent tissues and normal mammary epithelium. This was verified at protein and mRNA levels (Miao et al., 2007; Wani et al., 2014; Li et al., 2015; Behnam Azad et al., 2016). High expression was also detected in metastatic tissues (Wu et al., 2016; Yu et al., 2017b). Notably, ACKR3 overexpression correlates with poor prognosis and lung metastasis–free survival (Behnam Azad et al., 2016; Wu et al., 2016; Yu et al., 2017b).
Furthermore, there is an association between higher levels of ACKR3 and worse outcomes in terms of disease-free survival (Hassan et al., 2009), and a positive correlation between ACKR3 expression level and lymph node metastasis: ACKR3 expression is higher in primary tumors of patients with lymphatic metastasis than in those of metastasis-free patients (Li et al., 2015). Taken together, these findings indicate that ACKR3 plays a role in breast cancer progression and metastasis occurrence.
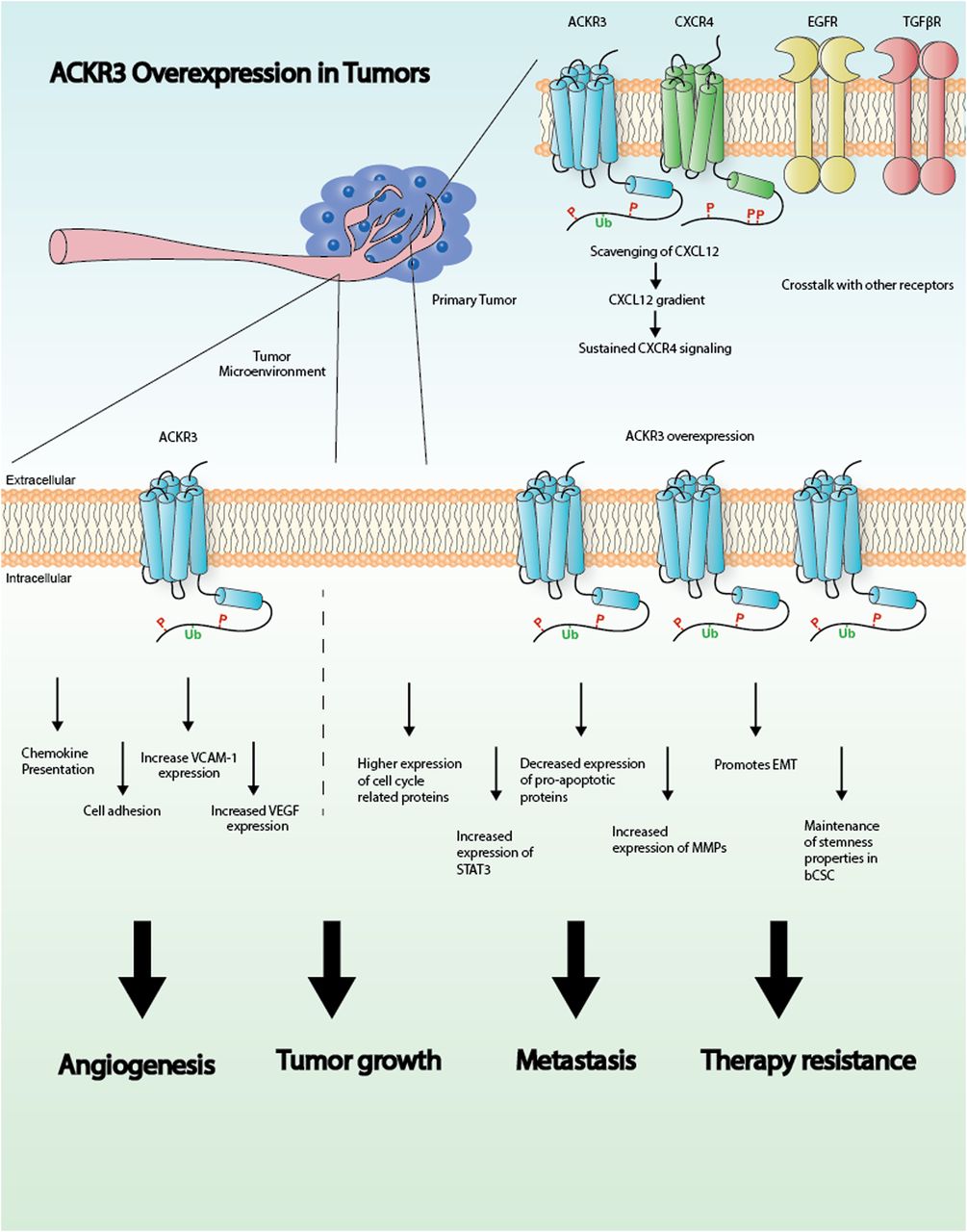
Several in vitro and in vivo studies have found that higher levels of ACKR3 result in increased cell proliferation and invasive migration, promoting tumor growth and metastasis (Miao et al., 2007; Xue et al., 2013; Gao et al., 2015; Li et al., 2015). However, the molecular mechanisms linking enhanced ACKR3 expression to these processes are not fully elucidated. Research performed in model cell lines with ectopically overexpressed ACKR3 and CXCR4 receptors or in different cell types with endogenous levels of these proteins has put forward several potential mechanisms. In mouse migrating interneurons, ACKR3 has been shown to be key for migration by scavenging CXCL12, thus preventing coexpressed CXCR4 from desensitization and degradation and allowing adequate levels of CXCL12-CXCR4 signaling (Sánchez-Alcañiz et al., 2011; Abe et al., 2014). These data suggest that high ACKR3 expression in breast cancer would foster CXCR4-dependent cascades. Such a scavenging role is related to constitutive and ligand-dependent ACKR3 internalization and recycling observed in different cell types (Naumann et al., 2010), including breast cancer cells (Luker et al., 2010). This allows receptor trafficking to lysosomes and degradation of receptor-bound CXCL12 (Luker et al., 2010; Naumann et al., 2010; Hoffmann et al., 2012). Consistent with this notion, uptake of CXCL12 by ACKR3-positive (ACKR3+) breast cancer cells increases proliferation and metastatic potential of CXCR4+ cells (Luker et al., 2012). Interestingly, ACKR3 ligands trigger β-arrestin recruitment to the receptor in model cell systems (Luker et al., 2009b; Rajagopal et al., 2010; Benredjem et al., 2017), and it has been reported that ACKR3 internalization and the resulting chemokine degradation are dependent on β-arrestin in breast cancer cells (Luker et al., 2010). However, constitutive receptor internalization and recycling can also take place in a ligand-independent way. Recent data show that β-arrestins are dispensable for chemokine scavenging (Montpas et al., 2018), suggesting that other ACKR3-interacting proteins might be involved in the internalization process, depending on the experimental conditions and cellular context.
In addition to its ligand-scavenging properties, it cannot be ruled out that the interaction of ACKR3 with β-arrestins (or other interacting proteins to be identified) may also trigger other downstream cascades in a cell-type-specific way (Sánchez-Alcañiz et al., 2011). ACKR3 recruitment of β-arrestins might also modify CXCR4 signaling by recruiting β-arrestin2 away from coexpressed CXCR4 (Coggins et al., 2014) or via ACKR3/CXCR4 cointernalization, as reported in MCF7 breast cancer cells (Sánchez-Martín et al., 2013).
Therefore, it is tempting to suggest that both CXCR4 and ACKR3 cooperate to promote tumor growth and metastasis through different interaction and cross-talk mechanisms (Sierro et al., 2007; Levoye et al., 2009; Luker et al., 2009a, 2010). While some reports have indicated that CXCR4 is overexpressed in breast cancer patient samples, with both receptors colocalized (Inaguma et al., 2015), others have shown that CXCR4 and ACKR3 are predominantly expressed on separated populations of breast cancer cells (Luker et al., 2012). Additionally, CXCR4 and ACKR3 coexpression in the MTLn3 breast cancer cell line decreases invasion in response to CXCL12 (Hernandez et al., 2011). Given the discrepancies in the available data, further investigating whether CXCR4 and ACKR3 are expressed in the same or distinct subpopulations of breast cancer cells is key to better understanding these potential cross-talk mechanisms.
In addition to CXCR4, ACKR3 may also functionally interact with other key signaling cascades in breast cancer, such as estrogen and epidermal growth factor receptors. Analysis of luminal-type breast cancer tumor samples revealed a positive correlation between ACKR3 and estrogen receptor (ER) levels. Overexpression of ACKR3 can result in increased estrogen signaling in ER+ breast tumor cells and is closely related to tamoxifen insensitivity, which would make ACKR3 a relevant target specifically in ER-positive breast cancer (Hao et al., 2018).
ACKR3 and epidermal growth factor receptor (EGFR) have also been found to be colocalized in ER+ breast cancer tissues. In vitro studies show that EGFR expression correlates with ACKR3 levels in breast cancer cell lines and, interestingly, that EGFR is absent in cells lacking ACKR3. Furthermore, partial depletion of ACKR3 in a breast cancer cell line results in decreased proliferation in response to epidermal growth factor and decreased activation of the extracellular signal-regulated kinase pathway and level of EGFR phosphorylation. These findings strongly suggest a cooperation between both receptors in tumor proliferation (Salazar et al., 2014).
In addition to the aforementioned roles involving other receptors, ACKR3 has an independent effect on cell cycle or apoptosis-related cascades in breast cancer cells. Decreasing ACKR3 levels alters expression of cell cycle–related proteins and leads to cell cycle arrest, hence reducing cell proliferation (Salazar et al., 2014). Additionally, knock down of the receptor in the MCF7 breast cancer cell line increased the expression of the proapoptotic caspase 3 and 8 proteins (Gao et al., 2015). In addition, ACKR3 expression enhances breast cancer proliferation through signal transducer and activator of transcription 3 activation, which has previously been linked to apoptosis resistance and cell cycle progression in invasive breast cancer (Wani et al., 2014; Li et al., 2015).
Another key event in breast cancer is cell migration and invasion, ultimately leading to metastasis occurrence, which is the primary cause of death of the majority of breast cancer patients. Migration and invasion events require degradation of basement membrane extracellular matrix proteins by matrix metalloproteinases (MMPs). ACKR3 expression is associated with the secretion of MMP2, MMP3, and MMP9 (Zabel et al., 2010; Gao et al., 2015). ACKR3 overexpression also accompanies mesenchymal-to-epithelial transition, a pathway leading to metastatic events (Leontovich et al., 2012).
In addition to metastasis, breast cancer stem cells (bCSCs), a population of cells located within the tumor with high proliferative capacity and exhibiting resistance to therapeutics, are a current challenge for breast cancer–targeted therapies. It was demonstrated that ACKR3 is crucial for tumorigenicity and maintenance of stem cell properties of bCSCs (Tang et al., 2016). Given the role of this receptor in both metastasis and bCSCs within the primary tumor, it appears to be an attractive target for new therapeutic approaches for breast cancer.
Accumulating evidence suggests that ACKR3 also plays a pivotal role in tumor-associated vasculature. One study assessing ACKR3 expression in tumor samples (Behnam Azad et al., 2016) showed an intense vascular staining specifically in the endothelium of tumor-associated blood vessels. In contrast, the receptor was not expressed in healthy tissue blood vessels. Another report demonstrated that concomitant expression of ACKR3 in tumor and tumor-associated vasculature promotes cell-cell interaction (Miao et al., 2007), suggesting a role for this receptor in modulating cell adhesion in the tumor microenvironment. ACKR3 also enhances vascular cell-adhesion molecule-1 expression (Wani et al., 2014). Since tumor cell adhesion to the basal membrane is crucial for the invasive process, these data suggest that ACKR3 mediates this step. Furthermore, a recent study showed that ACKR3 downregulation decreases vascular endothelial growth factor production and tube formation (Qian et al., 2018), suggesting a role for this receptor in angiogenesis, an important event that leads to tumor cell survival and migration. Nevertheless, another report using a different experimental model suggested that vascular ACKR3 suppresses tumor proliferation and decreases metastatic potential of tumor cells (Stacer et al., 2016). These discrepancies may be explained by the different models used in each study or might suggest a context-dependent role for ACKR3 that requires further investigation. On the other hand, it is worth noting that ACKR3 also increases tumor-promoting macrophages and recruitment to the tumor site (Wani et al., 2014), a key event in determining the tumor microenvironment.
In summary, these studies point to a pivotal role for ACKR3 in breast cancer progression and metastasis by promoting survival, proliferation, and migration of malignant cells. Moreover, ACKR3 modulates angiogenic pathways and helps to shape the tumor microenvironment, thus further controlling tumor growth (Fig. 1). Although more studies are needed to clarify the exact mechanisms by which ACKR3 operates, accumulating evidence strongly indicates that ACKR3 is a potential therapeutic target in breast cancer.
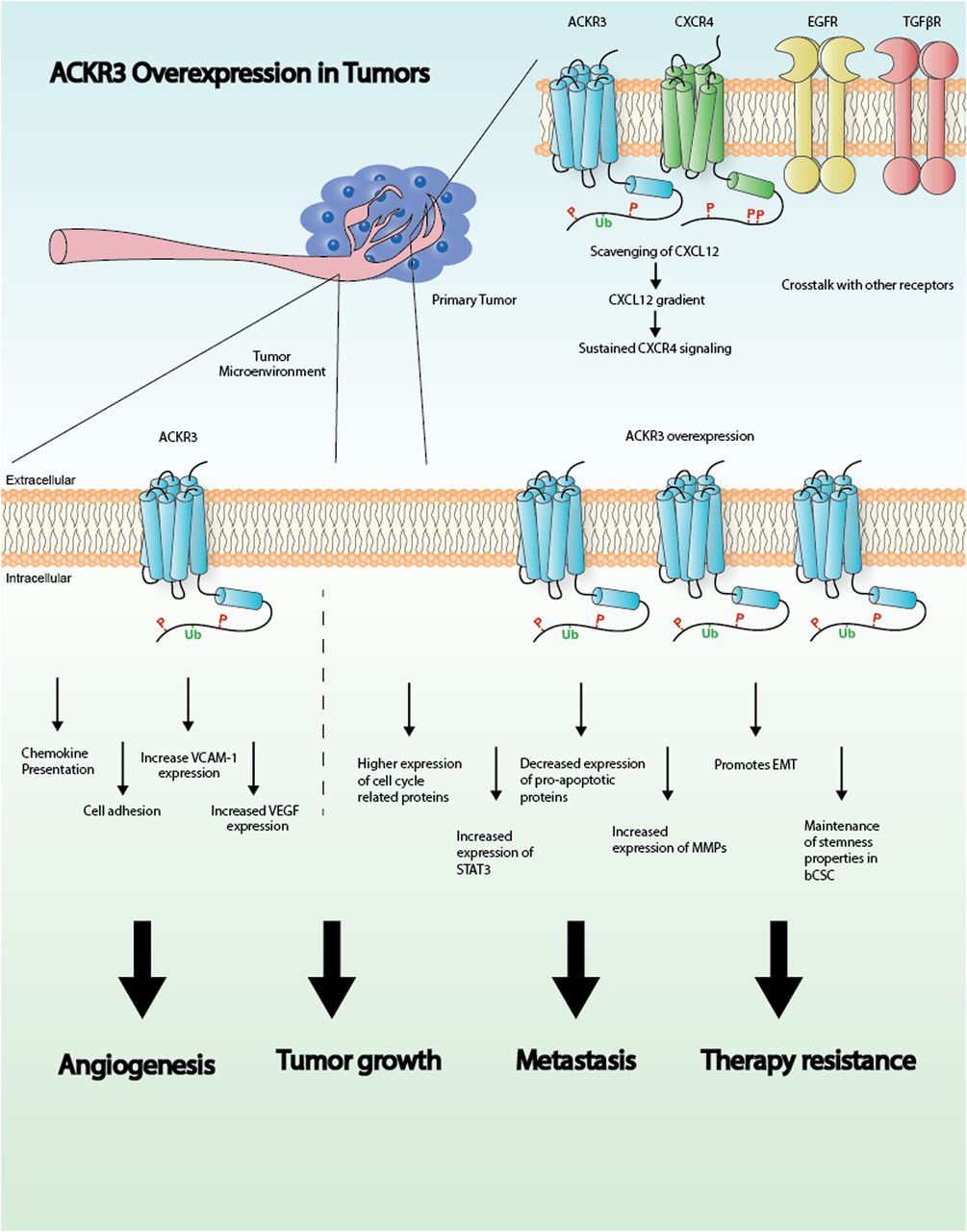
Graphical representation illustrating the role of ACKR3 in cancer. See text for details. EMT, epithelial-mesenchymal transition; STAT3, signal transducer and activator of transcription 3; VCAM-1, vascular cell-adhesion molecule-1; VEGF, vascular endothelial growth factor.
ACKR3 in Lung Cancer.
Lung cancer is the most frequent cause of cancer-related death in men and the second most frequent in women. Initial studies on ACKR3 reported an increased expression of ACKR3 in malignant lung carcinoma biopsy material when compared with healthy human lung samples (Burns et al., 2006). Further analysis of ACKR3 expression in multiple cancer types, including lung squamous cell carcinoma, showed localized ACKR3 staining on the tumor vasculature. These first findings suggested a role for ACKR3 in lung cancer (Miao et al., 2007), which was further investigated in vivo using mice engrafted with mouse Lewis lung carcinoma cells or human lung carcinoma cells. These studies demonstrated that both diminishing ACKR3 expression and inhibiting ACKR3 signaling by the ACKR3 ligand CCX754 resulted in decreased tumor growth. Interestingly, the extent to which CCX754 decreased tumor growth was almost similar to the marketed chemotherapy drug melphalan, showing the potential for targeting ACKR3 in lung cancer (Burns et al., 2006; Miao et al., 2007).
ACKR3 expression is found in many types of lung cancer. Investigation of human biopsy material indicates expression in small-cell lung cancer (Imai et al., 2010) and the non–small-cell lung cancer (NSCLC) subtypes adenocarcinoma, squamous cell carcinoma, and large-cell carcinoma (Miao et al., 2007; Goldmann et al., 2008; Iwakiri et al., 2009; Imai et al., 2010; Franco et al., 2012).
Within the Human Protein Atlas program, a recent initiative to define the oncological proteome, mRNA expression levels of ACKR3 were investigated in 794 lung cancer biopsies (Uhlen et al., 2017). Comparison of the expression of ACKR3 with overall survival rate of patients showed an unfavorable outcome for high ACKR3 expression. This trend did not have the statistical power to be used for prognostic purposes, but it highlights the relevance of ACKR3 expression in lung cancer (Uhlen et al., 2017).
Surgical removal of lung tumors is one of the standard treatment procedures for lung cancer. However, tumor recurrence is often observed and is related to poor prognosis (Pearson, 1999). A study of ACKR3 mRNA expression in the surgical specimens from 79 patients who underwent complete resection showed that ACKR3 mRNA expression could be used as a postoperative 5-year disease-free prognostic marker in stage I NSCLC patients. Furthermore, there was a significantly higher ACKR3 expression in pathologic stage I NSCLC patients with postoperative recurrence of secondary tumors compared with recurrence-free patients (Iwakiri et al., 2009). These results suggest that ACKR3 may be related to the process by which NSCLC forms secondary tumors.
A factor that has been linked to metastasis and, thus, the formation of secondary tumors of lung cancer is transforming growth factor β1 (TGF-β1) (Massagué, 2008). The mRNA and protein expression of ACKR3 is regulated by TGF-β1, and the combination of high TGF-β1 and ACKR3 expression correlates with poor patient survival (Wu et al., 2016). TGF-β1–mediated functional effects, including cancer cell motility and epithelial-to-mesenchymal transition, appear to depend on ACKR3 (Massagué, 2008; Wu et al., 2016). Furthermore, epithelial-to-mesenchymal transition of cancer cells leads to the formation of cancer stem cells which show high metastatic properties (Singh and Settleman, 2010), and TGF-β1 induces cancer stem cell formation in an ACKR3-dependent manner (Wu et al., 2016). Collectively, these findings suggest a key role of ACKR3 in cancer stem cell formation, what might underlie ACKR3-mediated formation of secondary tumors.
In lung cancer, mutations in the EGFR, KRAS, and ALK genes are mutually exclusive and can be used to define responsiveness to different chemotherapies. A study on the presence of EGFR mutations showed that ACKR3 mRNA expression is higher in lung cancer patients with mutated EGFR compared with wild-type EGFR patients, suggesting a role for ACKR3 in EGFR-mediated lung cancer (Iwakiri et al., 2009). Furthermore, an in vivo study in which Lkb1 and Pten were conditionally knocked down in mice showed that this leads to development of the lung cancer subtype squamous cell carcinoma (Xu et al., 2014). Transcriptomics data of the Lkb1 (−/−)/Pten (−/−) squamous cell carcinoma tumor showed an increase in ACKR3 expression when compared with healthy lung tissue, suggesting a role of ACKR3 in Lkb1/Pten-mediated lung cancer (Xu et al., 2014). Altogether, these observations suggest a universal role of ACKR3 in multiple lung cancer subtypes.
ACKR3 in Glioma.
The term glioma encloses a variety of intrinsic central nervous system tumors. These tumors were initially classified based on their presumed cellular origin (astrocytoma, oligodendroglioma, oligoastrocytoma) and extension of infiltration (diffuse or nondiffuse glioma). Regardless of the subclassification, glioma are divided into three malignancy grades (II, III, and IV) considering their mitotic activity, necrosis, and florid microvascular proliferation. Glioblastoma is the most malignant one (grade IV) and is categorized as either “secondary” or “primary,” depending on whether there is evidence of a progression from a lower-grade glioma. However, in 2016, the World Health Organization (WHO) introduced a genotypic classification based on the presence of recurrent point mutations in isocitrate dehydrogenase 1 and 2 (IDH1/IDH2) genes, dividing glioblastoma into glioblastoma-IDH-wild type and glioblastoma-IDH-mutant. Interestingly, the majority of secondary glioblastoma are IDH-mutants, whereas primary glioblastoma are typically IDH-wild type (Parsons et al., 2008). Recently, it was proposed that ACKR3 influences prognosis in human glioma, depending on the IDH classification (Birner et al., 2015). In fact, ACKR3 expression in tumor-associated vessels predicts a better prognosis in IDH1-wild-type glioma, whereas it has opposite consequences in the IDH1-mutant subtype. In addition, mRNA as well as protein levels of ACKR3 were found to be upregulated in glioma (Calatozzolo et al., 2011) and to positively correlate with WHO grade in several studies (Hattermann et al., 2010; Walters et al., 2014; Bianco et al., 2015; Birner et al., 2015). ACKR3 localization also changes with WHO grade. In fact, in grade II glioma, ACKR3 is mainly expressed in cancer cells. In grade III, it is present primarily in tumor vascular endothelial cells and only marginally in cancer cells. In glioblastoma, ACKR3 is found in cancer cells, in pseudopalisades nearest to necrotic areas, and in the tumor endothelium (Bianco et al., 2015). Standard care for the treatment of WHO grade III and IV glioma consists of surgery followed by chemotherapy (temozolomide) that can be combined with intermediate-frequency alternating electric fields (Touat et al., 2017). However, glioma exhibit both radio (Han et al., 2017) and drug resistance (Stavrovskaya et al., 2016), making the current therapies ineffective, with a median survival ranging from 12 to 16 months after diagnosis (Gilbert et al., 2014). Accumulating evidence suggests that ACKR3 might play a role in these acquired resistances. In fact, CXCL12 reverses the antiproliferative effect of nontoxic concentrations of temozolomide in C6 rat cells isolated from murine glioma (Hattermann et al., 2012). Accordingly, CXCL12 also reduces temozolomide-induced apoptosis. These CXCL12 effects might involve ACKR3, which is expressed at a much higher level than CXCR4 in C6 cells. Corroborating these findings, the presence of the ACKR3 modulator CCX771 after irradiation provokes tumor regression in nude mice injected with U251 glioma cells (Walters et al., 2014). In addition, treatment with the ACKR3 ligand CCX662 (able to induce β-arrestin recruitment to ACKR3 and inhibit CXCL12-dependent migration) extends the survival of rats with nitrosourea-induced brain tumors after irradiation. However, in a more aggressive model consisting of the injection of C6 glioma cells in rats, only the combination of irradiation with CCX662 treatment extends survival.
Contradictory results are emerging regarding the role of ACKR3 in glioma cell proliferation. In fact, in the U373 glioblastoma cell line and in human fetal astrocytes, CXCL12-induced proliferation is prevented by the ACKR3 ligand CCX733 (Calatozzolo et al., 2011). On the other hand, CXCL12 does not influence either proliferation or migration of A764 and U343 glioma cells (Hattermann et al., 2010). Moreover, in cocultures of U87 cells and human brain microvascular endothelial cells, the receptor was found to have no trophic effect (Rao et al., 2012). Long noncoding RNA X-inactive-specific transcript is upregulated in glioma endothelial cells forming the blood-tumor barrier (Yu et al., 2017a) and increases glioma angiogenesis by indirectly controlling ACKR3 expression. In fact, the study showed that downregulation of X-inactive-specific transcript increases miRNA-137 expression, which in turn inhibits the expression of forkhead box C1, a transcription factor that promotes ACKR3 expression.
The common view of ACKR3 as an atypical chemokine receptor unable to activate G proteins (Naumann et al., 2010) was challenged in 2012 when it was suggested that the receptor can signal through Gi/o proteins in astrocytes and human glioma cells (Odemis et al., 2010). This study showed that CXCL12 induces activation of G proteins and Ca2+ influx in rat astrocytes in primary culture. Both effects persisted in cultures obtained from CXCR4 knockout mice and were abolished by pretreatment of cells with an ACKR3 small interfering RNA. Moreover, both CXCL12-induced proliferation and migration of rat astrocytes did not require CXCR4 expression and were inhibited by pertussis toxin, suggesting that ACKR3 influences migration and proliferation of astrocytes in a G protein–dependent fashion. On the other hand, in the same study, CXCL11 did not activate G proteins, suggesting that the ability of ACKR3 to activate G proteins in astrocytes depends on the nature of the agonist. So far, the ability of ACKR3 to activate G proteins has only been found in glial cells and not in other cell types (Levoye et al., 2009; Rajagopal et al., 2010; Kumar et al., 2012). This unique capability of glial ACKR3 to activate G proteins certainly warrants further exploration, providing that it is confirmed in independent studies. In another study, CXCL12 also triggered mitogen-activated protein kinase phosphorylation in glioma cells via phospholipase C but not AKT and p38 (Hattermann et al., 2010). Whether this effect requires ACKR3 coupling to G proteins has not been investigated.
Collectively, previously published data show a high ACKR3 expression in glioma and glioma cell lines. Although its role in the progression of this cancer type and the underlying signaling mechanisms need to be fully characterized, available data indicate that this receptor plays an important role in glioma drug/radio resistance and angiogenesis.
Conclusions
Extended literature analysis points to a pivotal role of ACKR3 in breast, lung, and brain cancers. Expression of this atypical chemokine receptor, both at mRNA and protein levels, is higher in tumor samples when compared with healthy tissues. Moreover, increasing evidence indicates that receptor expression positively correlates with poor prognosis, disease grade, and reoccurrence, making it a valuable biomarker for diagnosis and prognosis in certain breast, lung, and brain cancer types. ACKR3 is also emerging as a key player of cancer progression and metastasis occurrence via actions on both tumor cells (favoring cell proliferation and invasion) and the tumor microenvironment by modulating angiogenesis (Fig. 1). The use of relevant animal or cellular models with endogenous levels of chemokine receptors will allow a better knowledge of the molecular mechanisms involved, the stimuli leading to ACKR3 overexpression in cancer contexts, and the cross-talk of ACKR3 with key oncogenic signaling cascades. Insight into these mechanisms is essential to help develop new strategies to limit tumor progression and avoid resistance to therapy.
Acknowledgments
We thank all of our colleagues from the ONCORNET consortium for the continuous scientific discussions and insights. This review is part of the mini review series “From insight to modulation of CXCR4 and ACKR3 (CXCR7) function.”
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Neves, Fumagalli, van den Bor, Marin, Smit, Mayor.
Footnotes
- Received November 21, 2018.
- Accepted January 30, 2019.
↵1 A.F. and J.v.d.B. contributed equally to this work.
This work was supported by the European Union H2020-MSCA Program [Grant Agreement 64183], ONCORNET to P.M., M.J.S., and F.M.; Ministerio de Economía, Industria y Competitividad of Spain [Grant SAF2017-84125-R] to F.M.; CIBERCV-Instituto de Salud Carlos III, Spain [Grant CB16/11/00278] to F.M.; cofunded with European FEDER contribution, Comunidad de Madrid [B2017/BMD-3671-INFLAMUNE] to F.M.; Fundación Ramón Areces to F.M.; Portuguese Foundation for Science and Technology [Grant SFRH/BD/136574/2018] to M.N.; Netherlands Organization for Scientific Research NWO: Vici [Grant 016.140.657] to M.J.S.; and grants from CNRS, INSERM, Université de Montpellier and Fondation pour la Recherche Médicale (FRM) to P.M.
Abbreviations
- ACKR3
- atypical chemokine receptor 3
- bCSC
- breast cancer stem cell
- CXCL11
- C-X-C motif chemokine 11
- CXCL12
- C-X-C motif chemokine 12
- CXCR4
- C-X-C chemokine receptor type 4
- EGFR
- epidermal growth factor receptor
- ER
- estrogen receptor
- IDH1/IDH2
- isocitrate dehydrogenase 1 and 2
- MMP
- matrix metalloproteinase
- NSCLC
- non–small-cell lung cancer
- TGF-β1
- transforming growth factor β1
- WHO
- World Health Organization
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}