


Abstract
We have shown previously that different chemical classes of small-molecule antagonists of the human chemokine CXCR2 receptor interact with distinct binding sites of the receptor. Although an intracellular binding site for diarylurea CXCR2 antagonists, such as N-(2-bromophenyl)-N′-(7-cyano-1H-benzotriazol-4-yl)urea (SB265610), and thiazolopyrimidine compounds was recently mapped by mutagenesis studies, we now report on an imidazolylpyrimidine antagonist binding pocket in the transmembrane domain of CXCR2. Using different CXCR2 orthologs, chimeric proteins, site-directed mutagenesis, and in silico modeling, we have elucidated the binding mode of this antagonist. Our in silico-guided mutagenesis studies indicate that the ligand binding cavity for imidazolylpyrimidine compounds in CXCR2 is located between transmembrane (TM) helices 3 (Phe1303.36), 5 (Ser2175.44, Phe2205.47), and 6 (Asn2686.52, Leu2716.55) and suggest that these antagonists enter CXCR2 via the TM5-TM6 interface. It is noteworthy that the same interface is postulated as the ligand entry channel in the opsin receptor and is occupied by lipid molecules in the recently solved crystal structure of the CXCR4 chemokine receptor, suggesting a general ligand entrance mechanism for nonpolar ligands to G protein-coupled receptors. The identification of a novel allosteric binding cavity in the TM domain of CXCR2, in addition to the previously identified intracellular binding site, shows the diversity in ligand recognition mechanisms by this receptor and offers new opportunities for the structure-based design of small allosteric modulators of CXCR2 in the future.
Introduction
The chemokine receptor CXCR2 belongs to the rhodopsin-like family of G-protein coupled receptors (GPCRs) (Murphy et al., 2000). It is expressed on endothelial cells and on different inflammatory cells such as eosinophils, natural killer cells, neutrophils, macrophages, mast cells, and monocytes (Murphy et al., 2000; Kraneveld et al., 2011). CXCR2 interacts with the chemokines CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8. Upon production of these chemokines, CXCR2 is responsible for the transmigration of inflammatory cells toward sites of inflammation. Several in vivo studies have shown an important role for CXCR2 in inflammatory diseases such as atherosclerosis (Boisvert et al., 1998), chronic obstructive pulmonary disease (Chapman et al., 2007), rheumatoid arthritis (Podolin et al., 2002), inflammatory bowel disease (Ajuebor et al., 2004; Buanne et al., 2007), and multiple sclerosis (Liu et al., 2010). Furthermore, the expression of CXCR2 is increased in psoriatic epidermis (Kulke et al., 1998) and in bronchial biopsies of patients with chronic obstructive pulmonary disease (Qiu et al., 2003). These studies indicate that CXCR2 is an interesting drug target. In view of this therapeutic potential, different CXCR2 antagonists are being developed (see, for example, Li et al., 2003; Widdowson et al., 2004; Baxter et al., 2006; Ho et al., 2006). Among them, 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1-enylamino}-benzamide (SCH-527123) and repertaxin are currently in phase II clinical trial (http://www.clinicaltrials.gov/ct2/show/NCT01006616; http://www.clinicaltrials.gov/ct2/show/NCT00224406).
We have investigated in detail the mechanism of action of CXCR2 antagonists that belong to the diarylurea, imidazolylpyrimidine, and thiazolopyrimidine classes (de Kruijf et al., 2009). Using tritium-labeled diarylurea N-(2-bromophenyl)-N′-(7-cyano-1H-benzotriazol-4-yl)urea (SB265610), we discovered that small-molecule CXCR2 antagonists of different classes bind to distinct binding sites. The imidazolylpyrimidine compounds (such as compound 1; see Fig. 1) were not able to displace [3H]SB265610, whereas the diarylurea (such as SB255610; see Fig. 1) and thiazolopyrimidine compounds could compete for [3H]SB265610 binding. Two groups have provided evidence that compounds belonging to the diarylurea and thiazolopyrimidine classes most likely bind intracellularly to CXCR2 (Nicholls et al., 2008; Salchow et al., 2010). The binding site of imidazolylpyrimidine CXCR2 antagonists remained to be investigated. The first chemokine receptor crystal structures have been solved (Wu et al., 2010). These structures show overlapping but distinct binding pockets in the TM binding domain of CXCR4 for the small molecule antagonist 1,3-dicyclohexyl-2-(3-methyl-6,6-dimethyl-5,6-dihydroimidazo[1,2-b] thiazole)-2-thiopseudourea (IT1t) and the cyclic peptide CVX15. Furthermore, site-directed mutagenesis studies with various chemokine receptors demonstrated that compounds can bind either in the minor subpocket [between transmembrane (TM) helices 1, 2, 3, and 7] or in the major subpocket [between TM3, 4, 5, 6, and 7] (Scholten et al., 2011).The primary aim of the current study was to probe the binding site of compound 1 in the TM binding domain.

The structures of nonpeptidergic CXCR2 antagonists compound 1 [(S)-2-(2-(1H-imidazol-1-yl)-6-(octylthio)pyrimidin-4-ylamino)-N-(3-ethoxypropyl)-4-methylpentanamide] and SB265610 [1-(2-bromophenyl)-3-(4-cyano-1H-benzo[d] [1,2,3]triazol-7-yl)urea] are shown.
Animal models are still crucial to investigate the therapeutic potential of compounds. Translating the results of animal studies to humans, however, can be hampered by differences between receptor orthologs. Species-related differences in the sequences of protein targets (“natural mutants”) have previously been used to identify the molecular determinants of ligand binding to GPCRs (Reinhart et al., 2004; Lim et al., 2010; Milligan, 2011). Species differences have also been observed for CXCR2. Although imidazolylpyrimidine compounds such as compound 1 (Fig. 1) are potent antagonists of human CXCR2, these compounds have no affinity for rodent CXCR2 receptors (J. C. H. M. Wijkmans, M. L. Webb, D. S. Auld, unpublished data). Human CXCR2 has a significantly higher homology to primate CXCR2 orthologs (89–100%) (Fig. 3A) than to rodent CXCR2 receptors (71–81%). Therefore, to investigate the possibility of circumventing species difference problems in preclinical animal studies, CXCR2 orthologs of different primate species (rhesus monkey, cynomolgus monkey, vervet, baboon, chimpanzee, gorilla, and orangutan) were cloned and pharmacologically characterized. We were surprised to find that some of the primate orthologs also have a significantly lower affinity for compound 1, despite their high sequence homology to human CXCR2. To investigate the molecular determinants of the binding of imidazolylpyrimidine compound 1 in more detail, we used various chimeric constructs of the human and baboon CXCR2 and site-directed mutagenesis in combination with in silico modeling to propose the TM binding mode for the imidazolylpyrimide compound 1. The current study describes the elucidation of the binding mode of the imidazolylpyrimide compound 1 at the human CXCR2 receptor by this systematic, in silico guided mutagenesis approach.
Materials and Methods
Materials.
Dulbecco's modified Eagle's medium, penicillin, and streptomycin were obtained from PAA Laboratories (Pasching, Austria). Fetal bovine serum was purchased from Integro B.V. (Dieren, The Netherlands). Linear 25-kDa polyethylenimine (PEI) was obtained from Polysciences (Warrington, PA), whereas branched 750-kDa PEI was purchased from Sigma (St. Louis, MO). Bovine serum albumin fraction V (BSA) was obtained from Roche (Mannheim, Germany). Na125I and [35S]GTPγS were purchased from PerkinElmer Life Sciences (Waltham, MA), and CXCL8 was either obtained from PeproTech (Rocky Hill, NJ) or was a kind gift from Dr. T. Sparer (University of Tennessee, Knoxville, TN). The CXCR2 antagonists 1-(2-bromophenyl)-3-(4-cyano-1H-benzo[d] [1,2,3]triazol-7-yl)urea [SB265610 (Bizzarri et al., 2006)] and (S)-2-(2-(1H-imidazol-1-yl)-6-(octylthio)pyrimidin-4-ylamino)-N-(3-ethoxypropyl)-4-methylpentanamide [compound 1 (Erickson et al., 2004; Ho et al., 2006)] were synthesized at Merck Research Laboratories (MSD, Oss, The Netherlands). Oligonucleotide primers for polymerase chain reaction (PCR) were synthesized by Biolegio (Nijmegen, The Netherlands). Pfu DNA polymerase, T4 DNA ligase and endonuclease restriction enzymes were all from MBI Fermentas (St. Leon-Rot, Germany).
DNA Constructs and Site-Directed Mutagenesis.
The wild-type human CXCR2 cloned in pcDNA3.1+ (Genbank accession no. P25025) was purchased from the Missouri S&T cDNA Resource Center (Rollo, MO). The CXCR2 receptors from baboon (Genbank accession no. Q8HZN3), chimpanzee (Genbank accession no. Q28807), gorilla (Genbank accession no. Q8HZN7), orangutan (Genbank accession no. Q8HZN6), rhesus monkey (GenBank accession no. Q8HZN5), and vervet (Genbank accession no. Q8HZN4) were cloned and characterized as described by Horlick et al. (2006). In brief, baboon, chimpanzee, gorilla, and orangutan genomic DNA were isolated from lymphoblast cells, whereas vervet genomic DNA was isolated from a liver biopsy specimen obtained from Dr. Frank Ervin (Caribbean Primates Ltd., Caribbean Primates Ltd., Madison). Next, PCR was used to amplify the coding sequences of these orthologs. Rhesus monkey genomic DNA was purchased from Clonetech (Palo Alto, Calif). The cDNA of cynomolgus monkey CXCR2 (HipKin et al., 2004) was made by PCR mutagenesis of rhesus monkey CXCR2 residues, including E2Q/L9F/K11E/G12N in the N terminus and L228F at the bottom of transmembrane V.
Chimeric receptor constructs were created by exchanging the domains between residue Lys1203.26 (EcoN1 restriction site in the cDNA) and residue Arg2125.39 (BamHI restriction site in the cDNA) of the human CXCR2 (see Fig. 3B) with that of the baboon CXCR2 or vice versa, resulting in the chimeric receptors HHB (human-human-baboon), BBH (baboon-baboon-human), HBB (human-baboon-baboon), BHH (baboon-human-human), HBH (human-baboon-human) and BHB (baboon-human-baboon). Site-directed mutagenesis was performed by PCR with the use of DNA primers with single-, double- or triple-base mismatches, resulting in a codon change for the desired amino acid substitution. The correct sequence of all DNA constructs was confirmed by sequencing analysis (Macrogen, Amsterdam, The Netherlands).
Cell Culturing and Transfection.
COS-7 cells were grown at 5% CO2 and 37°C in Dulbecco's modified Eagle's medium supplemented with 5% (v/v) fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin. Cells were transiently transfected (per 10-cm dish) with 2.5 μg of cDNA encoding the receptor supplemented with 2.5 μg of pcDNA by using linear PEI with a molecular mass of 25 kDa. In brief, 5 μg of total DNA was diluted in 250 μl of 150 mM NaCl. Next, 30 μg of PEI in 250 μl of 150 mM NaCl was added to the DNA solution. This mixture was incubated for 5 to 10 min at room temperature before it was added onto subconfluent COS-7 monolayer.
Membrane Preparation.
Two days after transfection, cells were detached from the plastic surface using ice-cold phosphate-buffered saline and centrifuged at 1500g for 10 min at 4°C. Next, the pellet was resuspended in the ice-cold phosphate-buffered saline and centrifuged again at 1500g for 10 min at 4°C. Later, cells were resuspended in ice-cold membrane buffer (15 mM Tris, 1 mM EGTA, 0.3 mM EDTA, and 2 mM MgCl2, pH 7.5), followed by homogenization by 10 strokes at 1100 to 1200 rpm using a Teflon-glass homogenizer and rotor. The membranes were subjected to two freeze-thaw cycles using liquid nitrogen, followed by centrifugation at 40,000g for 25 min at 4°C. The pellet was rinsed once with ice-cold Tris-sucrose buffer (20 mM Tris and 250 mM sucrose, pH 7.4) and subsequently resuspended in the same buffer and frozen at −80°C until use. Protein concentration was determined using a BCA-protein assay (Thermo Fisher Scientific, Waltham, MA).
Radioligand Binding Assays.
Membranes were incubated in 96-well plates in binding buffer (50 mM Na2HPO4 and 50 mM KH2PO4, pH 7.4) supplemented with 0.5% BSA, approximately 100 pM 125I-CXCL8 and indicated concentrations of CXCL8 or CXCR2 antagonist in a final volume of 100 μl. The reaction mixtures were incubated for 1 h at room temperature, harvested with rapid filtration through Unifilter GF/C 96-well filterplates (PerkinElmer Life and Analytical Sciences pretreated with 0.3% polyethylenimine and washed three times with ice-cold wash buffer (50 mM Na2HPO4 and 50 mM KH2PO4, pH 7.4). Bound radioactivity was determined using a MicroBeta (PerkinElmer Life and Analytical Sciences). Binding data were evaluated by a nonlinear curve fitting procedure using Prism 4.0 (GraphPad Software, San Diego, CA). Ligand affinities (pKi) from competition binding experiments were calculated from binding IC50 using the Cheng-Prusoff equation (Cheng and Prusoff, 1973).
[35S]GTPγS Binding Assay.
Membranes were incubated in 96-well plates in assay buffer (50 mM HEPES, 10 mM MgCl2, and 100 mM NaCl, pH 7.2) supplemented with 5 μg of saponin/well, 3 μM GDP, and approximately 400 pM [35S]GTPγS and indicated concentrations of CXCL8 in a final volume of 100 μl. When CXCR2 antagonists were applied to EC80 concentration of CXCL8, membranes were preincubated for 30 min at room temperature. The reaction mixtures were incubated for 1 h at room temperature, harvested with rapid filtration through Unifilter GF/B 96-well filterplates (PerkinElmer Life and Analytical Sciences), and washed three times with ice-cold wash buffer (50 mM Tris-HCl and 5 mM MgCl2, pH 7.4). [35S]GTPγS incorporation was determined using a MicroBeta scintillation counter (PerkinElmer Life and Analytical Sciences). Functional data were evaluated by a nonlinear curve-fitting procedure using Prism 4.0.
Whole-Cell–Based Enzyme-Linked Immunosorbent Assay.
One day after transfection, cells were trypsinized and seeded in poly-l-lysine-coated 48-well plates (5 × 104 cells/well). After 24 h, cells were fixed with 4% formaldehyde in TBS (50 mM Tris and 150 mM NaCl, pH 7.5). Next, cells were washed with TBS and permeabilized with 0.5% Nonidet P40 in TBS for 30 min (or TBS was added when permeabilization was not required). After blocking of 4 h with 1% nonfat milk in 0. 1M NaHCO3, pH 8.6, cells were incubated overnight at 4°C with anti-hCXCR2 antibody (R&D Systems, Minneapolis, MN) in TBS containing 0.1% BSA. The next day, cells were washed three times with TBS and incubated with goat anti-mouse horseradish peroxidase secondary antibody (Bio-Rad Laboratories, Hercules, CA) for 2 h. Subsequently, cells were incubated with substrate buffer (2 mM o-phenylenediamine, 35 mM citric acid, 66 mM Na2HPO4, and 0.015% H2O2, pH 5.6). The coloring reaction was stopped by adding 1 M H2SO4. Finally, the absorbance at 490 nm was determined using a Powerwave X340 absorbance plate reader (BioTek Instruments, Winooski, VT).
Residue Numbering and Nomenclature.
The Ballesteros-Weinstein residue number (Ballesteros and Weinstein, 1995) is given as superscript throughout the article for the enumeration of GPCR transmembrane helix residues in addition to the UniProt number (UniProt, 2011). The residues inside the extracellular loops are enumerated based on the Ballesteros and Weinstein (1995) numbering of the preceding transmembrane helix.
Construction CXCR2 In Silico Model.
A three dimensional model of the CXCR2 receptor was built with MOE version 2009.10 (Chemical Computing Group Inc., Montreal, ON, Canada) based on the recently elucidated crystal structure of its family member, the chemokine receptor CXCR4 solved with a small molecule [Protein Data Bank code 3ODU (Wu et al., 2010)]. To this end, the primary sequence of CXCR2 (Genbank accession no. P25025) was aligned to that of the crystal structure of CXCR4, disregarding the fused T4 lysozyme (Supplemental Fig. 1). The N-terminal residues 1 to 37 were omitted from model construction because of a lack of crystal data. Furthermore, as a result of the disordered region in the CXCR4 structure, the C-terminal residues 315 to 330 corresponding to half of TM7 and intracellular helix 8 of CXCR2 were modeled based on the crystal structure of human adrenergic β2 [Protein Data Bank code 2RH1 (Cherezov et al., 2007)].
Protein-Ligand Interactions.
Compound 1 was docked into the CXCR2 model using the program GOLD v4 (Verdonk et al., 2003). The docking was specifically aimed at the pocket between TMs 3, 4, 5, 6, and 7, delineating the major pocket within the TM domain, because this region was identified to determine the binding of compound 1 to CXCR2 in the chimera study. Protein-ligand interactions were optimized in MOE by energy minimization, during which the heavy atoms of the pocket residues were tethered with a 10.0 kcal/mol restraint, and the remainder of the receptor was fixed in space. Compound 1 was docked and optimized in a similar manner for the binding pose on the outside of the transmembrane region.
Results
Cloning and Pharmacological Characterization of CXCR2 Orthologs.
To elucidate the binding mode of compound 1 at human CXCR2, we started the study with different CXCR2 orthologs. The CXCR2 receptor cDNAs of rhesus monkey, vervet, baboon, chimpanzee, gorilla, and orangutan were cloned and amplified by using PCR (Horlick et al., 2006), whereas the cynomolgus monkey ortholog was derived from rhesus monkey CXCR2 by site-directed mutagenesis. Sequence analysis confirmed that the sequences indeed correspond to the CXCR2 ortholog sequences (Fig. 2). Comparison of the protein sequences of the seven CXCR2 orthologs and human CXCR2 revealed that all have a high homology with each other ranging from 89 to 100% (Fig. 3A). As a result of the high homology of the CXCR2 orthologs, we have used them as “natural mutants” to identify which amino acids are involved in the binding of compound 1. To determine the binding affinity of compound 1 to all CXCR2 orthologs, membranes of COS-7 cells transiently transfected with human CXCR2 or a CXCR2 ortholog were incubated with 125I-CXCL8 and indicated concentrations of CXCL8, SB265610 or compound 1 (Fig. 4A; Table 1). Figure 4 clearly shows that the binding affinity of compound 1 is significantly abolished at baboon CXCR2 compared with human CXCR2, whereas the binding affinity for both CXCL8 and SB265610 are not different. Analysis of homologous displacement with CXCL8 to all CXCR2 orthologs revealed binding of 125I-CXCL8, with a pKd ranging from 9.1 to 9.6. SB256510 dose-dependently displaces 125I-CXCL8 from all CXCR2 orthologs with pKi values from 7.3 to 7.7, whereas compound 1 only displaces 125I-CXCL8 from human, chimpanzee, gorilla, and orangutan with pKi values from 6.4 to 7.2 (Table 1). Differences of amino acids between the CXCR2 orthologs that cannot bind compound 1 (rhesus monkey, cynomolgus monkey, vervet, and baboon) and the orthologs that bind compound 1 (human, chimpanzee, gorilla, and orangutan) are indicated in Fig. 2. Four residues are located within the TM domain (Leu922.48, Ser1072.63, Ser2175.44 and Asn2686.52), two intracellular (Tyr751.59 and Ile3317.80), five extracellular (Val1924.73, Asn2065.32, Leu2105.36, Val2816.65, and Glu2876.71) and four in the N terminus (Ser10, Leu34, Ala36, and Glu40) (Figs. 2 and 3B). In the remainder of the study, we will denote the nonbinders as class A orthologs and the binders as class B orthologs, respectively.

Alignment of the sequences of various CXCR2 orthologs. The boxed residues differ all between the orthologs that are not able to bind compound 1 (rhesus monkey, cynomolgus monkey, vervet, and baboon; class A) and the orthologs that are able to bind compound 1 (human, chimpanzee, gorilla, and orangutan; class B). Residues indicated in bold differ between the human and baboon CXCR2.

Homology (percentage) of protein sequences of the CXCR2 receptor of human (Hm), baboon (Bb), chimpanzee (Ch), cynomolgus monkey (Cy), gorilla (Gr), orangutan (Ou), rhesus monkey (Rh), and vervet (Vv) calculated by ClusterW. *, chimpanzee CXCR2 lacks first five amino acids of human CXCR2 receptor. Besides that, all amino acid residues are identical (A). Snake plot of the human CXCR2 receptor. The residues indicated in bold and with a bigger font differ between human and baboon receptor. Residues indicated in a thick circle have been mutated, whereas residues indicated in gray are the conserved residues for the Ballesteros-Weinstein numbering scheme (B).

Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing human CXCR2 (A) or baboon CXCR2 (B). Membranes were incubated with indicated concentrations of CXCL8 (●), SB265610 (○), or compound 1 (■) and approximately 100 pM 125I-CXCL8 for 1 h at room temperature. The error bars indicate the S.E.M. of results of at least three independent experiments.
Affinity (pKd or pKi) of CXCR2 ligands at various CXCR2 orthologs
Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing indicated CXCR2 orthologs. The data are presented as mean ± S.E.M. of at least three independent experiments.
Chimeric Human-Baboon CXCR2 Approach.
To investigate which part of the human CXCR2 receptor is involved in the binding of compound 1, we choose baboon CXCR2 as a representative of the class A orthologs. Next, using the restriction sites EcoNI and BamHI (Fig. 3B), we created the chimeric constructs HHB, BBH, HBB, BHH, HBH, and BHB. The binding affinity of 125I-CXCL8 is equal to human and baboon CXCR2 at the chimeras BBH, BHH, HBH, and BHB, whereas the chimeras HHB and HBB are unable to bind the chemokine (Table 2). HHB and HBB are expressed on the cell surface as indicated by enzyme-linked immunosorbent assay (see Supplemental Figure 2). SB265610 displaces 125I-CXCL8 from the chimeras with almost equipotent affinity. Compared with the wild-type baboon CXCR2 receptor, the chimera BBH gains affinity for compound 1, whereas compound 1 has no affinity for the chimera BHB. Other chimeric receptors show results in line with this observation. Thus, we hypothesized that the region from TM5 to the C terminus is important for the binding of compound 1 to human CXCR2.
Affinity (pKd or pKi) of CXCR2 ligands at human-baboon chimeras
Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing indicated chimeric receptors HHB, BBH, HBB, BHH, HBH, and BHB. The data are presented as mean ± S.E.M. of at least three independent experiments.
Site-Directed Mutagenesis of the CXCR2 Receptor to Probe Baboon-Human Species Differences.
Following the results of the chimeric approach, we decided to continue with a site-directed mutagenesis approach to further pinpoint the amino acids involved in the binding of compound 1 at the human CXCR2 receptor. The amino acids from the human and baboon CXCR2 receptor differ at 35 positions; 13 of them are located in the region from TM5 to the C terminus (Fig. 2 and 3B). Only the residues that differ between the class A and B orthologs were subsequently mutated from the human to the respective baboon counterparts. Thus, we generated the mutants S2175.44T, N2686.52H, V2816.65L, and E2876.71Q (Fig. 3B). To extensively investigate differences between the human and baboon receptor in the extracellular loop 3, we also created the mutants Q2806.64R, Q2836.67N, and R2947.33Q. Because the binding site for SB265610 was previously identified to be intracellular (Nicholls et al., 2008; Salchow et al., 2010), and [3H]SB265610 was not displaced by compound 1 (de Kruijf et al., 2009), we suggested that the intracellular Ile3317.80 is not involved in the binding of compound 1 and thus did not mutate this residue.
All human-to-baboon single-point mutant cDNAs were transiently transfected in COS-7 cells. Competition binding analysis showed that all mutants were expressed and bind 125I-CXCL8 with nanomolar affinity (i.e., comparable with wild-type human CXCR2) (Table 3). SB265610 displaces 125I-CXCL8 from the mutants with almost equal binding affinity. It is noteworthy that compound 1 shows a significantly decreased affinity for the mutants S2175.44T and N2686.52H (Table 3; Fig. 5, A and B), whereas the affinity for the other mutants is unaltered. The N2686.52H mutant causes a greater decrease in affinity for compound 1 than S2175.44T, indicating that Asp2686.52 has a more important role in the binding of compound 1 to human CXCR2 than Ser2175.44.
Affinity (pKd or pKi) of CXCR2 ligands at site-directed mutagenesis CXCR2 human or baboon mutants to probe human-baboon species differences
Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing indicated human-to-baboon or baboon-to-human CXCR2 mutants. The data are presented as mean ± S.E.M. of at least three independent experiments.

Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing human CXCR2 (●), baboon CXCR2 (■), or indicated mutants by compound 1 (A–C) or SB265610 (D–F). Membranes were incubated with indicated concentrations of compound 1 or SB265610 and approximately 100 pM 125I-CXCL8 for 1 h at room temperature. The error bars indicate the S.E.M. of results of at least three independent experiments.
To confirm the important role of the serine and asparagine residues, we constructed the reciprocal baboon to human mutants T2125.44S and H2636.52N. As clearly shown in Fig. 5, A and B, mutation of the baboon residues into the human residues results in a gain of affinity of compound 1 (Table 3). In line with the results with the human to baboon mutant receptors, the baboon CXCR2 mutant H2636.52N causes a greater increase in binding affinity for compound 1 than T2125.44S. To investigate the role of these two residues in more detail, we created the double mutants S2175.44T/N2686.52H and T2125.44S/ H2636.52N. The human CXCR2 double mutant S2175.44T /N2686.52H, like the wild-type baboon receptor, showed no 125I-CXCL8 displacement by compound 1, whereas the baboon CXCR2 double mutant T2125.44S/ H2636.52N showed equal affinity for compound 1 as the wild-type human receptor (Fig. 5C; Table 3). Taken together, compound 1 shows only a markedly decreased affinity at the human CXCR2 mutants S2175.44T, N2686.52H, and S2175.44T/N2686.52H. Given these results, we conclude that compound 1 binds to the TM regions 5 and 6 of the CXCR2 receptor.
To investigate whether the (double mutant) receptors display the same properties in functional studies as observed in 125I-CXCL8 displacement assays, we performed [35S]GTPγS binding assays on membranes of COS-7 cells transiently transfected with CXCR2. As shown in Fig. 6A, CXCL8 shows a higher potency at baboon CXCR2 compared with human CXCR2 (Table 4). Mutations in either the human or baboon receptor do not alter the potency of CXCL8 compared with WT receptors. This is in agreement with the 125I-CXCL8 displacement studies. To test the dose-dependent antagonism of CXCL8-induced signal by compound 1 and SB265610, indicated concentrations of antagonists (Fig. 6) were used in combination with approximate EC80 concentration of CXCL8 (Fig. 6, B and C). SB255610 is able to inhibit the CXCL8-induced [35S]GTPγS binding dose-dependently at all receptors tested with pKb values of approximately 8.5 (Fig. 6B; Table 4). At the highest tested concentrations, SB265610 displays inverse agonism at the baboon constructs. In agreement with our 125I-CXCL8 displacement studies, compound 1 inhibits the CXCL8-induced [35S]GTPγS binding at human WT and baboon double mutant T2125.44S/H2636.52N with pKb values of 7.5 and 6.9, respectively. Furthermore, compound 1 barely inhibits CXCL8-induced [35S]GTPγS binding at baboon WT and human double mutant S2175.44T/N2686.52H (Fig. 6C; Table 4).

Stimulation of [35S]GTPγS by CXCL8 from COS-7 membranes expressing indicated receptors. Membranes were incubated with 3 μM GDP and approximately 400 pM [35S]GTPγS for 1 h at room temperature (A). The CXCL8-induced [35S]GTPγS binding was inhibited by SB265610 (B) or compound 1 (C). Membranes were preincubated with indicated concentrations of CXCR2 antagonists for 30 min at room temperature, followed by stimulation of approximately EC80 concentration of CXCL8 for 1 h at room temperature. Data are expressed as percentages, in which 0% represents basal signal and 100% CXCL8 signal without presence of antagonist (B and C). The error bars indicate the S.E.M of results of at least three independent experiments.
Affinity (pKd or pKi) and inhibition of CXCL8-induced [35S]GTPγS binding (pKb) of CXCR2 ligands at key constructs
Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing indicated key receptors. The data are presented as mean ± S.E.M. of at least three independent experiments. CXCL8 stimulated [35S]GTPγS binding to COS-7 membranes expressing indicated receptors and inhibition of CXCL8-induced [35S]GTPγS after 30 min before incubation with SB265610 and compound 1. The data are presented as mean ± S.E.M. of at least three independent experiments.
In Silico-Based Site-Directed Mutagenesis of the CXCR2 Receptor.
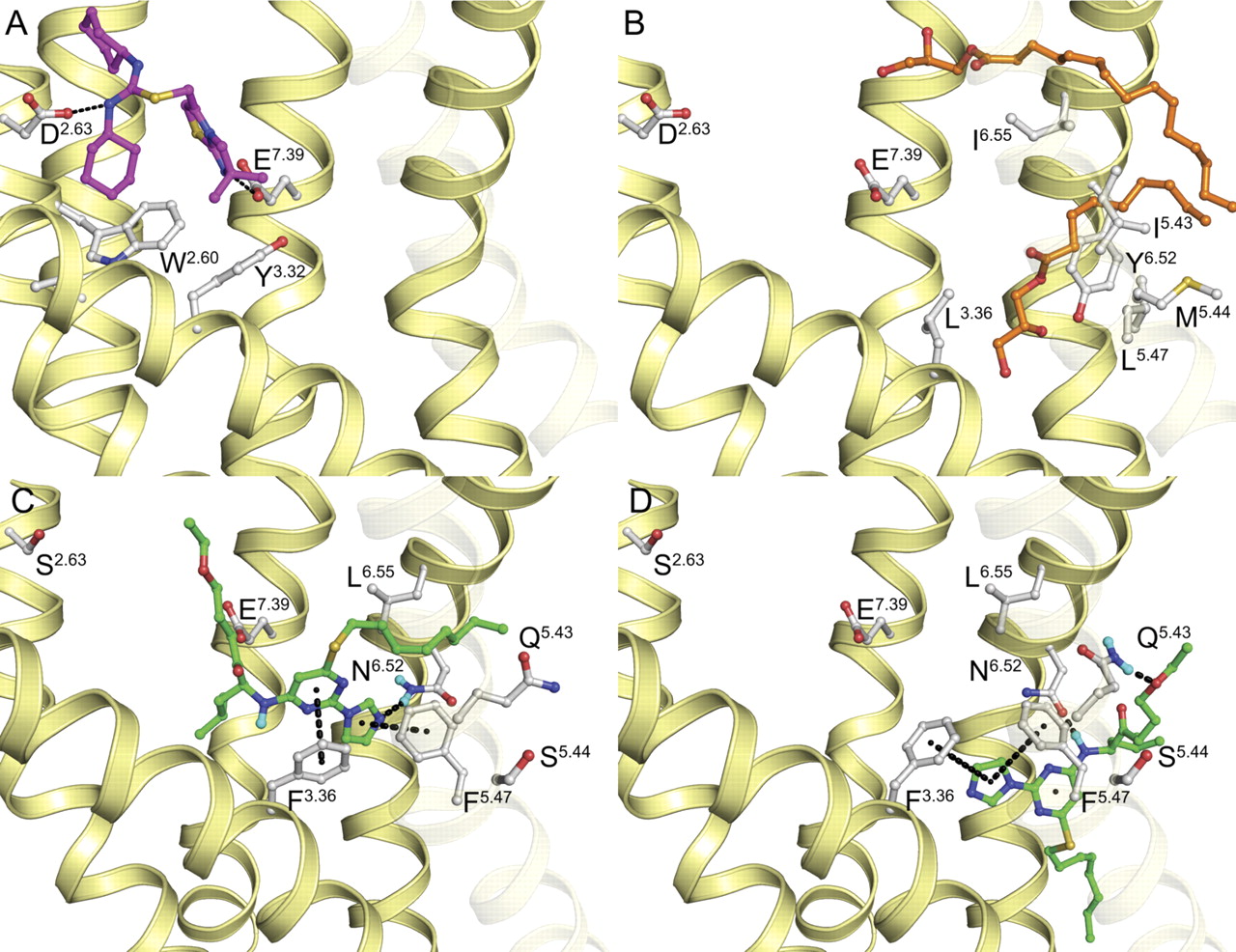
Because the chimeric and site-directed mutagenesis data provided only a rough indication that compound 1 is a TM-binder, we characterized the binding mode of compound 1 in more detail by constructing additional mutations guided by in silico modeling. A CXCR2 model was constructed based on the recently resolved structure of the chemokine receptor CXCR4 (Wu et al., 2010). Wu et al. (2010) cocrystallized the CXCR4 receptor with the ligand IT1t in the minor subpocket of the TM bundle encompassing TM1, TM2, TM3, and TM7, where it interacts with residues Asp972.63 and Glu2887.39 (Fig. 7A). Furthermore, the crystal structure shows two lipids in the major subpocket of the TM bundle encompassed by TM3, TM4, TM5, TM6, and TM7 (Fig. 7B). The same major subpocket is occupied by the peptide ligand CVX15 in another CXCR4 crystal structure (Wu et al., 2010).

Positioning of the small ligand IT1t (magenta) in the minor subpocket of the CXCR4 receptor (Protein Data Bank code 3ODU) (A), and the lipids (orange) in the major subpocket of the CXCR4 receptor (Protein Data Bank code 3ODU) (B). Suggested binding modes of compound 1 (green) in the major subpocket of the human CXCR2 receptor with the octane moiety pointing into the lipid membrane between TM5 and TM6 (C), and in the lipid membrane before entering the transmembrane major subpocket (D). The interactions between compound 1 and Phe3.36, Gln5.43, Phe5.47, Asn6.52, and Leu6.55 are in line with experimental data; they are annotated with dotted lines.
On the basis of our chimera and point mutant data, we hypothesized its binding site to be part of the major subpocket (Fig. 7, C and D). To confirm this, we created the mutants S1072.63A, E3007.39Q, and E3007.390S because these residues interact with ligand IT1t in the CXCR4 structure in the minor subpocket between TM2, TM3, and TM7 (Fig. 7A) (Wu et al., 2010). Ser1072.63 was mutated to an alanine residue present in baboon CXCR2 (S1072.63A). Glu3007.39 was mutated to a glutamine to prevent ionic interactions and to a serine (E3007.39S) because the inverse S7.39E mutation in CXCR3 enables binding of CXCR4 antagonists (Rosenkilde et al., 2004). The affinity of compound 1 at the mutants S1072.63A, E3007.39Q, and E3007.39S is unchanged compared with the human wild-type receptor. In addition, 125I-CXCL8 and SB265610 bind with the same affinity as that of the wild-type (Fig. 7; Table 5). These results indicate that compound 1 indeed does not bind to the minor subpocket.
Affinity (pKd or pKi) of CXCR2 ligands at in silico based site-directed mutagenesis human CXCR2 mutants
Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing indicated human single point mutants. The data are presented as mean ± S.E.M. of at least three independent experiments.
Subsequently, molecular docking results of compound 1 in the CXCR2 model suggested the residues Phe1303.36, Gln2165.43, Phe2205.47, Asn2686.52, and Leu2716.55 to interact with compound 1 (Fig. 7, C and D). We were surprised to find that Ser2175.44 showed no direct interaction with compound 1. Because of the high conservation of the TM structure, none of the currently known crystal structures possess residue 5.44 pointing into the major pocket. As such, this is also true in our CXCR2 model. Therefore, we believe that our model is justified, and we propose that the influence of this residue is of indirect nature. Our model suggests two binding modes representing different states along the same ligand-binding pathway. In the binding mode presented in Fig. 7C, the pyrimidine and imidazole moieties of compound 1 interact with Phe1303.36 and Phe2205.47 through π-π stacking, whereas the imidazole moiety forms a hydrogen bond with Asn2686.52. In Fig. 7D, the imidazole moiety of compound 1 interacts with Phe1303.36 and Phe2205.47 through π-π interactions, the aminopyrimidine forms hydrogen bonds with N2686.52, and the ether moiety interacts with Gln2165.43. In both binding modes, the octane moiety protrudes through TM5 and TM6 between residues Gln2165.43 and Leu2716.55 similar to the CXCR4 lipids (Fig. 7B). The interactions of compound 1 in both models are in line with the structure-activity relationship investigation described by Ho et al. (2006), which emphasizes the importance of the long hydrophobic octane moiety as well as the geometry and directionality of the imidazole acceptor group. To assess the involvement of these five residues, we constructed the mutants F1303.36A, Q2165.43E, F2205.47A, and L2716.55A (Asn2686.52 has already been mutated; see Site-Directed Mutagenesis of the CXCR2 Receptor to Probe Baboon-Human Species Differences). However, because F2205.47A did not bind 125I-CXCL8, we created the additional mutant F2205.47L. Compared with wild type CXCR2 (pKi = 7.2, Table 5) the affinity of compound 1 is slightly decreased at Q2165.43E (pKi = 6.5) and significantly decreased for F2205.47L (pKi = 6.1) and L2716.55A (pKi = 6.2). Figure 8 clearly shows that compound 1 binding is completely abolished in the mutant F1303.36A (Table 5). None of the mutants affected the binding affinity of 125I-CXCL8. In line with the binding studies, compound 1 barely inhibits the CXCL8-induced [35S]GTPγS binding at F1303.36A (pKb is <4; see Supplemental Fig. 3).

Displacement of 125I-CXCL8 binding to COS-7 cell membranes expressing indicated human CXCR2 mutants by compound 1 (A) or SB265610 (B). Membranes were incubated with indicated concentrations of SB265610 or compound 1 and approximately 100 pM 125I-CXCL8 for 1 h at room temperature. The error bars indicate the S.E.M. of results of at least three independent experiments.
Discussion
CXCR2 is a potential drug target because of its involvement in several inflammatory diseases, such as atherosclerosis, chronic obstructive pulmonary disease, psoriasis, rheumatoid arthritis, inflammatory bowel disease, and multiple sclerosis (Boisvert et al., 1998; Podolin et al., 2002; Ajuebor et al., 2004; Buanne et al., 2007; Chapman et al., 2007; Liu et al., 2010). In view of their therapeutic potential, the search for CXCR2 antagonists has been extensive and has led to the identification of different classes of CXCR2 antagonist (Li et al., 2003; Widdowson et al., 2004; Baxter et al., 2006; Ho et al., 2006). In our previous publication (de Kruijf et al., 2009), we reported that CXCR2 antagonists of the diarylurea (such as SB265610; see Fig. 1) and the thiazolopyrimidine classes bind to a distinct site compared with compounds of the imidazolylpyrimidine class (such as compound 1; see Fig. 1). Studies show that diarylurea and thiazolopyrimidine compounds most likely bind intracellularly to CXCR2 (Nicholls et al., 2008; Salchow et al., 2010). In this study, we identified a binding site for the imidazolylpyrimidine compound 1 within the TM region.
Identification of a Novel Allosteric Binding Site in CXCR2 by Natural Mutants.
Different monkey CXCR2 orthologs (Fig. 2, 3) were used as natural mutants to identify key residues involved in the binding of compound 1 to human CXCR2. In all experiments, we defined the binding affinity of SB25610 as a control. Because this compound seemed to bind intracellularly (Salchow et al., 2010), we expected no significant change in binding affinity at the different CXCR2 receptors. Our results show that the rhesus monkey, cynomolgus monkey, vervet, and baboon orthologs are devoid of affinity for compound 1, whereas the human, chimpanzee, gorilla, and orangutan orthologs bind this compound with high affinity (Fig. 4; Table 1). We have labeled these species as class A and B, respectively. Using human-baboon chimeric proteins and subsequently human-to-baboon reciprocal single-point mutants, we identified Ser2175.44 and Asn2686.52 as key residues distinguishing class A and B orthologs. Imidazolylpyrimidine compounds have also no affinity for the mouse, rat, and rabbit CXCR2 orthologs (J. C. H. M. Wijkmans, M. L. Webb, D. S. Auld, unpublished data). Because the rodent orthologs all possess Asn6.52, but do contain Thr5.44 instead of Ser5.44 (Supplemental Fig. 4.), we hypothesize that in these rodent receptors, Thr5.44 and possibly other residues are responsible for the lack of affinity for compound 1.
Investigation of the CXCR2 Minor Subpocket.
In the recently resolved crystal structure of the CXCR4 receptor, the small ligand IT1t interacts with the residues Asp972.63 and Glu2887.39 in the minor subpocket (Wu et al., 2010). Furthermore, studies with CXCR1, which has 77% sequence homology to CXCR2, revealed that both (R)-ketoprofen and repertaxin interact also with residues located in the minor subpocket, such as Ile431.36, Tyr461.39, Lys992.64, Val1133.28, and Glu2917.39 (Bertini et al., 2004; Allegretti et al., 2005; Moriconi et al., 2007). Moreover, Glu7.39 plays a role in binding of antagonists at various other chemokine receptor as reviewed by Scholten et al. (2011). Because the binding affinity of compound 1 was not changed at the mutants S1072.63A, E3007.39S, and E3007.39Q (Fig. 8; Table 5), we conclude that compound 1 does not bind to the minor subpocket at CXCR2. From the chimeric studies, we already expected that Ser1072.63 was not a key residue in the binding of compound 1 to human CXCR2, because TM2 was not essential for the difference between the class A and B orthologs.
In Silico Guided Mutagenesis of the CXCR2 Major Subpocket to Elucidate Antagonist Binding Mode.
A CXCR2 model was constructed on the basis of the recently resolved CXCR4 crystal structure (Wu et al., 2010). Molecular docking results of compound 1 in this model suggested that the residues Phe1303.36, Gln2165.43, Phe2205.47, Asn2686.52, and Leu2716.55 could interact with compound 1 (Fig. 7, C and D). Although we showed that Ser2175.44, like Asn2686.52, was essential for compound 1 binding, it showed no direct interaction with compound 1 in our model. Threonine (class A orthologs) and serine (class B orthologs) residues in combination with proline residues are known to cause kinks in TM helixes (Deupi et al., 2010), as demonstrated, for example, by the T2.56XP2.58-induced kink in TM2 in chemokine receptors (Govaerts et al., 2001; Wu et al., 2010). The presence of an extra proline residue at position 5.42 might even emphasize the induction of TM5 kinks. The involvement of serine 2175.44 in compound 1 binding might be explained by a different rotation state of the TM5 helix compared with a threonine, thereby affecting the binding of compound 1 by changing the shape of the ligand binding pocket and entrance channel between TM5 and TM6 (Fig. 7, C and D). To investigate how compound 1 binds to the major subpocket, we constructed additional mutants based on in silico modeling.
Our binding studies with the mutants F1303.36A, Q2165.43E, F2205.47L, and L2716.55A revealed that these residues, like Asn2686.52, interact with compound 1 (Fig. 8; Table 5). To the best of our knowledge, this is the first investigation in which the imidazolylpyrimidine CXCR2 antagonists have been characterized to bind inside the TM domain of the CXCR2 receptor (major pocket). As shown in Fig. 7C, the proposed binding location is similar to the location of the fatty acids cocrystallized with the CXCR4 receptor (Protein Data Bank code 3ODU) pointing partially into the lipid bilayer. The binding mode is also in line with the structure-activity relationship investigation described by Ho et al. (2006), which emphasizes the importance of the long hydrophobic octane moiety (which lies between TM5 and TM6) as well as the geometry and directionality of the imidazole acceptor group. The data from the F1303.36A and F2205.47L mutants can be explained by a loss of aromatic interactions with the pyrimidine and imidazole moieties, the N2686.52H mutant by the loss of a hydrogen bonding interaction with the imidazole moiety, and the L2716.55A mutant by a loss of hydrophobic matching with the octane moiety of compound 1. No direct indication for the small loss of activity by Q2165,43E mutation could be derived.
CXCR2 Ligand Entry Mechanism.
We propose as well a binding mode on the interface of the major pocket and the lipid bilayer (Fig. 7D), which is in line with the structure-activity data. This binding mode involves interactions for Phe1303.36, Phe2205.47, and Asn2686.52 similar to those for the binding mode in the major pocket (Fig. 7C) but ones in which Gln2165.43 forms a hydrogen bonding interaction with the ether group and the Leu2716.55 shows no direct interaction. We postulate that compound 1 first binds to the interface coming from the lipid bilayer (Fig. 7D), followed by a rotation to get into the major pocket (Fig. 7C), in analogy with sn-2-arachidonoylglycerol in the cannabinoid 2 receptor (Hurst et al., 2010). During ligand entry, Ser2175.44 might play a crucial role by adapting the conformation of TM5 to open the entry channel for compound 1, whereby Asn2686.52 supports ligand entry to the major subpocket. An entry channel between these TMs has been earlier described for the opsin receptor (Park et al., 2008). In this receptor, the residues Ile2055.40, Phe2085.43, Phe2736.56, and Phe2766.59 accommodate an entry channel for 11-cis-retinal between TM5 and TM6.
Although it may seem counterintuitive that a ligand fits partially in the entrance channel of the TM domain, recent advances in CXCR2-ligand binding pocket elucidations have shown that an intracellular binding site for CXCR2 antagonists exists as well (Nicholls et al., 2008; Salchow et al., 2010), a result seen only for two other GPCRs, the chemokine receptors CCR4 and CCR5 (Andrews et al., 2008). As such, this does not only imply that CXCR2 behaves differently from other GPCRs, but it could also mean that we should be thinking outside of the metaphoric box that is the TM domain.
Conclusions
Taken together, using different CXCR2 orthologs, chimeras, site-directed mutagenesis, and in silico modeling, we have identified binding modes in the transmembrane domain for the imidazolylpyrimidine compound 1 at the human CXCR2 receptor. Our in silico guided mutagenesis studies indicate a new ligand binding cavity in CXCR2 located between TM helices 3, 5, and 6 and suggest that the imidazolylpyrimidine antagonist enters CXCR2 via the TM5-TM6 interface. A ligand entry channel at the same interface was already described for the opsin receptor (Park et al., 2008). Furthermore, lipid molecules in the recently solved crystal structure of the CXCR4 receptor (Wu et al., 2010) are encompassed by the same interface. This suggests a general ligand entrance mechanism for nonpolar ligands to GPCRs. Our identification of a novel allosteric binding cavity in the TM domain of CXCR2, in addition to the previously identified intracellular binding site, shows the diversity in ligand recognition mechanisms by this receptor and offers new opportunities for the structure-based design of small allosteric modulators of CXCR2 in the future.
Authorship Contributions
Participated in research design: de Kruijf, Lim, Roumen, Webb, Auld, Wijkmans, Zaman, Smit, de Graaf, and Leurs.
Conducted experiments: de Kruijf, Lim, Roumen, Renjaän, Zhao, and de Graaf.
Contributed new reagent or analytic tools: Wijkmans and Zaman.
Performed data analysis: de Kruijf, Lim, Roumen, and Renjaän.
Wrote or contributed to the writing of manuscript: de Kruijf, Lim, Roumen, Zaman, Smit, de Graaf, and Leurs.
Acknowledgments
We acknowledge Annechien Hengeveld for constructing the chimeric proteins.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This study was performed within the framework of the Dutch Top Institute Pharma [projects T101-3, D1-105].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.073825.
-
ABBREVIATIONS:
- CXCR
- CXC chemokine receptor
- GPCR
- G-protein coupled receptor
- SB265610
- N-(2-bromophenyl)-N′-(7-cyano-1H-benzotriazol-4-yl)urea
- SCH-527123
- 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1-enylamino}-benzamide
- IT1t
- 1,3-dicyclohexyl-2-(3-methyl-6,6-dimethyl-5,6-dihydroimidazo[1,2-b] thiazole)-2-thiopseudourea
- ketoprofen
- 2-(3-benzoylphenyl)propanoic acid
- CVX15
- Arg-Arg-Nal-Cys-Tyr-Gln-Lys-dPro-Pro-Tyr-Arg-Cit-Cys-Arg-Gly-dPro
- TM
- transmembrane
- PEI
- polyethylenimine
- BSA
- bovine serum albumin fraction V
- GTPγS
- guanosine 5′-O-(3-thio)triphosphate; compound 1, (S)-2-(2-(1H-imidazol-1-yl)-6-(octylthio)pyrimidin-4-ylamino)-N-(3-ethoxypropyl)-4-methylpentanamide
- PCR
- polymerase chain reaction
- H
- human
- B
- baboon
- TBS
- Tris-buffered saline
- WT
- wild type.
- Received May 27, 2011.
- Accepted September 23, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}