


Visual Overview
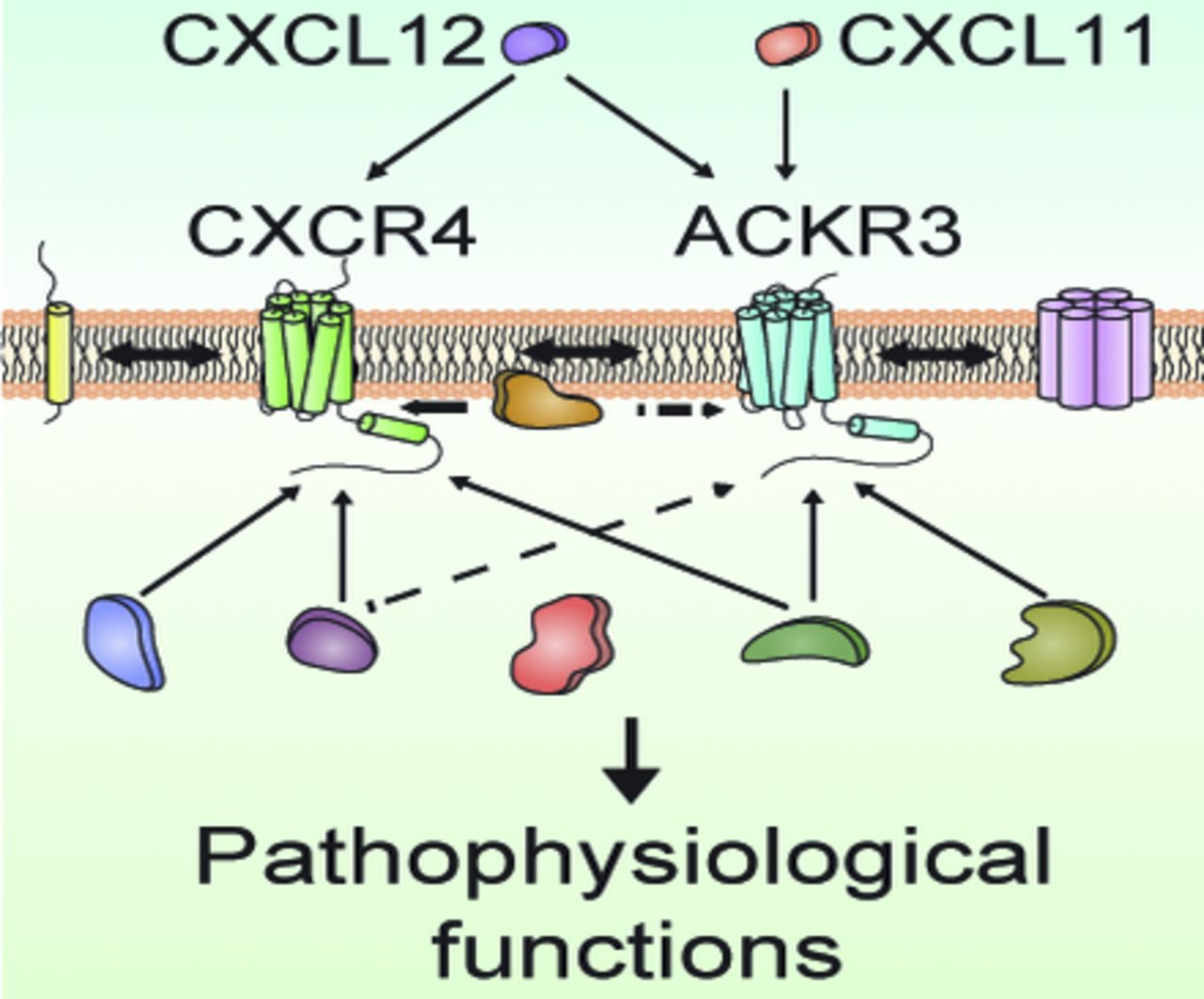
Abstract
The C-X-C motif chemokine receptor type 4 (CXCR4) and the atypical chemokine receptor 3 (ACKR3/CXCR7) are class A G protein-coupled receptors (GPCRs). Accumulating evidence indicates that GPCR subcellular localization, trafficking, transduction properties, and ultimately their pathophysiological functions are regulated by both interacting proteins and post-translational modifications. This has encouraged the development of novel techniques to characterize the GPCR interactome and to identify residues subjected to post-translational modifications, with a special focus on phosphorylation. This review first describes state-of-the-art methods for the identification of GPCR-interacting proteins and GPCR phosphorylated sites. In addition, we provide an overview of the current knowledge of CXCR4 and ACKR3 post-translational modifications and an exhaustive list of previously identified CXCR4- or ACKR3-interacting proteins. We then describe studies highlighting the importance of the reciprocal influence of CXCR4/ACKR3 interactomes and phosphorylation states. We also discuss their impact on the functional status of each receptor. These studies suggest that deeper knowledge of the CXCR4/ACKR3 interactomes along with their phosphorylation and ubiquitination status would shed new light on their regulation and pathophysiological functions.
Introduction
The C-X-C chemokine receptor type 4 (CXCR4) and the atypical chemokine receptor 3 (ACKR3), earlier referred to as CXCR7, are class A G protein-coupled receptors (GPCRs). Stromal cell–derived factor-1/C-X-C motif chemokine 12 (CXCL12) binds to both CXCR4 and ACKR3 receptors, whereas C-X-C motif chemokine 11 (CXCL11) binds only to the latter and to the C-X-C chemokine receptor type 3. CXCR4 and ACKR3 are coexpressed in various cell types [e.g., endothelial cells (Volin et al., 1998; Berahovich et al., 2014), neurons (Banisadr et al., 2002; Sánchez-Alcañiz et al., 2011); and glial cells (Banisadr et al., 2002, 2016; Odemis et al., 2010)], where they play a pivotal role in migration, proliferation, and differentiation. They are also overexpressed in various tumors and control invasion and metastasis (Sun et al., 2010; Zhao et al., 2015; Nazari et al., 2017).
There is now considerable evidence indicating that GPCRs do not operate as isolated proteins within the plasma membrane. Instead, they physically interact with numerous proteins that influence their activity, trafficking, and signal transduction properties (Bockaert et al., 2004; Ritter and Hall, 2009; Magalhaes et al., 2012). These include proteins canonically associated with most GPCRs, such as G proteins, G protein-coupled receptor kinases (GRKs) and β-arrestins, specific partner proteins, and even GPCRs themselves. In fact, in comparison with monomers, GPCRs can form homomers and heteromers with specific pharmacological and signal transduction properties (Ferré et al., 2014).
Phosphorylation is another key mechanism contributing to the regulation of GPCR functional status and signal transduction (Tobin, 2008). Both canonical GRKs and other specific protein kinases are able to phosphorylate GPCRs at multiple sites (Luo et al., 2017), generating the so-called GPCR phosphorylation barcode that determines β-arrestin recruitment, receptor intracellular fate, and signaling outcomes (Reiter et al., 2012).
This review will describe recent data highlighting the influence of the CXCR4 and ACKR3 interactome on their functional activity and signal transduction properties. A special focus will be given to the reciprocal influence of the interactome on CXCR4/ACKR3 phosphorylation and its impact on the functional status and pathophysiological functions of each receptor.
Methods for the Identification of GPCR-Interacting Proteins
Considerable evidence suggests that GPCRs recruit GPCR-interacting proteins (GIPs) (Maurice et al., 2011). This prompted investigations aimed at identifying GIPs and at characterizing GPCR-GIP interactions, using either unbiased or targeted approaches. In unbiased methods, no knowledge of the GIPs is required beforehand and the GPCR of interest is used as bait to purify unknown GIPs. Meanwhile, targeted methods are devoted to the validation and characterization of previously identified GPCR-GIP interactions. Methods for identifying GIPs or characterizing GPCR-GIP interactions include genetic, biophysical, biochemical, or proteomic approaches and are summarized in Table 1.
Principal methods used to identify GPCR-interacting proteins and phosphorylated residues.
GPCR-interacting proteins (1–4) and phosphorylated residues (5).
Genetic Methods.
The first method belonging to this class is the yeast two-hybrid assay (Fields and Song, 1989). In this method, the protein of interest (the bait protein) is expressed in yeast as a fusion to the DNA-binding domain of a transcription factor lacking the transcription activation domain. To identify partners of this bait, a plasmid library that expresses cDNA-encoded proteins fused to a transcription activation domain is introduced into the yeast strain. Interaction of a cDNA-encoded protein with the bait protein results in the activation of the transcription factor and expression of a reporter gene, enabling growth on specific media or a color reaction and the identification of the cDNA encoding the target proteins. A first disadvantage is the loss of spatial-temporal localization of the interaction; in fact, the yeast two-hybrid assay only captures a snapshot of potential interactions in an artificial biologic system. A second disadvantage is that it is not possible to investigate membrane-anchored proteins since the two proteins must cross the nuclear membrane to carry the reconstituted transcription factor to the DNA. To overcome this issue, the membrane yeast two-hybrid assay (Stagljar et al., 1998) was developed. In this assay, the ubiquitin protein is split into two fragments that are fused to the two proteins of interest. The ubiquitin C-terminal fragment is then conjugated to a transcription factor that is released when the interaction occurs, and the ubiquitin protein is reformed. However, as in the yeast two-hybrid assay, the spatial-temporal localization of the interaction is lost. A second limitation is that the ubiquitin C-terminus carrying the transcription factor cannot be fused to soluble proteins because they could diffuse into the nucleus. Therefore, a mammalian version of the assay called mammalian membrane two-hybrid (Petschnigg et al., 2014) was developed. The kinase substrate sensor assay (Lievens et al., 2014), using the signal transducer and activator of transcription 3 (STAT3) as transcription factor, can also be used for investigating protein-protein interactions, including those involving cytosolic and membrane proteins in mammalian cells. However, the kinase substrate sensor assay cannot be used for studying GPCR interaction with proteins involved in the STAT3 cascade.
Biophysical Methods.
Energy transfer–based methods, such as bioluminescence and fluorescent resonance energy transfer [BRET (Xu et al., 1999) and FRET (Clegg, 1995)] assays, are targeted methods that are generally used to investigate previously reported interactions. The basis of both is the transfer of energy from a donor to a nearby acceptor (<100 Å). Their high sensitivity allows the study of weak and transient interactions. The high spatial-temporal resolution permits accurate kinetic studies for investigating interaction dynamics.
Another biophysical method, with FRET as a basis and employed for the study of protein-protein interaction, is fluorescent lifetime imaging microscopy (Sun et al., 2012). The fluorescence lifetime is the average time that a molecule spends in the excited state before returning to the ground state. Since in FRET the energy transfer from the acceptor to the donor depopulates the excited state energy of the latter, it also shortens its lifetime. Fluorescent lifetime imaging microscopy can accurately measure the shorter donor lifetime that results from FRET; thus, it allows mapping of the spatial distribution of protein-protein interactions in living cells (Sun et al., 2011). Its main advantage over intensity-based FRET is the more accurate measurement of FRET, because only donor signals are measured, which eliminates the corrections for spectral bleed-through (Sun et al., 2011). Its main disadvantages are the necessity of a live specimen and the complexity of data recording and analysis.
Biochemical Methods.
The proximity ligation assay (Fredriksson et al., 2002) is another powerful targeted method. In the direct version of the technique, two DNA oligonucleotide-conjugated antibodies are used against the proteins of interest. In the indirect version, secondary DNA-conjugated antibodies are used after the proteins of interest are targeted with an appropriate primary antibody. If the two conjugated antibodies are close enough (30–40 nm), they can bind together. The DNA connecting the two probes is then amplified and hybridized with fluorophores. This allows the visualization of the interaction in the place where it occurs, at a single-molecule resolution. The main disadvantages of the approach are the high costs and the necessity for specific antibodies, which are not always available.
In the bimolecular fluorescent complementation assay (Hu et al., 2002; Hu and Kerppola, 2003), a fluorescent protein is divided into two nonfluorescent fragments that are fused to the proteins of interest. Interaction reconstitutes the entire fluorescent protein. This method allows the direct visualization of the interaction and can be used for soluble or membrane-bound proteins. In addition, several interactions can be investigated in parallel through the use of different fluorescent proteins. Since there is a delay in fluorescence formation upon reconstitution of the fluorescent proteins, and the fluorophore formation is irreversible, these methods are usually not well suited for kinetic studies. To overcome these limitations, a novel assay called NanoBiT was developed. In this assay the nanoluciferase enzyme is divided in two subunits (LgBiT and SmBiT), with low affinity for each other, that can be brought together by the two interacting proteins. The low affinity makes the interaction reversible and therefore suitable for the investigation of kinetics (Duellman et al., 2017).
Proteomics Methods.
Proteomic methods aim to identify GIPs of a receptor of interest via the use of affinity purification coupled with mass spectrometry (AP-MS). This approach is usually employed as an unbiased method for screening virtually all the GIPs of a GPCR of interest. Targeted versions of the method also exist and rely on GIP identification by Western blotting. However, the requirement for specific antibodies seriously limits its application. Several strategies can be used for the affinity purification step. In co-immunoprecipitation (Co-IP), specific antibodies against the protein of interest are used for precipitating the bait from a protein lysate. As specific GPCR antibodies providing high immunoprecipitation (IP) yields are rarely available, epitope-tagged versions of the receptor of interest are often expressed in the cell type or the organism of interest and precipitated using antibodies against the tag. The main advantages of Co-IP are the purification of proteins interacting with the entire receptor (whenever possible the native receptor) in living cells or tissues and its ability to purify the entire associated protein complex. The main limitations are the necessity for specific antibodies to precipitate GPCRs, the loss of spatial-temporal information, and the use of detergents for cell lysis that might denaturate the GPCR of interest and, accordingly, disrupt interactions with their protein partners. For this reason, special attention must be paid to lysis conditions that efficiently solubilize the receptor while conserving the receptor’s native conformation and its interactions with GIPs. For instance, detergents such as Triton and NP-40 completely denaturate CXCR4 (Palmesino et al., 2016), whereas 3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate, also called CHAPSO (Babcock et al., 2001), and n-dodecyl-β-d-maltopyranoside, also called DDM (Palmesino et al., 2016), yield the highest proportion of receptor in the native conformation. Despite this limitation, several CXCR4-interacting proteins have been identified using Co-IP approaches (see Table 2).
CXCR4- and ACKR3-interacting proteins described in the literature.
Alternatively, pull-down assays can be performed to purify GPCR partners from a cell or tissue lysate. This approach uses the receptor (or one of its domains) fused with an affinity tag (e.g., glutathione-S-transferase) and immobilized on beads as bait. Such in vitro binding assays can also be used to prove direct physical interaction between two protein partners. In this case, the bait is incubated with a purified protein instead of a cell or tissue lysate. In all methods, affinity purified proteins are systematically identified by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). A two-step version, named tandem affinity purification (Puig et al., 2001), has also been reported (Daulat et al., 2007) and applies to both Co-IPs or pull-downs. Although tandem affinity purification methods drastically reduce the number of false-positive identifications, they require larger amounts of starting material.
In the proximity-dependent biotin identification method (Roux et al., 2013), the bait protein is fused to a prokaryotic biotin ligase molecule that biotinylates proteins in close proximity once expressed in cells. The method can detect weak and transient interactions occurring in living cells, and detergents do not affect the results. Though the fusion of biotin ligase to the bait might alter its targeting or functions, proximity-dependent approaches were recently applied to identify a GPCR-associated protein network with a high temporal resolution. Specifically, engineered ascorbic acid peroxidase was employed in combination with quantitative proteomics to decipher β-2 adrenergic (Lobingier et al., 2017; Paek et al., 2017) and angiotensin II type 1 (Paek et al., 2017) receptor–interacting proteins.
Methods for the Identification of GPCR Phosphorylated Sites
GPCR phosphorylation is a key regulatory mechanism of receptor function (Lefkowitz, 2004). Over the past years, numerous techniques have appeared with increasing resolution to pinpoint phosphorylated residues (summarized in Table 1), which consist of serines, threonines, or tyrosines.
Radioactive Labeling.
The first method that was introduced for deciphering the phosphorylation status of GPCRs is a radioactivity-based technique, consisting of culturing cells in a medium in which phosphate is replaced with its radioactive counterpart, 32P, resulting in radioactive phosphorylated residues (Meisenhelder et al., 2001). After culture, cells are lysed and receptors are immunoprecipitated using specific antibodies, and then resolved by SDS-PAGE. Receptors can then be digested using an enzyme such as trypsin, and the resulting peptides are separated by two-dimensional migration using electrophoresis and chromatography. Radioactive peptides are then detected in-gel by autoradiography or using a phosphorimager, yielding a phosphorylation map for a given receptor in a given cell line (Chen et al., 2013). This method is very sensitive but does not give precise information on the number of phosphorylated sites nor their position. Radioactive labeling was initially employed to characterize CXCR4 phosphorylation upon agonist stimulation (Haribabu et al., 1997). These studies characterized the C-terminal domain as the preferred site for phosphorylation (Haribabu et al., 1997) and identified a serine cluster present in the C-terminal domain and containing two residues (Ser338, Ser339) phosphorylated following CXCL12 challenge (Orsini et al., 1999).
LC-MS/MS.
More recently, radioactive labeling-based methods have been progressively supplanted by the identification of phosphorylated residues by LC-MS/MS. In this method, the GPCR of interest is digested enzymatically, with one or several proteases, to generate peptides that cover a large part of the receptor sequence. The resulting peptides are then analyzed by LC-MS/MS (Dephoure et al., 2013). Although this approach can pinpoint any phosphorylated residue with high confidence, a few limitations complicate phosphorylated residue identification. First, phosphorylation can be lost during fragmentation. Second, since phosphorylation sites have a limited level of phosphorylation, only a small percentage of peptide is actually phosphorylated (Wu et al., 2011). Third, the identification of the phosphorylated residues in peptides with multiple adjacent phosphorylated residues can be challenging. For each modified site, a phosphorylation index can be estimated by dividing the ion signal intensity corresponding to the phosphorylated peptide by the sum of the ion signals of the phosphorylated peptide and its nonphosphorylated counterpart. Absolute quantification, and thus the stoichiometry of phosphorylation, can also be determined for each modified residue by spiking the sample with a known concentration of high-purity, heavy isotope–labeled peptides (AQUA peptides) that correspond to the phosphorylated peptide and not phosphorylated one. The respective ion signals of unlabeled and labeled peptides are then compared (Gerber et al., 2003). This powerful technology allowed a first comprehensive phosphorylation map of CXCR4, stimulated or not with CXCL12 in human embryonic kidney (HEK)293 cells: LC-MS/MS analyses identified six phosphorylated residues: Ser321, Ser324, Ser325, one between Ser338/339/341, one between Ser346/347/348, and either Ser351 or Ser352 (Busillo et al., 2010).
Mutagenesis.
Another approach that can be used as a stand-alone technique or in complement with the previously described methods is to mutate potential or predicted phosphorylated residues (into alanine or aspartate to inhibit or mimic their phosphorylation, respectively) and assess functional differences between mutated and wild-type receptor (Okamoto and Shikano, 2017). Nevertheless, introducing those mutations can potentially alter expression of the receptor, its conformation, or its cellular localization. Despite these limitations, mutagenesis approaches have shown unequivocal efficiency in identifying or validating several phosphorylated residues on CXCR4 (Orsini et al., 1999; Mueller et al., 2013) in combination with a radioactive-labeling method or use of phospho-specific antibodies. Furthermore, mutating all the serine and threonine residues to alanine in the ACKR3 C-terminus abolished β-arrestin recruitment and receptor internalization, suggesting that receptor trafficking depends on the phosphorylation of some of these residues (Canals et al., 2012).
Phospho-Specific Antibody.
To be able to detect and assess phosphorylation of residues in cells or tissues, antibodies that specifically target previously identified phosphorylated residues of GPCRs can be generated by immunizing animals with purified synthetic phosphorylated peptides encompassing the phosphorylated residues (Chen et al., 2013). After selection and functional validation, those antibodies can be used in Western blot or immunohistochemistry experiments. Phosphorylation can also be indirectly detected using antibodies specific to the unphosphorylated GPCR, showing decreased binding to the target when residues are phosphorylated, and recovery of the binding by using a protein phosphatase to dephosphorylate the receptor (Hoffmann et al., 2012). Antibodies that recognize several CXCR4 phosphorylated residues [Ser324/325, Ser330, Ser339, Ser338/339, and Ser346/347 (Woerner et al., 2005; Busillo et al., 2010; Mueller et al., 2013)] have been generated and used to investigate the receptor phosphorylation status in various conditions. To our knowledge, such phospho-specific antibodies are still lacking for ACKR3.
Association of CXCR4 and ACKR3 with Canonical GPCR-Interacting Proteins
G proteins, GRKs, and β-arrestins are the protein families considered canonical GPCR-interacting proteins controlling receptor activity or being involved in signal transduction. GPCR activity is a result of a tightly regulated balance between activation, desensitization, and resensitization events. After receptor activation and interaction with G proteins, several mechanisms integrate to trigger GPCR desensitization and/or modulate additional signaling cascades, including phosphorylation by GRKs and recruitment of β-arrestins (Penela et al., 2010b; Nogués et al., 2018).
G Proteins.
CXCR4 is known to couple to the pertussis toxin–sensitive Gαi protein family that mediates most of its signaling pathways (Busillo and Benovic, 2007). However, CXCR4 can also couple to other G proteins such as Gα12/13 (Tan et al., 2006; Kumar et al., 2011) and Gαq (Soede et al., 2001). Tan and colleagues (2001) observed that both Gαi and Gα13, as well as Gβγ subunits are involved in the CXCL12-dependent migration of Jurkat T cells. Specifically, Gαi proteins promote migration through the activation of Rac, whereas Gα12/13 proteins activate Rho. Though CXCR4 is coupled to both Gα12/13 and Gαi proteins in Jurkat cells, such a dual coupling has not been observed in other cell lines where the receptor specifically activates one or the other G protein family (Yagi et al., 2011). In fact, pertussis toxin inhibited the migration of nonmetastatic breast cancer cells (MCF-7), indicating that Gαi activation is required. However, it did not prevent the migration of metastatic breast cancer cells such as MDA-MB-231 and SUM-159. In those cell lines, Gα12/13 activation mediates CXCL12-induced migration via the activation of the Rho signaling axis (Yagi et al., 2011). Therefore, CXCR4 coupling to one or the other G protein family might depend on the abundance of GPCRs, G proteins, and downstream targets.
As an atypical chemokine receptor, ACKR3 lacks the DRYLAIV (Asp-Arg-Tyr-Leu-Ala-Ile-Val) motif necessary for interaction with G proteins. Nevertheless, using BRET, a study showed interaction of the receptor with G proteins in transfected HEK293 cells (Levoye et al., 2009). Yet, this interaction did not lead to the activation of G proteins (Levoye et al., 2009), reinforcing the common consensus that ACKR3 is unable to activate G proteins. Consistently, other studies showed that ACKR3 signals independently of G proteins through a mechanism requiring β-arrestins (Rajagopal et al., 2010; Canals et al., 2012).
Although these findings clearly indicate that ACKR3 cannot activate G proteins in most of the cell types, a report suggested that ACKR3 might activate G proteins in two specific cellular contexts, namely primary rodent astrocytes and human glioma cells (Ödemis et al., 2012). Using [35S]GTPγS-binding assay, calcium mobilization, and pertussis toxin-dependent activation of downstream signaling pathways [extracellular signal–regulated kinases (ERKs 1/2 and AKT phosphorylation), this group showed an ACKR3-dependent activation of Gαi/o proteins in primary astrocytes (Ödemis et al., 2012). They also reported pertussis toxin-dependent migration, proliferation, and activation of downstream signaling effectors in two human glioma cell lines (A764 and U343), further suggesting an ACKR3-dependent activation of Gαi/o. So far, this is the only report suggesting a possible coupling of ACKR3 with G proteins. Though these data must be further confirmed, one possibility is that such a coupling is cell type–specific. Since ACKR3 was shown to form a heterodimer with CXCR4 in transfected cell lines (Levoye et al., 2009) and CXCR4 is well known for its coupling with G proteins (vide supra), the authors also investigated the possibility that the ACKR3-dependent activation of Gαi/o proteins was mediated by the ACKR3/CXCR4 complex. However, constitutive suppression of CXCR4 expression in primary astrocytes did not influence the ability of ACKR3 to activate G proteins in the [35S]GTPγS-binding assay (Ödemis et al., 2012). Consistently, transient suppression of CXCR4 expression did not influence the ACKR3-dependent calcium mobilization in primary cultures of astrocytes. This suggests that ACKR3 coupling to G proteins in astroglial cells, if any, occurs independently of the ACKR3/CXCR4 complex assembly.
Although in that specific case CXCR4 did not influence ACKR3 signaling, accumulating evidence supports the hypothesis that ACKR3 might conversely influence CXCR4 signaling. Specifically, the organization of CXCR4 and ACKR3 in heterodimers appears to inhibit CXCR4 interaction with G proteins in transfected HEK293T cells, as assessed by saturation BRET (Levoye et al., 2009). In accordance with a possible influence of ACKR3 on CXCR4-dependent Gαi activation, Sierro and colleagues (2007) showed that the coexpression of ACKR3 with CXCR4 hindered the fast and G protein-dependent ERK activation triggered by CXCL12 exposure. In spite of these observations demonstrating that ACKR3 influences CXCR4-dependent G protein signaling, a direct proof of the role of the physical interaction between both receptors is still missing.
GRKs.
Agonist-occupied GPCRs are specifically phosphorylated by different GRKs, a family of seven serine/threonine kinases (Ribas et al., 2007; Penela et al., 2010a). GRKs 2, 3, 5, and 6 phosphorylate CXCR4 in the C-terminus, which contains 15 serine and three threonine residues that are potential phosphorylation sites (Fig. 1). At least six of these residues were shown to be phosphorylated following receptor activation by CXCL12 (Busillo et al., 2010; Barker and Benovic, 2011; Mueller et al., 2013). In HEK293 cells, Ser321, Ser324, Ser325, Ser330, Ser339, and two sites between Ser346 and Ser352 were shown to be phosphorylated in response to CXCL12 in the CXCR4 C-terminus using LC-MS/MS and phosphosite-specific antibodies (Busillo et al., 2010). GRK6 is able to phosphorylate Ser324/5, Ser339 and Ser330, the latter with slower kinetics, whereas GRK2 and GRK3 phosphorylate residues between Ser346 and Ser352 (Fig. 2) (Busillo et al., 2010), and specifically Ser346/347 (Mueller et al., 2013). Interestingly, the latter study suggested a hierarchy in such phosphorylation events, since Ser346/347 phosphorylation is achieved faster and is needed for the subsequent phosphorylation of Ser338/339 and Ser324/325. Notably, ligand washout resulted in rapid Ser324/325 and Ser338/339 dephosphorylation, whereas Ser346/347 residues did not exhibit major dephosphorylation during the 60-minute period studied (Mueller et al., 2013). Phosphorylation of CXCR4 by different GRKs can elicit several molecular responses, such as fluctuations in intracellular calcium concentration and phosphorylation of ERKs 1 and 2, leading to integrated cellular responses. In HEK293 cells, calcium mobilization is negatively regulated by GRK2, GRK6, and β-arrestin2. On the other hand, GRKs 3 and 6 together with β-arrestins act as positive regulators of ERK1/2 (Busillo et al., 2010). Overall, these studies show nonoverlapping roles for the different GRKs in the regulation of CXCR4 signaling. These differential roles may explain distinct cell type–dependent responses to CXCL12. However, what dictates the specific GRK subtype recruitment still needs to be investigated. Changes in the normal CXCR4 phosphorylation pattern as a result of receptor mutations or altered GRK activity can lead to abnormal receptor expression and/or responsiveness that promotes aberrant cell signaling and thus can contribute to several pathologies. Deletion of Ser346/347 leads to a gain of CXCR4-function and decreases receptor internalization and subsequent desensitization, indicating that mutations in the far C-terminus affect CXCR4-mediated signaling (Mueller et al., 2013). In this regard, a subpopulation of patients affected by WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis) syndrome, a rare primary immunodeficiency disease, display C-terminally truncated CXCR4, leading to refractoriness to desensitization and enhanced signaling (Balabanian et al., 2008). On the contrary, increased CXCR4 phosphorylation at Ser339 is associated with poor survival in adults with B-cell acute lymphoblastic leukemia and correlates with poor prognosis in acute myeloid leukemia patients (Konoplev et al., 2011; Brault et al., 2014). Altered GRK expression/activity can also impair CXCR4 phosphorylation patterns. GRK3 suppression may contribute to abnormally sustained CXCR4 signaling in classic types of glioblastoma (Woerner et al., 2012), some WHIM patients (Balabanian et al., 2008), and in triple negative breast cancer, thus potentiating CXCR4-dependent migration, invasion, and metastasis (Billard et al., 2016; Nogués et al., 2018). It is interesting to note that, although GRKs 2 and 3 share a high homology and are able to phosphorylate the same residues in CXCR4 in model cells, their function is not redundant. Whereas both CXCR4 and GRK2 levels are increased in breast cancer patients, GRK3 is decreased, suggesting a differential role for both GRKs in a cancer context (Billard et al., 2016; Nogués et al., 2018). In fact, deregulation of GRK2 potentiates several malignant features of breast cancer cells, and its level positively correlates with tumor growth and increased metastasis occurrence (Nogués et al., 2016), but whether these roles involve changes in CXCR4 modulation is still under investigation. On the other hand, impaired chemotaxis of T and B cells toward CXCL12 is noted in the absence of GRK6, whereas GRK6 deficiency potentiates neutrophil chemotactic response to CXCL12 (Fong et al., 2002; Vroon et al., 2004), suggesting that the occurrence of highly cell type–specific mechanisms in the control of the CXCL12-CXCR4-GRK6 axis. Overall, these data indicate the complexity of CXCR4 modulation by GRKs and support the need for a better characterization of cell type– or disease-specific CXCR4-GRKs interactions.
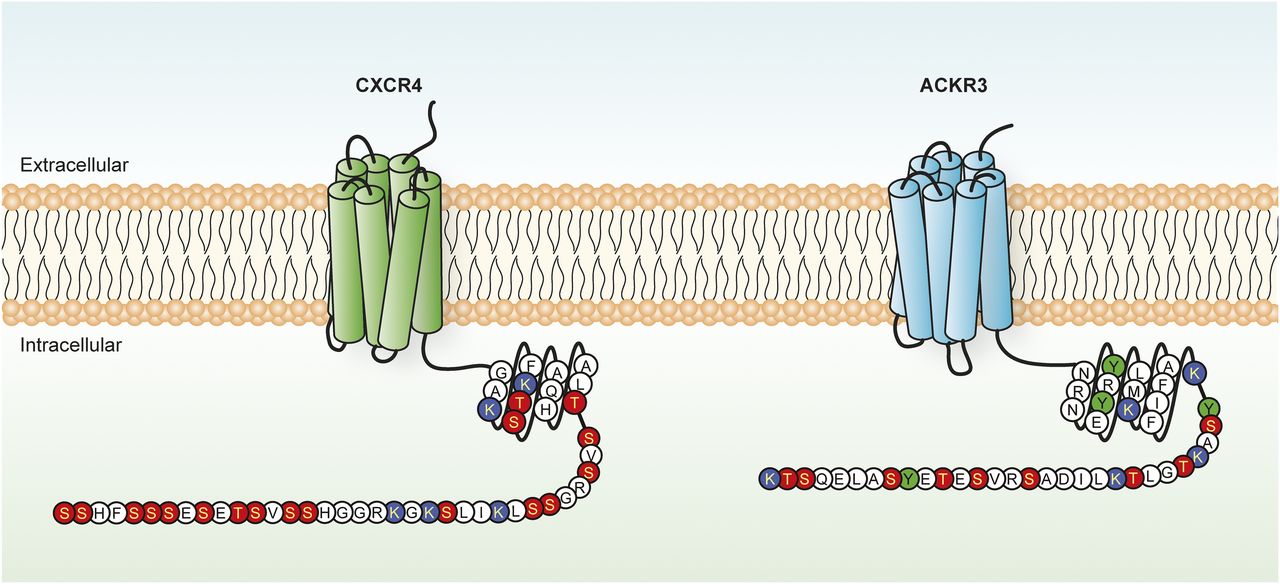
CXCR4 and ACKR3 residues potentially subjected to post-translational modifications. Schematic representation of the C-terminal tail of CXCR4 and ACKR3, in which serine/threonine (red), tyrosine (green), and lysine (blue) residues potentially subjected to post-translational modifications are highlighted.

CXCR4 C-terminus phosphosites. Schematic representation of the C-terminal tail of CXCR4, in which serine residues known to be phosphorylated are highlighted in light blue. The kinases or the extracellular stimuli responsible for the phosphorylation are also specified. Hrg, heregulin.
ACKR3 has lately been the focus of many studies, in particular because of its role in cancer progression and metastasis. However, the mechanisms underlying its regulation are still not well understood, although this receptor has been shown to interact with GRKs and arrestins and to associate with other partners. The C-terminus of ACKR3 contains five serine and four threonine residues that can potentially be phosphorylated (Fig. 1). Unlike for CXCR4, little is known about their actual phosphorylation status during the activation of the receptor, as no mass spectrometry data are available to date and only few mutational studies have been conducted (Canals et al., 2012; Hoffmann et al., 2012). In fact, only one study conducted in astrocytes showed that ACKR3 is phosphorylated by GRK2, but not other GRKs, and that this phosphorylation is essential for subsequent ACKR3-operated activation of ERK1/2 and AKT pathways (Lipfert et al., 2013). This study suggests that ACKR3 is indeed phosphorylated by GRKs, but the isoform(s) involved and subsequent responses are probably cell type–dependent and remain to be investigated in detail.
Arrestins.
A study revealed that site-specific phosphorylation of CXCR4 by GRK isoforms has contrasting effects upon β-arrestin recruitment: Although receptor phosphorylation at its extreme C-terminus [two residues between Ser346 and Ser352 (Busillo et al., 2010), and specifically Ser346/347 (Mueller et al., 2013)] by GRK2/3 is a necessary step in β-arrestin binding; its phosphorylation by GRK6 at upstream residues [Ser324/5, Ser330, and Ser339 (Busillo et al., 2010)] appears to inhibit arrestin recruitment to CXCR4 or results in a receptor/arrestin complex that adopts a conformation that is distinct from that induced by phosphorylation of extreme C-terminal residues (Oakley et al., 2000; Busillo et al., 2010; Mueller et al., 2013). Further supporting the importance of Ser324/5 and Ser339 phosphorylation in β-arrestin recruitment, CXCR4 truncation mutants showing impaired phosphorylation at Ser324/325 and Ser338/339 also exhibit reduced CXCL12-induced receptor internalization (Mueller et al., 2013).
β-arrestins are also scaffold proteins for several signaling molecules, thus eliciting additional β-arrestin-dependent signaling pathways (Shenoy and Lefkowitz, 2011; Peterson and Luttrell, 2017). Following the recognition of β-arrestin-dependent signaling, the notion of biased ligands that preferentially induce G protein-dependent or independent signaling has emerged (Reiter et al., 2012). Biased signaling at chemokine receptors has been exhaustively reviewed elsewhere (Steen et al., 2014). For instance, a CXCR4-derived pepducin, ATI-2341, acts as a biased CXCR4 agonist that promotes Gαi signaling but not β-arrestin signaling, in contrast to CXCL12, which activates both G protein-dependent and independent pathways (Quoyer et al., 2013; Steen et al., 2014).
Upon activation by its cognate ligands, CXCL11 and CXCL12, ACKR3 recruits β-arrestin2 both in vitro (Rajagopal et al., 2010; Benredjem et al., 2016) and in vivo (Luker et al., 2009), a process leading to receptor internalization (Canals et al., 2012), transport to lysosomes, and degradation of the receptor-bound chemokine (Luker et al., 2010; Naumann et al., 2010; Hoffmann et al., 2012). The receptor is then mainly recycled back to the plasma membrane (Luker et al., 2010) even if a partial degradation of ACKR3 can be observed (Hoffmann et al., 2012). Interestingly, the rate of receptor internalization is faster and recycling is lower in the presence of CXCL11, compared with CXCL12 (Montpas et al., 2018).
As previously mentioned, systematic mutation of C-terminal serine/threonine residues to alanine abolished ligand-induced β-arrestin2 recruitment to ACKR3, as monitored by BRET (Canals et al., 2012) and decreased ACKR3 internalization and subsequent degradation of radiolabeled CXCL12 in HEK293 cells (Hoffmann et al., 2012). Selective mutations of the two C-terminal serine/threonine clusters to alanine revealed differences in their functional properties. Mutating Ser335, Thr338, and Thr341 (first cluster) or Ser350, Thr352, and Ser355 (second cluster) to alanine decreased CXCL12 internalization only after a 5-minute challenge but not following longer agonist receptor stimulation. Yet, only mutation of the second cluster prevented CXCL12 degradation. Furthermore, ACKR3 appears to undergo ligand-independent internalization to a much greater extent than CXCR4 (Ray et al., 2012), and residues 339–362 (the two serine/threonine clusters) are essential for this peculiar cell fate in HEK293 cells. Although numerous studies have shown that ACKR3 internalization and the resulting chemokine degradation are dependent on β-arrestin, recent findings have been challenging this consensus (Montpas et al., 2018). Specifically, this study shows that β-arrestins are dispensable to chemokine degradation, suggesting that other scaffold proteins might be involved in this process.
Association of CXCR4 with Noncanonical GPCR-Interacting Proteins
Functional Interaction of CXCR4 with Second Messenger–Dependent Kinases and Receptor Tyrosine Kinases.
Accumulating evidence indicates that phosphorylation of GPCRs by second messenger–dependent kinases such as protein kinase A and protein kinase C (PKC) (Lefkowitz, 1993; Ferguson et al., 1996; Krupnick and Benovic, 1998), as well as by members of the receptor tyrosine kinase family (Delcourt et al., 2007), participates in the regulation of GPCR signaling. CXCR4 is phosphorylated by PKC at Ser324/5 upon CXCL12 stimulation (Busillo et al., 2010), and this kinase has also been involved in Ser346/7 phosphorylation (Luo et al., 2017), even though these results are not entirely consistent with a previous study using different PKC inhibitors (Mueller et al., 2013). In some glioblastoma cell types, CXCR4 is phosphorylated at Ser339 in response to the PKC activator phorbol myristate acetate (Woerner et al., 2005). This suggests that Ser339 is also a PKC phosphorylation site, and that this phosphorylation event may serve as a crosstalk mechanism between CXCR4 and GPCRs that activate Gαq-PKC signaling. Sphingosine 1-phosphate receptors, neurokinin-1 and lysophosphatidic acid receptors may possibly be involved in glioblastoma progression via this means (Cherry and Stella, 2014). Nevertheless, the functional impact of Ser339 phosphorylation in glioblastoma remains to be established. Likewise, epidermal growth factor (EGF) through activation of its receptor can also promote CXCR4 phosphorylation at Ser339 in glioblastoma cells (Woerner et al., 2005), and both EGF and heregulin trigger Ser324/325 and Ser330 phosphorylation in the breast cancer T47D cell line (Sosa et al., 2010). Interestingly, in MCF7 breast cancer cells, heregulin also promotes CXCR4 phosphorylation on tyrosine residues via epidermal growth factor receptor (EGFR), leading to β-arrestin2 association with CXCR4 and downstream activation of the PRex1/Rac1 axis. However, it is still unclear whether the EGFR-CXCR4 functional interaction is direct or depends on other kinases (Sosa et al., 2010). In another breast cancer line, BT-474, CXCR4 is phosphorylated on tyrosine residues in response to CXCL12 and through activation of ErbB2/ErbB3 and EGFR (Sosa et al., 2010). Although the specific Tyr residue(s) phosphorylated were not identified, it is worth noting that CXCR4 displays four intracellular Tyr residues (Ahr et al., 2005). Tyr157 in the third intracellular loop has been be involved in CXCR4-dependent STAT3 signaling (Ahr et al., 2005), whereas Tyr135, within the conserved DRY motif, might be involved in receptor coupling to G proteins (Rovati et al., 2007). Consistent with this hypothesis, EGFR-mediated phosphorylation of the equivalent Tyr in another GPCR (the μ-opioid receptor) has been reported to reduce coupling to G proteins (Clayton et al., 2010). Therefore, identification of tyrosine residues phosphorylated in CXCR4 might add some insight into the mechanisms by which growth factor–receptor tyrosine kinases modulate CXCR4 activity. The crosstalk between CXCR4 and ErbB2/ErbB3 and EGFR remains an interesting avenue for future research, given the involvement of both receptors in cancer.
Physical Interaction with Noncanonical GPCR-Interacting Proteins.
Beside canonical GIPs, CXCR4 has been shown to interact with additional proteins that modulate CXCR4 trafficking, subcellular localization and signaling, and proteins whose functions are still unknown. CXCR-interacting proteins, the methods used for the identification of these proteins, the site of their interaction in the receptor sequence, and their functional impact are summarized in Table 2.
Proteins controlling CXCR4 localization or trafficking.
Filamin A directly interacts with CXCR4 and stabilizes the receptor at the plasma membrane by blocking its endocytosis (Gómez-Moutón et al., 2015). CXCR4 association with the E3 ubiquitin ligase atrophin–interacting protein 4 (AIP4) has opposite consequences: ubiquitination of CXCR4 by AIP4 targets the receptor to multivesicular bodies, which is followed by receptor degradation. In addition, agonist treatment increases CXCR4/AIP4 interaction, as assessed by Co-IP and FRET experiments (Bhandari et al., 2009), indicating that this interaction is dynamically regulated by a receptor conformational state. In addition, the authors identified Ser324 and Ser325 as critical sites for the formation of the CXCR4/AIP4 complex upon CXCL12 exposure. The mutation of both residues to alanine drastically reduces association of AIP4 with CXCR4, whereas their mutation to aspartic acid increases this interaction. Since Ser324/325 are phosphorylated by GRK6 (Busillo et al., 2010), these results suggest that CXCR4 activation by CXCL12 triggers recruitment of GRK6, which in turn phosphorylates the receptor at Ser324/325 to promote its interaction with AIP4. AIP4 then ubiquitinates CXCR4 and mediates its degradation (Bhandari et al., 2009). Reticulon-3 is another CXCR4-interacting protein that promotes its translocation to the cytoplasm (Li et al., 2016).
Proteins modulating CXCR4 signaling and functions.
HLA class II histocompatibility antigen gamma chain (CD74), a single-pass type II membrane protein that shares with CXCR4 the ability to bind to the macrophage migration inhibitory factor (MIF), was also shown to interact with CXCR4. The CXCR4/CD74 complex is involved in AKT activation (Schwartz et al., 2009). In fact, blocking either CXCR4 or CD74 inhibits MIF-induced AKT activation. Using FRET, an interaction between CXCR4 and the Toll-like receptor 2 was observed in human monocytes upon activation by Pg-fimbria (fimbriae produced by the major pathogen associated with periodontitis named Porphyromonas gingivalis). Analysis of a possible crosstalk between the two receptors showed that Pg-fimbria directly binds to CXCR4 and inhibits Toll-like receptor 2–induced nuclear factor-κB activation by P. gingivalis (Hajishengallis et al., 2008; Triantafilou et al., 2008). In Jurkat cells, endolyn (CD164) coprecipitates with CXCR4 in the presence of CXCL12 presented on fibronectin (Forde et al., 2007). CXCR4-CD164 interaction participates in CXCL12-induced activation of AKT and protein kinase C zeta (PKCζ). In fact, the downregulation of CD164 reduces the activation of both kinases measured upon exposure of Jurkat cells to CXCL12. CXCR4/CD164 interaction has been detected in additional cell lines, such as primary human ovarian surface epithelial cells stably expressing CD164 (Huang et al., 2013).
The ability of CXCR4 to promote cell migration requires deep cytoskeletal rearrangements that can be modulated by CXCR4-interacting proteins. In Jurkat J77 cells, CXCR4 constitutively associates with drebrin (Pérez-Martínez et al., 2010), a protein known to bind to F-actin and stabilize actin filaments. Drebrin is also involved in CXCR4- and CD4-dependent HIV cellular penetration (Gordón-Alonso et al., 2013). CXCR4 interacts with diaphanous-related formin-2 (mDIA2). This interaction induces cytoskeletal rearrangements that lead to nonapoptotic blebbing. mDIA2-CXCR4 interaction is only detected during nonapoptotic amoeboid blebbing and is confined to nonapoptotic blebs upon CXCL12 stimulation (Wyse et al., 2017), suggesting a fine spatiotemporal regulation of the interaction. CXCR4 also constitutively associates with the motor protein nonmuscle myosin H chain (NMMHC) via its C-terminus (Rey et al., 2002). The authors showed that NMMHC and CXCR4 are colocalized in the leading edge of migrating lymphocytes, suggesting that this association might have a role in lymphocyte migration. The PI3-kinase isoform p110γ coprecipitates with CXCR4 in CXCL12-stimulated human myeloid cells. This interaction contributes to receptor-operated integrin activation and chemotaxis of myeloid cells (Schmid et al., 2011). Finally, CXCR4 was found to be part of a junctional mechano-sensitive complex through its interaction with the platelet endothelial cell adhesion molecule (PECAM-1) (dela Paz et al., 2014).
Proteins with unknown functions.
Other potential CXCR4-interacting proteins have been identified using unbiased methods. These include the lysosomal protein cation-transporting ATPase ATP13A2 (Usenovic et al., 2012) and the nuclear protein Myb-related protein B that is involved in cell cycle progression (Wang et al., 2011). In a study aimed at characterizing the human interactome by Co-IP of 1125 green fluorescent protein–tagged proteins and LC-MS/MS analysis, CXCR4 was found to coprecipitate with the potassium channel subfamily K member 1, the CSC1-like protein 2, and the vesicle transport protein GOT1B (Hein et al., 2015). In another study, CXCR4 was found to interact with the eukaryotic translation initiation factor 2B complex in an acute lymphoblastic leukemia cell line (pre-B NALM-6 cells) but not in primary lymphocytes (Palmesino et al., 2016). The interaction was negatively regulated by CXCL12 exposure and confirmed by colocalization analysis. The same study showed that CXCR4 recruits parafibromin, SH2 domain binding protein, hypothetical protein PD2, nucleophosmin, cyclin-dependent kinase 11B, receptor-type tyrosine-protein phosphatase S and galectin (Palmesino et al., 2016).
Association of ACKR3 with Noncanonical GPCR-Interacting Proteins
Unlike for CXCR4, only few proteins are described as ACKR3-interacting proteins. Given the described role of ACKR3 in cancer, several studies have addressed ACKR3 crosstalk with well known pro-oncogenic growth factor receptors. ACKR3 colocalizes with and phosphorylates EGFR in breast and prostate cancer cells (Singh and Lokeshwar, 2011; Salazar et al., 2014; Kallifatidis et al., 2016), via cell type–specific mechanisms. However, a potential role for EGFR in ACKR3 crossactivation was not assessed in these studies. Some reports also suggest a possible functional interaction between ACKR3 and transforming growth factor beta (Rath et al., 2015) or vascular endothelial growth factor (Singh and Lokeshwar, 2011) receptors, but whether they involve physical interaction with ACKR3 and/or ACKR3 phosphorylation and activation was not assessed. ACKR3 weakly interacts with the MIF receptor CD74 (Alampour-Rajabi et al., 2015). Moreover, ACKR3 colocalizes with PECAM-1, the cell adhesion molecule required for leukocyte transendothelial migration in human coronary artery endothelial cells (dela Paz et al., 2014). Using a membrane yeast two-hybrid assay screen, cation-transporting ATPase ATP13A2 was identified as a putative ACKR3-interacting protein (Usenovic et al., 2012). In the study aimed at characterizing the human interactome of 1125 green fluorescent protein–tagged proteins, ACKR3 was found to interact with the gap junction β-2 protein, the 54S ribosomal protein L4, 54S ribosomal protein L4, mitochondrial (MRPL4), different ATP synthases (ATP5H, ATP5B, ATP5A1, ATP50), ACKR3 itself, the caspase separin ESPL1, the probable E3 ubiquitin-protein ligase HECTD2, and the putative E3 ubiquitin-protein ligase UBR7 (Hein et al., 2015). Ubiquitination is an essential mechanism of receptor regulation (Marchese and Benovic, 2001; Shenoy, 2007). ACKR3 can undergo ubiquitination in an agonist-dependent and -independent manner to regulate receptor trafficking. Ubiquitination is promoted by three enzymes, E1, E2, and E3, that ubiquitinate proteins on lysine residues (Dores and Trejo, 2012; Alonso and Friedman, 2013). Unexpectedly, ACKR3 is ubiquitinated by E3 ubiquitin ligase (E3) in the absence of an agonist and undergoes deubiquitination upon CXCL12 activation (Canals et al., 2012). Mutation of the five lysines in the receptor C-terminus to alanine, to prevent ubiquitination, impaired ACKR3 cell-trafficking and decreased ACKR3-mediated CXCL12 degradation (Hoffmann et al., 2012).
Conclusions
The identification of GPCR-interacting proteins and residues subjected to post-translational modification is of utmost importance. Several techniques are currently available to decipher the GPCR interactome and phosphorylation profile. These techniques have been successfully applied to CXCR4, revealing important interacting proteins as well as key residues involved in the regulation of receptor-mediated signal transduction. In contrast, ACKR3 interactome and phosphorylation sites have not been systematically investigated. Unbiased studies of the ACKR3 interactome and its phosphorylated residues and their control by ACKR3 ligands should open new avenues in the understanding of ACKR3 pathophysiological functions and the underlying signaling mechanisms.
Acknowledgments
This review is part of the minireview series “From insight to modulation of CXCR4 and ACKR3 (CXCR7) function.” We thank all our colleagues from the ONCORNET consortium for continuous scientific discussions and insights.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Fumagalli, Zarca, Neves, Caspar, Hill, Mayor, Smit, Marin.
Footnotes
- Received November 28, 2018.
- Accepted February 21, 2019.
↵1 A.Z. and M.N. contributed equally to this work.
This research was funded by a European Union’s Horizon2020 MSCA Program [Grant agreement 641833 (ONCORNET)]; A.F. and P.M. are also supported by CNRS, INSERM, Université de Montpellier and Fondation pour la Recherche Médicale (FRM); F.M. laboratory is also supported by grants from Ministerio de Economía; Industria y Competitividad (MINECO) of Spain [Grant SAF2017-84125-R]; CIBERCV-Instituto de Salud Carlos III, Spain [Grant CB16/11/00278] to F.M., cofunded with European FEDER contribution); Comunidad de Madrid-B2017/BMD-3671-INFLAMUNE; and Fundación Ramón Areces.
Abbreviations
- ACKR3 or CXCR7
- atypical chemokine receptor 3
- AIP4
- E3 ubiquitin ligase atrophin-interacting protein 4
- BRET
- bioluminescence resonance energy transfer
- CD164
- endolyn
- CD74
- HLA class II histocompatibility antigen gamma chain
- CHAPSO
- 3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate
- Co-IP
- co-immunoprecipitation
- CXCL12
- C-X-C motif chemokine 12
- CXCR4
- C-X-C motif chemokine receptor 4
- DDM
- n-dodecyl-β-d-maltopyranoside
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal–regulated kinases
- FRET
- fluorescence resonance energy transfer
- GIP
- G protein-coupled receptor–interacting protein
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- HEK
- human embryonic kidney
- IP
- immunoprecipitation
- LC-MS/MS
- liquid chromatography coupled to tandem mass spectrometry
- MIF
- macrophage-migration inhibitory factor
- PKC
- protein kinase C
- STAT
- signal transducer and activator of transcription
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}