


Abstract
Chemokine receptors belong to the class A of G protein–coupled receptors (GPCRs) and are implicated in a wide variety of physiologic functions, mostly related to the homeostasis of the immune system. Chemokine receptors are also involved in multiple pathologic processes, including immune and autoimmune diseases, as well as cancer. Hence, several members of this GPCR subfamily are considered to be very relevant therapeutic targets. Since drug discovery efforts can be significantly reinforced by the availability of crystal structures, substantial efforts in the area of chemokine receptor structural biology could dramatically increase the outcome of drug discovery campaigns. This short review summarizes the available data on chemokine receptor crystal structures, discusses the numerous applications from chemokine receptor structures that can enhance the daily work of molecular pharmacologists, and describes the challenges and pitfalls to consider when relying on crystal structures for further research applications.
SIGNIFICANCE STATEMENT This short review summarizes the available data on chemokine receptor crystal structures, discusses the numerous applications from chemokine receptor structures that can enhance the daily work of molecular pharmacologists, and describes the challenges and pitfalls to consider when relying on crystal structures for further research applications.
Introduction
G protein–coupled receptors (GPCRs) are one of the biggest families of transmembrane proteins in the human genome (Lander et al., 2001). Their relevance is reflected by the large proportion of marketed drugs targeting GPCRs (Hopkins and Groom, 2002; Hauser et al., 2018) and the dramatic increase in the number of GPCR structures deposited in the Protein Data Bank (PDB) in the last decade (Katritch et al., 2013; Hauser et al., 2018). To illustrate, 337 GPCR-ligand complexes have been solved to date, including 63 unique receptor proteins, according to data from the GPCR database (GPCRdb) (Pándy-Szekeres et al., 2018). These GPCR structures have been obtained by binding a wide variety of ligands and show a range of conformational GPCR states. This wave of structural knowledge is stimulating virtual screening and structure-based drug design (SBDD) approaches, as well as the elucidation of the molecular mechanisms of receptor activation and functional selectivity (Venkatakrishnan et al., 2016; Erlandson et al., 2018).
Chemokine receptors are a subfamily of class A GPCRs with a number of key physiologic roles. These roles include a variety of developmental functions, homeostasis of the immune system by controlling the homing of hematopoietic stem cells, and regulation of the activation, differentiation, migration, and survival of leukocytes (Scholten et al., 2012; Koenen et al., 2019). Chemokine receptors are therefore key in inflammatory processes and are also involved in a number of immune and autoimmune diseases, including psoriasis, atherosclerosis, and allergies, among others (Murdoch and Finn, 2000); they are also considered to be important in cancer (Neves et al., 2019). Some chemokine receptors are also used by the human immunodeficiency virus (HIV) to enter into target cells (Scarlatti et al., 1997). Consequently, several chemokine receptors are very interesting drug targets (Kufareva et al., 2017). To date, the U.S. Food and Drug Administration has approved three chemokine receptor–based therapeutics: plerixafor (Mozobil; Sanofi) targeting C-X-C chemokine receptor CXCR4, indicated for stem cell mobilization in non-Hodgkin lymphoma and multiple myeloma (Sancho et al., 2016); maraviroc (Celsentri; ViiV Healthcare) targeting C-C chemokine receptor CCR5, indicated for HIV-1 infection (Meanwell and Kadow, 2007); and the monoclonal antibody mogamulizumab (Poteligeo; Kyowa Kirin, Inc.) targeting CCR4, indicated for cutaneous T-cell lymphoma (Ishii et al., 2010).
As mentioned, structural knowledge on the family of GPCRs has substantially increased over the past years, and both NMR and X-ray crystallography approaches have been successfully applied for solving chemokine receptor structures as well (Wu et al., 2010; Park et al., 2012; Tan et al., 2013; Burg et al., 2015; Qin et al., 2015; Oswald et al., 2016; Zheng et al., 2016, 2017; Miles et al., 2018; Peng et al., 2018; Apel et al., 2019). This review will only focus on X-ray structures; for a recent overview of the application of NMR to GPCR structure elucidation, the reader is referred to (Shimada et al., 2019). To date, crystal structures of five different chemokine receptor complexes are available (Fig. 1): 1) CXCR4 binding the small molecule isotiourea-1t (IT1t) (Wu et al., 2010), the peptide-like ligand CVX15 (Wu et al., 2010), and the chemokine-like viral macrophage inflammatory protein 2 (vMIP-II) (Qin et al., 2015); 2) CCR5 binding the small molecule U.S. Food and Drug Administration–approved drug maraviroc (Tan et al., 2013), the C-C motif chemokine ligand CCL5 truncation 5P7 (Zheng et al., 2017), and the small molecule antagonist compounds 21 and 34 (Peng et al., 2018); 3) US28, a viral chemokine-like receptor, binding the human chemokine agonist C-X3-C chemokine ligand CX3CL1 (Burg et al., 2015), an engineered version of CX3CL1 with randomized N-terminal residues (namely CX3CL1.35; Miles et al., 2018), and intracellular nanobody Nb7 that keeps the receptor in an active-like state (Burg et al., 2015; Miles et al., 2018); 4) CCR2 binding small molecule ligands in two different binding sites, BMS-681 and CCR2-RA-[R] (Zheng et al., 2016), as well as the small molecule antagonist MK-0812 (Apel et al., 2019); and 5) CCR9 binding the intracellular small molecule vercirnon (Oswald et al., 2016). In addition, a CCR5 electron microscopy structure has been solved bound to the HIV envelope glycoprotein 160 (gp160) and the cluster of differentiation CD4 receptor (Shaik et al., 2019).

Overview of chemokines and chemokine receptor structures. All cocrystallized chemokine receptors are shown in gray cartoons and depicted according to their phylogenetic relationship within the chemokine receptors subfamily. The crystallized receptors include CCR5 (PDB IDs: 4MBS, 5UIW, 6AKX, and 6AKY), CCR2 (PDB IDs: 5T1A, 6GPS, and 6GPX), CCR9 (PDB ID: 5LWE), US28 (PDB IDs: 4XT1, 4XT3, 5WB1, and 5WB2), and CXCR4 (PDB IDs: 3ODU, 3OE0, 3OE6, 3OE8, 3OE9, and 4RWS). Ligands are colored based on chemical nature: small molecules (green), peptides (magenta), chemokines (cyan), and nanobody (purple). Examples of soluble chemokine structures are shown in the central panel, including CCL5 cocrystallized in complex with a heparin-derived disaccharide (PDB ID: 5DNF) (Liang et al., 2016), the CXCL12 dimer cocrystallized with a small molecule modulator (PDB ID: 4UAI) (Smith et al., 2014), and a CXCL11 NMR model (PDB ID: 1RJT) (Booth et al., 2004). For a more complete overview of standalone chemokine structures, the reader is referred to (Kufareva et al., 2017) and (Ziarek et al., 2017). The phylogenetic tree is based on the structure-based alignment in GPCRdb (Pándy-Szekeres et al., 2018) and calculated in JalView (Waterhouse et al., 2009) (average distance using percent identity).
The potential of using SBDD methods is reflected by the large amount of ligands found for various chemokine receptors (Kellenberger et al., 2007; Liu et al., 2008; Davies et al., 2009; Pérez-Nueno et al., 2009; Mysinger et al., 2012; Vitale et al., 2013; Yoshikawa et al., 2013; Wang et al., 2014; Das et al., 2015; Schmidt et al., 2015; Mishra et al., 2016; Arimont et al., 2017). Moreover, SBDD methods prove to be more effective when a crystal structure is available over the use of de novo techniques or homology models (Mysinger et al., 2012; Arimont et al., 2017). This illustrates one of the numerous advantages of relying on structural knowledge over the protein sequence only, as well as the need for new structures of other chemokine receptors. However, chemokine receptors, like any other GPCR and transmembrane proteins, are inherently challenging to crystallize (Kobilka, 2013; Piscitelli et al., 2015). The first prerequisite for experimentally solving a protein structure is obtaining large amounts of stable, purified, homogeneous protein (Piscitelli et al., 2015). Protein purification has proven challenging with highly dynamic GPCRs (Milić and Veprintsev, 2015). Multiple advances in the use of protein engineering, including mutagenesis, truncations, and chimeric constructs, as well as the use of stabilizing interaction partners, such as antibodies and nanobodies (Bobkov V. et al., submitted manuscript), have helped to overcome the challenges of GPCR instability (Ayoub et al., 2017; Manglik et al., 2017). To illustrate, CXCR4 and CCR2 have been crystallized fused to the T4 lysozyme (Wu et al., 2010; Qin et al., 2015; Zheng et al., 2016), CCR5 and CCR2 fused to rubredoxin (Tan et al., 2013; Peng et al., 2018; Apel et al., 2019), CCR9 with seven thermostabilizing mutations (Oswald et al., 2016), and US28 binding the intracellular nanobody Nb7 (Burg et al., 2015; Miles et al., 2018).
In this review, we will summarize the available data on chemokine receptor structures, discuss the numerous applications of chemokine receptor structures for drug discovery, and describe the challenges and pitfalls that one can encounter when relying on crystal structures for research applications.
Chemokine Receptor Structures
To date, 5 of 28 chemokine receptor family members have been crystallized (Fig. 1). Despite the low representation of cocrystallized chemokine GPCRs, the structures available offer rather representative information for this GPCR subfamily: cocrystallized ligands of different chemical nature, binding in different binding sites, and receptors in different conformational states have been crystallized. In addition, a complex of CCR5-CD4-gp160 of HIV-1 was recently solved by cryo-electron microscopy (cryo-EM) (Shaik et al., 2019).
Chemokine-bound crystal structures include CXCR4-vMIP-II (Qin et al., 2015), US28-CX3CL1 (Burg et al., 2015; Miles et al., 2018), and CCR5-5P7-CCL5 (Zheng et al., 2017) and support the two-steps/two-sides mechanism of chemokine binding (Kufareva et al., 2014). The sequences of the cocrystallized chemokines can be found in Fig. 2. According to the two-steps/two-sides model, the globular core of the chemokine binds first to the extracellular surface of the receptor (chemokine recognition site CRS1), which allows for a subsequent interaction between the N terminus of the chemokine and the orthosteric binding site within the seven-transmembrane (7TM) domain (or CRS2) (Scholten et al., 2012). This model was recently expanded in accordance with the most recent crystal structure of a chemokine-bound receptor, the CCR5-5P7-CCL5 complex (Zheng et al., 2017), and to extensive experimental evaluation of the contributions of amino acid residues in both CCR1 and its ligands to affinity and receptor activation (Sanchez et al., 2019). The CCR5-5P7-CCL5 complex shows a third site that the authors annotate as CRS1.5, where the conserved receptor motif P19-C20 is packed against the chemokine disulphide bond (Zheng et al., 2017). This third site would act as a pivot point between CRS1 and CRS2, allowing a specific arrangement of interactions in between receptor and ligand (Zheng et al., 2017). Experimental evaluation of CCR1 and its ligands has shown that CRS1 and CRS2 contribute to ligand binding but that full receptor activation cannot be explained solely by high-affinity ligand binding. This observation has led to the proposal of a third step for the model, now called the three-step model, in which a conformational change of the receptor-ligand complex results in receptor activation (Sanchez et al., 2019).

Overview of cocrystallized chemokine receptor ligands, including small molecules, peptides, and chemokines.
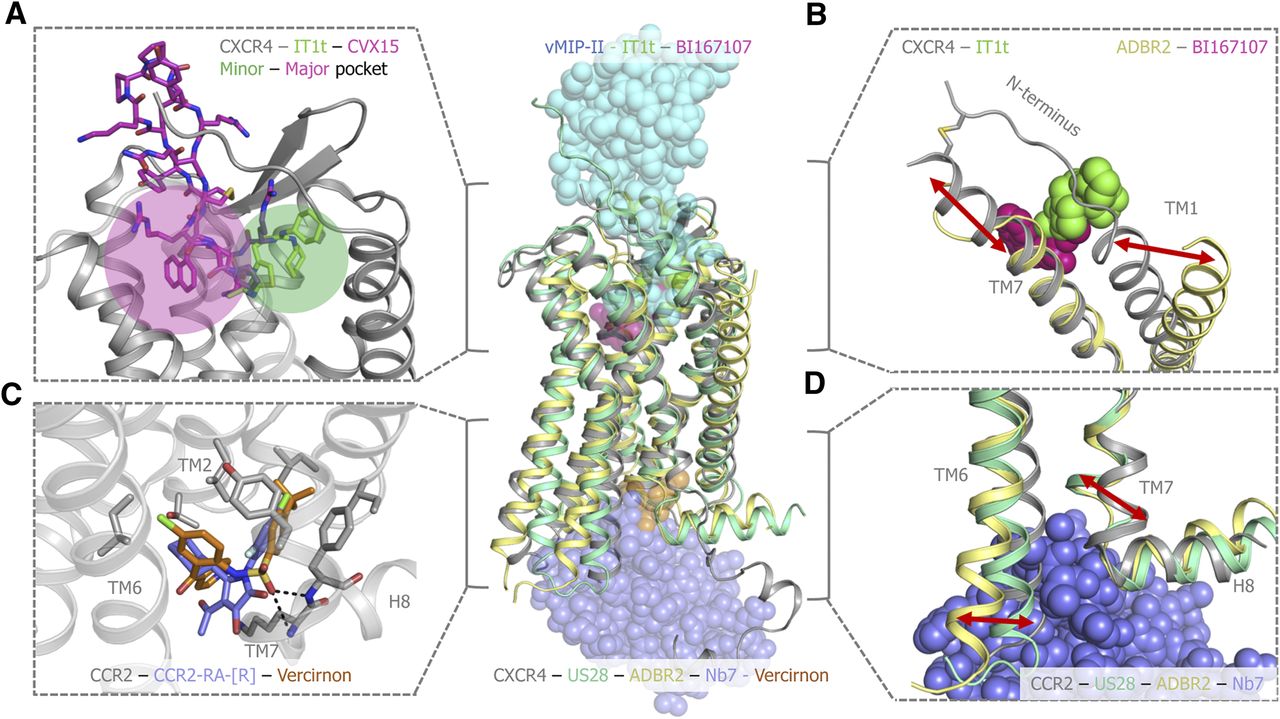
Despite the overall conserved geometry and stoichiometry of chemokine binding, the specific interactions that occur in CRS2 are chemokine receptor specific, as illustrated by the reported chemokine-bound X-ray structures (Burg et al., 2015; Qin et al., 2015; Zheng et al., 2017; Miles et al., 2018). Despite the knowledge on the conserved mechanism of binding and the specific pattern of interactions between the chemokine and its receptors, the redundancy and functional selectivity of the chemokine system is still poorly understood. Multiple chemokines are able to bind the same GPCR, whereas some GPCRs are able to bind multiple chemokines (Scholten et al., 2012). Chemokine receptors are also able to bind small molecule and peptide ligands (Fig. 2). The CVX15-bound CXCR4 structure shows the ability of peptide mimetics to mimic the binding of the large chemokines, interacting with multiple chemokine binding residues or hotspots (Wu et al., 2010; Arimont et al., 2017) (Fig. 3A). Small molecules occupy a space in the binding site that can (partially) overlap with the chemokine binding site, as has been reported for IT1t (Wu et al., 2010), maraviroc (Tan et al., 2013), compounds 21 and 34 (Peng et al., 2018), BMS-681 (Zheng et al., 2016), and MK-0812 (Apel et al., 2019) (Figs. 1, 2, and 3A). The (partial) overlap explains the different levels of chemokine displacement that are observed in experiments using different small molecule (potentially allosteric) modulators (Adlere et al., 2019). Moreover, the orthosteric binding site is divided into a minor subpocket and a major subpocket (Fig. 3A). Ligands can bind to either pocket exclusively (IT1t, minor pocket; and CVX15, major pocket; CXCR4 in Fig. 1, green and magenta, respectively) (Wu et al., 2010) or simultaneously (maraviroc and compounds 21 and 34; CCR5 in Fig. 1, green) (Tan et al., 2013). BMS-681and MK-0812 have been crystallized protruding from the TM domain toward the membrane interface (Zheng et al., 2016; Apel et al., 2019). Intriguingly, the CCR2 and CCR9 crystal structures show that small molecule modulators CCR2-RA-[R] (Zheng et al., 2016) and vercirnon (Oswald et al., 2016) are also able to bind at the intracellular side of chemokine receptors, showing the targetability of an intracellular pocket (Figs. 1, 2, and 3C).

Details of unique features observed in chemokine receptor structures. (A) CVX15 (magenta) and IT1t-bound (green) CXCR4 representation (gray cartoon). CVX15 binds exclusively to the so-called major pocket between TM3 and TM7, able to mimic to a greater extent the binding of a chemokine, whereas IT1t binds exclusively to the so-called minor pocket between TM1–TM3 and TM7. (B) Comparison of the positioning of TM1 in chemokine receptors (CXCR4, pale yellow cartoon) and other class A GPCRs (exemplified by ADBR2, gray cartoon). A disulphide bridge between the chemokine receptor’s N terminus and the top of TM7 (two helix turns longer than in ADBR2) positions TM1 toward TM7 and the binding site. (C) The intracellular binding site of chemokine receptors (represented by CCR2, gray cartoon) illustrates a conserved pharmacophore between intracellular ligand binders, where a key double backbone hydrogen bond occurs between residues 8.49 and 8.50 and the ligands. (D) Common structural rearrangements upon receptor activation in GPCRs. An outward movement of TM6 and an inward movement of TM7 allow for the accommodation of the stabilizing nanobody (purple spheres). ADBR2, β2 adrenergic receptor.
Most chemokine receptor crystal structures are cocrystallized with antagonists and resemble an inactive conformational state. However, the human cytomegalovirus–encoded viral receptor US28 is bound to human chemokine CX3CL1 and stabilizing nanobody Nb7 and resembles an active state (Burg et al., 2015; Miles et al., 2018). Yet the rearrangements do not completely resemble the fully active conformation observed in the G protein–bound β2 adrenergic receptor (Rasmussen et al., 2011) (Figs. 1 and 3D).
Applications of Chemokine Receptor Crystal Structures for Molecular Pharmacologists
The information that can be extracted from crystal structures goes far beyond the ligand interaction or protein structural analysis and is often neglected. In this section, we review multiple aspects of chemokine receptor crystal structures that can be useful for molecular pharmacologists. We will focus on the distinct aspects of chemokine receptor structures that can guide the understanding of the mechanistic details of GPCR function. Moreover, we will provide insights on how these structures can be used for the design of tools and experiments to explore different pharmacological aspects of chemokine receptors.
Structural Determinants of Ligand Binding.
Understanding the mechanism of binding of small molecule ligands and chemokine mimics is key to rationally modulate their pharmacology (Arimont et al., 2017). Crystal structures are key to reveal interactions of ligand-receptor complexes (Fig. 4, yellow), which can subsequently be used to 1) generate ligand analogs with improved interactions with key residues, 2) drive SBDD efforts, or 3) design site-directed mutagenesis (SDM) experiments and elucidate the interactions that contribute most to the free energy of binding. Medicinal chemistry efforts are often focused on modifying ligands to generate structure-activity relationships, exploring the different chemical features of the ligand that enhance the desired activity parameters (e.g., affinity, potency, selectivity, kinetics). However, this exercise requires significant synthetic efforts, as identifying these key features implies designing modifications in all possible chemical substituents of the ligands. These efforts are exponentially bigger as the ligand complexity increases, and chemokine receptor ligands are often rather complex, ranging from big and flexible small molecules to peptides and chemokines. The challenges of such a ligand-based approach can often be overcome by combining it with structure-based methods. Analysis of key interactions in crystal structures offers a rationale for designing ligand modifications. To illustrate, a recent study focused on the IT1t-bound CXCR4 crystal structure as a baseline to identify new fragment hits and analyzed the crystal structure to rationally design a fragment growing strategy (Adlere et al., 2019). In this study, the identification of a highly hydrophobic hotspot in the binding site of the cocrystallized ligand was used to generate analogs with increased lipophilicity to target the hotspot. Furthermore, experimentally validated ligand binding modes provided by crystal structures can be used for the rational design of ligand modifications. Such modifications include the attachment of radioactive or fluorescent probes (Iliopoulos-Tsoutsouvas et al., 2018), which are key tools for a wide variety of molecular pharmacology experiments, ranging from affinity determination (ligand displacement) and kinetics profiling to in vitro and in vivo imaging. To illustrate, the IT1t-bound CXCR4 crystal structure was successfully used to design fluorescent probes based on the chemical structure of IT1t and analogs (Dekkers et al., manuscript in preparation).

Overview of potential applications of chemokine receptor crystal structures. Chemokine receptor crystal structures may allow the pharmacologist to map ortholog variants that might potentially affect the pharmacological profile of ligands (orange), visualize and interpret clinically relevant SNPs (dark blue), understand the key features of ligand binding (yellow), critically analyze the potential dimerization interfaces of receptors (green), and understand the molecular mechanisms of receptor function based on key structural motifs (cyan).
SBDD requires detailed knowledge on the ligand-receptor interactions. Molecular docking predicts the conformation and interactions of the ligand in the binding site based on its shape and physicochemical properties. However, despite the rather successful application of ligand docking in SBDD for some GPCRs, relatively small conformational differences in protein binding sites (Coudrat et al., 2017a) and the availability of experimentally validated structural information, including SDM or ligand quantitative structure-activity relationships, can be critical for the success of virtual screening (Yoshikawa et al., 2013). The use of GPCR crystal structures has long proven to enhance hit identification and lead optimization (Kooistra et al., 2014, 2015; Kuhne et al., 2016; Coudrat et al., 2017a,b; Lee et al., 2018). In the case of chemokine receptors, multiple crystal structure-based virtual screening campaigns have led to the discovery of new chemokine receptor ligands (Arimont et al., 2017). Hence, the use of crystal structures is key for the discovery of new tool compounds and potential drug candidates targeting chemokine receptors. A major contribution from chemokine receptor crystal structures to the field of SBDD in GPCRs was the revelation of a novel druggable pocket in the intracellular side of the receptor (Oswald et al., 2016; Zheng et al., 2016), exposed to the cytosol, which partially overlaps with the binding sites for both G proteins (Rasmussen et al., 2011) and β-arrestins (Kang et al., 2015; Zhou et al., 2017). This allosteric binding site was explored previously in chemokine receptors (de Kruijf et al., 2011; Zweemer et al., 2014), but the ligand-bound crystal structures now clearly reveal a conserved pharmacophore that opens new doors to SBDD for chemokine receptors (Fig. 3C).
Crystal structures can also be used to guide SDM experiments of a receptor to identify the key residues that drive the ligand pharmacology of interest. To illustrate, multiple models of peptide ligand binding (e.g., T140, Trent et al., 2003; and FC131, Våbenø et al., 2006) were proposed before the release of the CVX15-bound CXCR4 structure. However, the release of the crystal structure revealed a different binding mode from that previously suggested. New peptide-bound CXCR4 models were proposed after the release of the crystal structure that consider the observed interaction pattern in the X-ray structure, and the pattern has been used to guide SDM experiments and validate the proposed new models (Thiele et al., 2014). The structural insights gained from this experimental validation allowed rational design strategies toward the discovery of new CXCR4 ligands (Våbenø et al., 2015; Di Maro et al., 2017) and even the ability to translate this information to other chemokine receptor ligands (Oishi et al., 2015).
Structural Determinants of Protein Function.
All GPCRs share a common seven-transmembrane (7TM) domain topology, despite the low sequence identity between class A subfamilies (∼20%–25%) (Katritch et al., 2013) and the different classes A–F (<20%) (Fredriksson et al., 2003). Furthermore, GPCRs possess highly conserved sequence motifs that have been shown to contribute to their structural integrity and folding, including a highly conserved disulphide bridge between TM3 and extracellular loop ECL2, which upon mutation induces protein misfolding (Rader et al., 2004). This disulphide bridge is also present in chemokine receptors, as well as an additional disulphide bridge between the N terminus and ECL3 uniquely conserved among chemokine receptors (Szpakowska et al., 2014) (Fig. 3B). Disruption of this covalent interaction through mutagenesis destabilizes the tertiary structure of CXCR4 and consequently also potentially decreases ligand binding affinity (Zhou and Tai, 2000). The crystal structures of chemokine receptors show the impact of the N terminus–ECL3 disulphide bridge on protein conformation and, specifically, on the orthosteric ligand binding site between the 7TM domains (Fig. 3B): TM7 is two α-helical turns longer than in other GPCRs, and the N terminus is repositioned toward TM7 (Arimont et al., 2017). The effects of these structural motifs must therefore be considered when designing chemokine or peptide ligands, small molecule fluorescent probes, and even protein constructs such as N-terminal probes. Another characteristic structural motif in chemokine receptors with a substantial impact regarding ligand binding and mutant design is the so-called S/T2.56xP2.58 motif in TM2. Mutation of residue Thr2.56 in CCR5 has a significant effect on the binding affinity and functional response of CCR5 to CCL5 (Govaerts et al., 2001). The available crystal structures of chemokine receptors show that this motif induces a unique helical kink in TM2 that places residues 2.60 and 2.63 toward the ligand binding site instead of toward the membrane interface as in other GPCRs (Arimont et al., 2017). Consequently, it is necessary to introduce two gaps in sequence alignments of chemokine receptors to translate this structural effect into the sequence space (Gonzalez et al., 2012).
Moreover, GPCRs share a conserved mechanism of signal transmission and activation (Venkatakrishnan et al., 2013), even though they bind a wide variety of ligands of different chemical natures. Identification of this conserved mechanism has been possible through the study of the multiple crystal structures released not only in the inactive state but also in the active and even intermediate states (Venkatakrishnan et al., 2016). This mechanism seems to extend to chemokine receptors as well, as proven by the conserved active-like conformation in the CX3CL1-bound US28 structures (Burg et al., 2015; Miles et al., 2018). The most notorious structural changes associated to the active state include a significant outward shift of the intracellular half of TM6, accompanied by an inward movement of the bottom of TM7, and a subtler lateral displacement of the bottom of TM5 (Fig. 3D).
GPCRs are intrinsically allosteric by nature, as signal transduction involves changes occurring at spatially distinct protein sites (Thal et al., 2018). Many sequence motifs conserved throughout class A GPCRs are long known to be key for GPCR signaling (Figs. 4, cyan, and 5), including the DRY motif (Rovati et al., 2007), the NPxxY motif (Audet and Bouvier, 2012), the sodium binding site (Katritch et al., 2014), the PIF motif or transmission switch (Venkatakrishnan et al., 2013), and the CWxP motif (Kobilka and Deupi, 2007). A study reporting on point mutations at all residues of CXCR4 proved that at least one of the key residues in each of the aforementioned structural motifs, upon mutation, significantly decreased CXCR4-mediated signaling (Wescott et al., 2016). However, only the analysis and comparison of inactive and active crystal structures has elucidated the molecular mechanism that triggers the aforementioned effects on signaling (Venkatakrishnan et al., 2016). With crystal structures at hand, the environment of these conserved motifs has been identified, supporting the rational design of specific mutant points to disrupt or enhance their structural or pharmacological effect. By these means, the role of the motifs has been thoroughly studied in CXCR4, where mutation in a residue on any of these motifs, with the exception of the sodium pocket, significantly impairs G-protein signaling without significantly affecting CXCL12 binding (Wescott et al., 2016) (Fig. 4).

Key sequence motifs conserved throughout class A GPCRs known to be key for GPCR signaling, including the DRY motif (Rovati et al., 2007) (blue), the NPxxY motif (Audet and Bouvier, 2012) (orange), the PIF motif or transmission switch (Venkatakrishnan et al., 2013) (green), the CWXP motif (Kobilka and Deupi, 2007) (red), and the sodium binding site (Katritch et al., 2014) (purple; the sodium ion is shown as a reference, but it has not been crystallized in any available chemokine receptor crystal structure). Residues mutated in CXCR4 that significantly reduce its signaling are highlighted in red boxes (Wescott et al., 2016).
Rational Evaluation of Chemokine Receptor Ortholog Variants and Polymorphisms.
The basis of most molecular pharmacology projects ultimately includes the evaluation of the effect of drugs or other chemical entities on a human protein target, often expressed in human-derived cell lines. Yet knowledge on the effect of drug candidates on a number of animal orthologs is crucial for proper translation of the pharmacological effects of a drug candidate from in vitro to in vivo settings, as protein sequence divergence between species can be substantial (Fig. 4, orange). Despite the high similarity between orthologs of chemokine receptors (90%–100% within mammals; data retrieved from GPCRdb and Pándy-Szekeres et al., 2018), there are receptor regions with lower conservation, especially in the loops. To illustrate, the mouse CXCR4 sequence presents a five-amino-acid insertion in ECL2, proven to be key for the binding of multiple ligands (Arimont et al., 2017). Crystal structures are useful to map sequence differences and to predict, based on their location, whether the ortholog variant may affect the ligand binding and signaling. By means of in silico methods, crystal structures can be used to predict the effect of such ortholog variants—for example, by a combination of homology modeling (if needed) and molecular docking.
Differences exist not only between species but within the human population (Fig. 4, blue). Natural genetic variations can cause differences in the individual response to drugs, including therapeutic efficacy and safety. To illustrate, eight of the nine residues in the binding site of maraviroc in CCR5 are polymorphic, which suggests that patients with HIV who carry one of these variants may show an altered response to the antiretroviral drug (Hauser et al., 2018). These naturally occurring genetic variations may therefore represent a challenge to the health care system and may be taken into consideration in early-stage drug development. This is key in the field of chemokine receptors as illustrated by the wide distribution of single nucleotide polymorphisms (SNPs) that have been reported for these GPCRs, covering residues from the binding site to key signaling structural motifs (Supplemental Table 1). To illustrate, GPCRdb reports ∼2500 SNPs for chemokine receptors, from which ∼43% corresponds to the C-C chemokine motif subfamily, 23.5% to the C-X-C chemokine motif subfamily, and ∼17.7% to the atypical chemokine receptor (ACKR) subfamily. Within these subfamilies, the receptors with the most reported SNPs are CCR5 (n = 175), CXCR1 (n = 160), and ACKR2 (n = 165), respectively. The average amount of SNPs observed per each transmembrane domain within the entire chemokine receptor family is rather conserved, with an average of two SNPs reported per residue (maximum TM5 = 2.21 SNPs/residue, minimum TM3 = 1.6 SNPs/residue) (Hauser et al., 2018; Pándy-Szekeres et al., 2018). Also, in this context, crystal structures can assist to map these genetic variations to predict potential pharmacological effects. In addition, crystal structures can guide drug design to prevent a negative effect of these polymorphisms, for example, avoiding key ligand interactions with the polymorphic amino acid when possible.
Analysis of Oligomerization Interfaces.
Many GPCRs, including chemokine receptors (Fig. 4, green) (Stephens and Handel, 2013), are known to form functional oligomers in the cell membrane (Bulenger et al., 2005; Cottet et al., 2012). Chemokine receptor oligomers cause negative cooperativity in the binding of their chemokine ligands and, in the case of some receptors, oligomerization can either enhance or inhibit receptor activation through allosteric communication (Percherancier et al., 2005; Stephens and Handel, 2013; Armando et al., 2014). The current methodologies that are able to probe dimerization are mainly based on fluorescence and bioluminescence resonance energy transfer approaches (Percherancier et al., 2005; Goddard and Watts, 2012; Fumagalli et al., 2019; Heuninck et al., 2019), fluorescence fluctuation spectroscopy (Isbilir et al., 2017; Briddon et al., 2018), and spatial intensity distribution analysis (Pediani et al., 2018). Such biophysical approaches are very sensitive to detect oligomers but are not able to identify the oligomerization interface between protomers, which is key for molecular understanding of the quaternary GPCR structures. Multiple chemokine receptors have been crystallized in an oligomeric state, including the IT1t-bound CXCR4 structures (Wu et al., 2010), the maraviroc-bound CCR5 structure (Tan et al., 2013), the compound 21–bound CCR5 structure (Peng et al., 2018), and the vercirnon-bound CCR9 structure (Oswald et al., 2016). Although the biologic relevance of GPCR oligomers cannot be deduced from crystal structures and the oligomeric states could originate from crystallographic artifacts, they provide a relevant basis for the design of validation experiments. The most commonly observed dimerization interface derived from GPCR crystal structures is the one involving TM5/TM6, also observed in the IT1t-bound CXCR4 structures (Ferré et al., 2014). This interface was suggested by use of synthetic peptides mimicking the sequence of the transmembrane domains (Hebert et al., 1996; Banères and Parello, 2003) and provides evidence that GPCR dimers may be biologically relevant. Another common method to probe oligomerization interfaces is the use of SDM experiments to disrupt the interactions needed for the formation of the complex. In this case, the use of crystal structures to guide the experimental design has been key. Formation of oligomers requires interactions between the membrane-oriented faces of transmembrane domains, which, by nature, are highly hydrophobic (Li and Deber, 1994; Deber and Li, 1995). The hydrophobic nature of the oligomeric interfaces suggests that the stabilization of the complexes may require significant hydrophobic packing. Consequently, the impact of a single mutation along the interface may not be enough to disrupt oligomer formation, but multiple mutations may be required. Alternatively, identification of the few key polar contacts (if present) only found in the top or bottom of the helices is key (Ferré et al., 2014). Crystal structures have provided a relevant guide toward the rational design of such mutations in strategic positions. This strategy has proven effective to map the CCR5 dimeric interface, where disulphide crosslinking experiments based on the dimeric interfaces observed in the CXCR4 and µ-opioid receptor crystal structures (Jin et al., 2018).
Another technique that relies on the knowledge of interaction interfaces is disulphide crosslinking or disulphide trapping. Disulphide trapping is an experimental approach in which the introduction of two cysteine residues in different protein partners at strategical positions of the interface generates (irreversible) covalently bound protein complexes (Kufareva et al., 2016). Consequently, disulphide trapping allows protein complexes to be stabilized in specific conformations preventing spontaneous dissociation, hence facilitating structural studies. To illustrate, crystallizing chemokine receptors in complex with endogenous chemokines has often proven very challenging, potentially due to the lower affinity of chemokines for detergent-solubilized receptors yielding lower-stability complexes and/or the high selectivity of chemokines for specific conformational states of their receptors to bind with high affinity (e.g., active state) (Rasmussen et al., 2011; Kufareva et al., 2016). Such high-affinity conformational states (e.g., an active state) are currently quite challenging to achieve under crystallization conditions and have thus far only succeeded for US28 with the aid of a stabilizing nanobody (Burg et al., 2015; Miles et al., 2018). Despite these challenges, the use of disulphide trapping has yielded the crystallization of CXCR4 in complex with the viral chemokine v-MIP-II (Qin et al., 2015). Disulphide crosslinking also facilitates the evaluation of the proximity between residues, providing insights into complex interactions when combined with molecular modeling. Using these methods, the interaction geometry of ACKR3 and its endogenous ligand CXCL12 has been mapped and proven to be consistent with the hypothesis of two sites of chemokine binding (Gustavsson et al., 2017).
Challenges and Pitfalls
Despite the great value of GPCR crystal structures, there are some challenges and pitfalls to be considered when interpreting them. X-ray structures as visualized are molecular models built to obtain as good a fit to an electron density map as possible. This fit is done by means of advanced computational methods, but sometimes assumptions (e.g., about missing data) are necessary (Piscitelli et al., 2015). One of the most used quality metrics is resolution measured in angstroms, which reports the highest angle reflections recorded in the X-ray diffraction pattern. Resolution is a measure of the data quality that is used to build the models and is not a measure of the model quality itself (Piscitelli et al., 2015). The lower the value of the resolution, the better the quality of the data. The resolution of GPCR crystal structures ranges from 1.7 to 7.7 Å, with an average of 2.89 Å at the time of this writing. Structures with a resolution lower than 2 Å are considered of very high quality; at this resolution, it is possible to accurately position water molecules and ions in the protein structures. The resolution of the solved chemokine receptor crystal structures ranges from 2.2 Å in the 5P7-CCL5–bound CCR5 structure to 3.8 Å in one of the US28 structures (PDB ID: 4XT3), with an average of 3.03 Å. Despite the comparable average resolution values, no high-quality structures have been reported for any chemokine receptor thus far and experimental finetuning is needed to improve the quality of chemokine receptor structures. A summary of the current chemokine receptor structure quality metrics can be found in Supplemental Table 2.
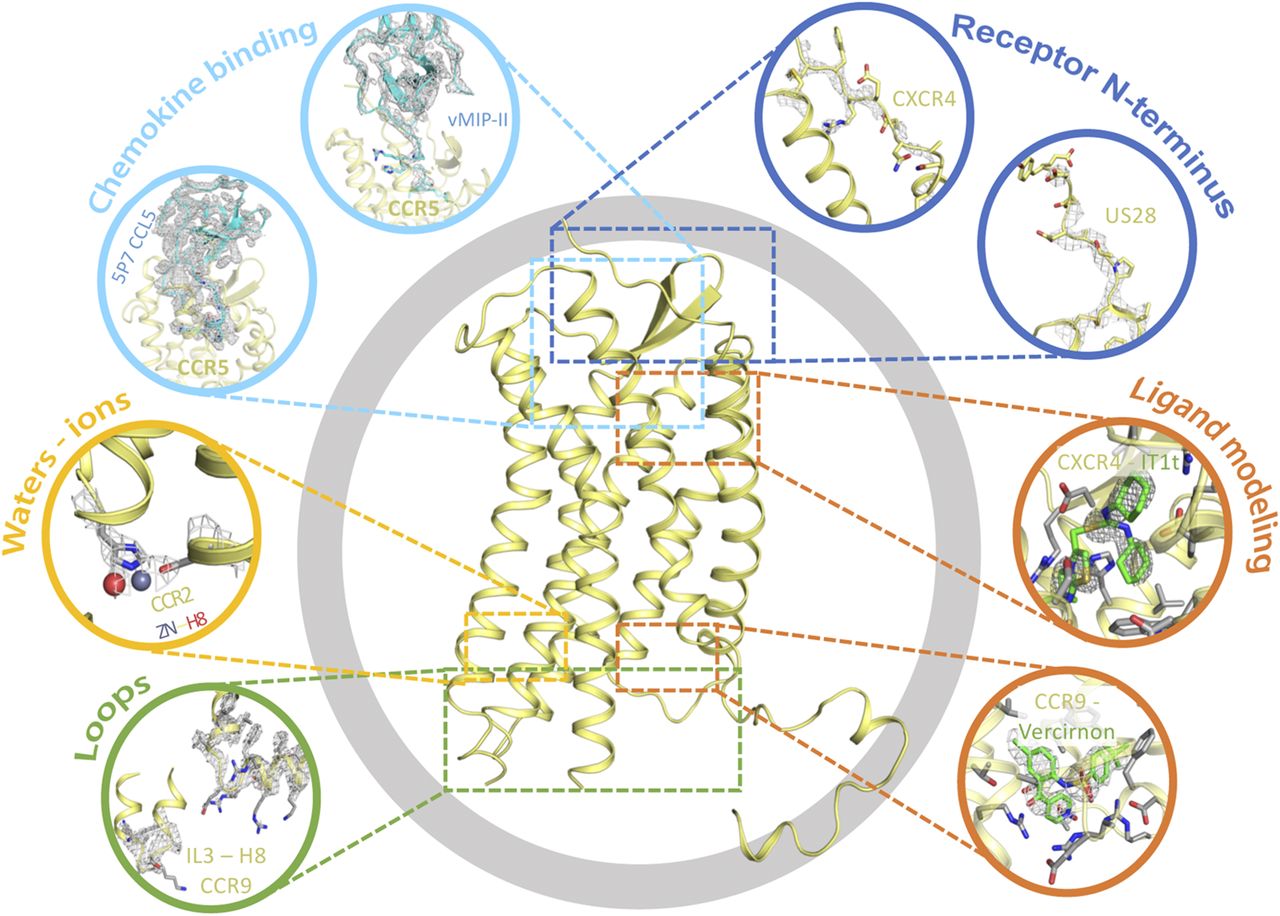
Next to differences in the overall resolution of protein structures, the quality of the electron density of a GPCR structure can vary significantly between the transmembrane domains, solvent-exposed loops, and ligand binding sites. The N and C termini, and sometimes the intracellular loops (ICLs) and ECLs, are often truncated or shortened for optimized protein crystallization and/or are not solved due to their flexibility. Therefore, it often occurs that only a few residues of such protein regions can be modeled (Piscitelli et al., 2015). In the case of chemokine receptors, the N terminus is key for binding chemokines, but only 1 (CCR2; PDB ID: 6GPX) to 10 (US28; PDB ID: 4XT1) residues can be modeled in the solved crystal structures (Fig. 6, dark blue). For example, no residues of the N terminus have been solved for CCR2 (PDB ID: 5T1A) and CCR9, limiting the value of the structure for designing probes or engineering ligands that interact with the N terminus (e.g., chemokines, antibodies, nanobodies) (Fig. 6, cyan).

Electron density challenges and pitfalls for crystal structures of chemokine receptors. Electron density maps of chemokine binding (cyan), receptor N termini (dark blue), cocrystallized ligands (orange), ICLs (green), and water molecules and ions (yellow).
Other receptor areas that are often not solved are the ICLs and ECLs (Fig. 6, green). When considering ECLs, ECL2 is usually the longest and most flexible loop in GPCRs, and therefore its resolution is often lower. However, ECL2 can exhibit different secondary structural elements, including α-helical or β-sheet elements (Woolley and Conner, 2017). In chemokine receptors, ECL2 has an organized secondary structure of two antiparallel β-sheets (Arimont et al., 2017) and is solved in all structures except CCR9. In the CCR9 X-ray, only ECL3 is resolved and a residual signal is observed for ECL2 C18745.50, involved in the highly conserved disulphide bridge with TM3 (Arimont et al., 2017). ECL1 is not fully solved in the US28 structures (PDB IDs: 4XT1 and 4XT3), and ECL3 is not fully solved in one CCR2 structure (PDB ID: 6GPS). Regarding ICLs, all structures fused to stabilizing proteins, including T4 lysozyme and rubredoxin, lack ICL3, as the fusions were engineered in ICL3. Furthermore, the CVX15-bound and vMIP-II–bound CXCR4 structures also lack ICL1, whereas a CCR2 structure misses ICL2 (PDB ID: 6GPX).
A challenging aspect when analyzing electron density maps is the interpretation of ligand densities (Fig. 6, orange). As mentioned, the average resolution of the solved GPCR structures is around 3 Å, which allows the visualization of electron density basic contours of amino acid side chains and ligands (Piscitelli et al., 2015). To illustrate, the symmetric thiourea group of IT1t is solved in the crystal structure in a tautomeric state in which the nitrogen N4 forms a salt bridge with Asp972.63 (Wu et al., 2010). However, the electron density does not exclude the existence of a very similar conformation with the thiourea group flipped, in which the nitrogen N3 forms the salt bridge with Asp972.63. These ambiguities in the interpretation of ligand electron density should be considered when designing new ligands, SDM experiments, or attachment points for fluorescent probes or bivalent ligands.
The role of water molecules in GPCRs is of great interest because waters play a critical role in the binding of drugs and the function of many proteins (Chaplin, 2006; Ball, 2008; Mason et al., 2012) (Fig. 6, yellow). Information extracted from crystal structures is key to identify the biologic functions of conserved structural waters and to exploit them in drug discovery. For example, modulating the presence of waters in a binding site by means of ligand scaffold modifications has proven successful to modulate ligand affinity (Mason et al., 2013) or predict ligand kinetics (Bortolato et al., 2013). On a structural level, water molecules are postulated to contribute to the functional plasticity needed for the GPCR conformational changes related to signal transmission and activation (Angel et al., 2009a,b). Importantly, structures with resolutions >2.5 Å do not provide clear electron density maps for waters and hence these can often not be modeled. This explains the scarce amount of water molecules in chemokine receptor crystal structures. In addition, to consider a water molecule as conserved and structurally relevant, it has to be visible in a number of crystal structures, and crystallized chemokine receptor-ligand complexes are thus far mostly unique. The receptor-ligand complexes crystallized multiple times include CXCR4-IT1t and US28-CX3CL1, for which only the PDB 3ODU and 4XT1 structures, respectively, contain water molecules. Therefore, at this stage no conclusions on relevant water-mediated interactions can be drawn based only on chemokine receptor crystal structures, and the role of water molecules in chemokine receptor function will have to be further explored experimentally.
Ions, specifically monovalent cations, are long known to be key for GPCR structure and function (Childers et al., 1979; Vickery et al., 2018). The most representative example is the Na+ ion, which in the inactive GPCR state binds in a pocket surrounding the most conserved aspartic acid in TM2 (Asp2.50), in the center of the TM domain (Liu et al., 2012). This sodium ion is coordinated directly by interactions with Asp2.50 and Ser3.39 and indirectly through a water network by Trp6.48, Asn7.49, Asn7.45, and Ser7.46. These coordinating residues are highly conserved throughout class A GPCRs, and numerous high-resolution crystal structures of different class A GPCRs demonstrate the presence of a sodium ion in this conserved site. Therefore, sodium binding in this pocket is assumed to be a conserved feature shared by the majority of class A GPCRs (Katritch et al., 2014; Vickery et al., 2018). Multiple studies reveal that mutation of the residues in the sodium binding pocket modulates agonist binding, signaling, and biased signaling (Liu et al., 2012; Fenalti et al., 2014; Massink et al., 2015). It has been proposed by other studies that, driven by electrochemical gradients, the sodium ion can traverse the receptor to the cytosol, coupled to the protonation of Asp2.50, and that this is a key step in GPCR activation (Vickery et al., 2018; White et al., 2018). No chemokine receptor crystal structure has been solved with a sodium ion in the sodium binding pocket, which may be explained by the lack of high-resolution structures. However, it is important to consider that the sodium binding site is not fully conserved in chemokine receptors, according to the GPCRdb sequence alignments (Pándy-Szekeres et al., 2018): Asp2.50, Trp6.48, and Asn7.49 are highly conserved, but residue 3.39 is a glycine in 48% of chemokine receptors, residue 7.45 is a histidine in 74%, and residue 7.46 is a cysteine in 74%. Therefore, the role of the sodium ion in chemokine receptors has to be addressed experimentally, and a study with mutagenesis of these residues in CXCR4 failed to severely affect agonist-mediated calcium flux (Wescott et al., 2016).
Often, other cocrystallized ions, especially cations, are solved in GPCR structures, including chemokine receptor structures. To illustrate, clear electron density of a metal ion is observed in the CCR2 structure (PDB ID: 5T1A), which was identified as zinc through X-ray fluorescence scans. This zinc is positioned between the bottom of TM3, ICL3, and the T4 lysozyme. However, the biologic relevance of such ions has to be further studied, as divalent cations such as zinc or magnesium are common components of the buffers used for crystallization and can induce artifacts in the crystal environment (Salom et al., 2013; Piscitelli et al., 2015).
Concluding Remarks
Members of the family of chemokine receptors are important therapeutic targets due to their involvement in numerous inflammatory and immune diseases as well as in cancer. Despite the lack of crystal structures for the majority of chemokine receptors (only 5 of 28 GPCRs of this subfamily have been crystallized to date), the available structures highlight several relevant characteristics of chemokine receptors and their structure, including the ability to bind ligands of different chemical natures in different druggable binding sites and the ability of the GPCRs to adopt multiple conformational states. Analysis of the crystal structures of chemokine receptors provides detailed understanding of chemokine receptor-ligand binding and insights into the molecular mechanisms that drive receptor function. Moreover, it allows the rational design of tools and experiments for a wide variety of applications. These applications may include the design of SDM experiments for the identification of key determinants of receptor pharmacology, receptor oligomerization interfaces, or the functional effect of conserved structural motifs, ortholog variants, and polymorphisms. However, despite the great impact of GPCR X-ray structures, there are some pitfalls to be considered as well. The quality of the electron density maps used to model receptor structures is a key determinant to confidently interpret them and to understand highly dynamic regions, such as loops, ambiguous ligand moieties, and side chain rotamers, as well as conserved water molecules and structurally relevant ions. Awareness of the strengths and pitfalls of crystal structure analysis is crucial to fully exploit the value of GPCR X-ray structures by medicinal and computational chemists and molecular pharmacologists.
Prospectively, acquisition of new chemokine receptor crystal structures would boost the field of chemokine receptor drug discovery. Chemokine receptor structures that have not yet been solved would be of great added value to drive drug discovery campaigns, but the resolution of the structure of an ACKR would be especially interesting and could shed some light on the still puzzling biased signaling of these chemokine receptors. New ligand-bound complexes, including different modulator types such as small molecules, peptides, or biologicals, would deepen the understanding of the different binding patterns and mechanism of action of the different ligand types, potentially enhancing the design of new ligands and tools. Furthermore, future small molecule agonist-bound structures may allow for the identification of activation-driving interactions that could be targeted to achieve a pharmacological effect of interest. Similarly, solving a G protein–bound complex or an β-arrestin–bound chemokine receptor complex would enhance the analysis of the geometry and interaction patterns of a fully active receptor. The field of cryo-EM has made great progress on the resolution of big protein complexes, including multiple GPCR/G-protein complexes. In the field of chemokine receptors, the cryo-EM structure of CCR5 bound to gp160 has provided very valuable insights on the binding mechanism of HIV (Shaik et al., 2019). However, a G protein–bound chemokine receptor structure is still missing. Understanding the mechanism of binding of G proteins or β-arrestins to chemokine receptors would provide valuable information that could potentially drive the exploration of new mechanisms of modulation of these intracellular partners, including, for example, the design of modulators targeting the intracellular side of the receptors.
Acknowledgments
We thank Abigail Horder for valuable contributions to data collection of chemokine receptor SNPs. This minireview is part of the minireview series “From Insight to Modulation of CXCR4 and ACKR3 (CXCR7) Function.”
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Arimont, Hoffmann, de Graaf, Leurs.
Footnotes
- Received May 9, 2019.
- Accepted June 21, 2019.
This work was supported by the European Union’s Horizon 2020 Marie Curie Innovative Training Network [Grant 641833 (ONCORNET)] and Eurostars [Grant 12132 (PROTEUS)] programs.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACKR
- atypical chemokine receptor
- CCL
- C-C motif chemokine ligand
- CCR
- C-C chemokine receptor
- CRS
- chemokine recognition site
- cryo-EM
- cryo-electron microscopy
- CX3CL
- C-X3-C chemokine ligand
- CXCL
- C-X-C chemokine ligand
- CXCR
- C-X-C chemokine receptor
- ECL
- extracellular loop
- GPCR
- G protein–coupled receptor
- GPCRdb
- GPCR database
- ICL
- intracellular loop
- IT1t
- isotiourea-1t
- PDB
- Protein Data Bank
- SBDD
- structure-based drug design
- SDM
- site-directed mutagenesis
- SNP
- single nucleotide polymorphism
- TM
- transmembrane
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}