


Abstract
The histamine H4 receptor (H4R) is the latest identified histamine receptor to emerge as a potential drug target for inflammatory diseases. Animal models are employed to validate this potential drug target. Concomitantly, various H4R orthologs have been cloned, including the human, mouse, rat, guinea pig, monkey, pig, and dog H4Rs. In this article, we expressed all these H4R orthologs in human embryonic kidney 293T cells and compared their interactions with currently used standard H4R ligands, including the H4R agonists histamine, 4-methylhistamine, guanidinylethyl isothiourea (VUF 8430), the H4R antagonists 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methylpiperazine (JNJ 7777120) and [(5-chloro-1H-benzimidazol-2-yl)carbonyl]-4-methylpiperazine (VUF 6002), and the inverse H4R agonist thioperamide. Most of the evaluated ligands display significantly different affinities at the different H4R orthologs. These “natural mutants” of H4R were used to study ligand-receptor interactions by using chimeric human-pig-human and pig-human-pig H4R proteins and site-directed mutagenesis. Our results are a useful reference for ligand selection for studies in animal models of diseases and offer new insights in the understanding of H4R-ligand receptor interactions.
The histamine H4 receptor (H4R) is the latest identified member of the four known histamine receptor subtypes (Hough, 2001), which all belong to the family of G-protein coupled receptors (GPCRs). In view of the success of the histamine H1 receptor (H1R) and the histamine H2 receptor as drug targets for the treatment of allergic conditions and gastric ulcers, respectively (Parsons and Ganellin, 2006), and the ongoing clinical trials of histamine H3 receptor (H3R) antagonists for central nervous system applications (Celanire et al., 2005), expectations for drugs targeting the H4R are high (Smits et al., 2009). The H4R is mainly present in leukocytes and mast cells, which are important components of the body's defense system (Oda et al., 2000; Liu et al., 2001a). A growing body of evidence implicates the H4R in the regulation of the immune system, such as chemotaxis of eosinophils, mast cells, and monocyte-derived dendritic cells and modulation of chemical mediator production, such as leukotriene B4, interleukin 16, and other interleukins (Takeshita et al., 2003; Thurmond et al., 2008). These preclinical studies support the view that H4R is a potential new drug target for inflammatory diseases such as allergic asthma and rheumatoid arthritis (Thurmond et al., 2008).
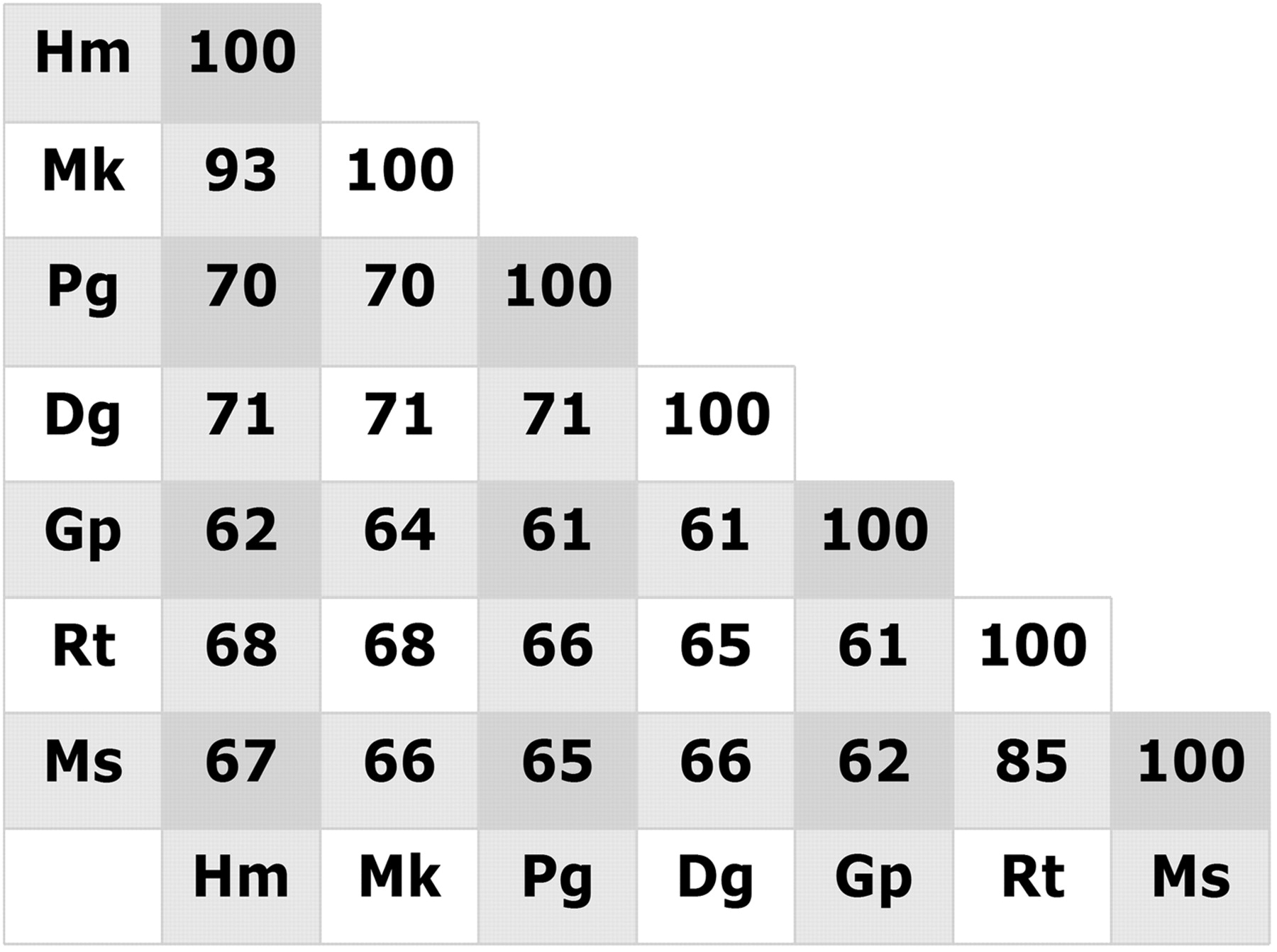
Translational preclinical animal models are still crucial to predict the therapeutic potential of newly developed ligands in humans. Therefore, to study therapeutic effects of H4R ligands in animal models of disease, it is important to characterize the H4R of the corresponding species. For several GPCRs, including the H3R, significant species differences are known and have seriously hampered drug discovery efforts (Oksenberg et al., 1992; Maconi et al., 2002; Reinhart et al., 2004; Hancock, 2006). Various species orthologs of H4R were promptly cloned based on their homology to the human H4R sequence (Oda et al., 2000), including those of mouse, rat, guinea pig, pig, monkey (Macaca fascilularis), and dog (Liu et al., 2001b; Oda et al., 2002, 2005; Jiang et al., 2008). The H4R species variants show relatively low homology to the human H4R (65–71%), except for the monkey H4R, which shows an overall amino acid homology of 93% (Fig. 1). The sequence differences of human, rat, and mouse H4R have been reported to result in significant differences in the affinity for the endogenous agonist histamine (Liu et al., 2001b). Detailed analysis of the differences in receptor structure resulted in the identification of Phe16945.55 in the second extracellular loop as one of the amino acid residues responsible for the mouse/human species difference in ligand binding (Lim et al., 2008).

Homology (%) of protein sequences of the histamine H4R of human (hm), M. fascicularis monkey (mk), pig (pg), dog (dog), guinea pig (gp), rat (rt), and mouse (ms).
In view of the relatively wide divergence in amino acid sequence among the various H4R species orthologs (which can be considered “natural mutagenesis”), we have extensively and systematically investigated this issue by expressing the human, monkey, pig, dog, guinea pig, mouse, and rat H4Rs in HEK 293T cells and evaluated the interactions of the various H4R proteins with a set of reference H4R ligands that have been used in H4R studies, including the H4R agonists histamine, 4-methylhistamine (Lim et al., 2005), VUF 8430 (Lim et al., 2006), clozapine, and clobenpropit (Buckland et al., 2003), the H4R antagonists JNJ 7777120 (Thurmond et al., 2004) and VUF 6002 (Terzioglu et al., 2004; Venable et al., 2005), and the H4R inverse agonist thioperamide (Takeshita et al., 2003). Using chimeric human-pig-human and pig-human-pig H4R proteins and site-directed mutagenesis (SDM) in combination with in silico modeling studies, we investigated the ligand-receptor interactions in detail and systematically identified key residues responsible for observed ligand-dependent species differences.
Materials and Methods
Materials.
Dulbecco's modified Eagle medium (DMEM), penicillin and streptomycin were purchased from Invitrogen (Merelbeke, Belgium). Cell culture plastic wares were obtained from Greiner Bio-One (Wemmel, Belgium). Tris base was purchased from AppliChem (Darmstadt, Germany). Linear 25-kDa polyethylenimine (PEI) was obtained from Polysciences, Inc. (Warrington, PA). Histamine dihydrochloride, clozapine, and branched 750-kDa PEI were purchased from Sigma (St. Louis, MO), whereas VUF 8430, thioperamide maleate, clobenpropit dihydrochloride, JNJ 7777120, and VUF 6002 were synthesized at the Department of Medicinal Chemistry, Vrije Universiteit Amsterdam. [3H]Histamine (18.10 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Oligonucleotide primers for PCR were synthesized by Isogen Bioscience (Maarsen, The Netherlands). Endonuclease restriction enzymes, T4 DNA ligase, and Pfu DNA polymerase were from MBI Fermentas (St. Leon-Rot, Germany).
DNA Constructs and Site-Directed Mutagenesis.
The wild-type human H4R cDNA cloned in pcDNA3.1 was purchased from the Missouri S&T cDNA Resource Center (Rollo, MO). The cDNA was subcloned into a mammalian expression vector pcDEF3 (a gift from Dr. J. Langer, Robert Wood Johnson Medical School, Piscataway, NJ) at BamHI and XbaI sites. The cDNA encoding the pig H4R that was a gift from Yamanouchi Pharmaceuticals (Tokyo, Japan) (Oda et al., 2002) was subcloned in pcDEF3. The cDNAs of the other H4R species variants were synthesized by HD Biosciences Co., Ltd. (Shanghai, China) according to the sequences in GenBank (XM_547634 for dog, AAK97379 for guinea pig, BAE16558 for monkey M. fascicularis, NP_694727 for mouse, NP_571984 for rat H4R, and cloned in pcDEF3. All the H4R cDNAs contain a Kozak sequence (GCCACC) before the start codon ATG. The plasmids were amplified in Escherichia coli JM109 (Promega, Madison, WI) and purified using Nucleobond AX columns (Macherey-Nagel, Düren, Germany). Chimeric receptor constructs were created by exchanging the domain between the DRY motif at the bottom of TM3 (ClaI restriction site in the cDNA) and residue Glu5.46 in TM5 (EcoRI restriction site in the cDNA) of the human H4R with that of the pig H4R ]resulting in the chimeric receptor HPH (human-pig-human)] and the corresponding domain of pig H4R with that of the human H4R [resulting in the PHP chimera (pig-human-pig)]. Because of the presence of an EcoRI restriction site in the pig H4R cDNA region corresponding to proximity of N and C termini, we used PCR to facilitate construction of the latter chimera. Site-directed mutagenesis was performed by PCR with the use of mutant oligonucleotide primers and verified by sequencing analysis.
Cell Culture and Transfection.
HEK 293T cells were maintained in DMEM supplemented with 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin in a 5% CO2 and 95% humidity at 37°C. For transfections, 5 μg of receptor plasmid was mixed in 0.5 ml of serum-free DMEM with 25 μl of 1 mg/ml 25-kDa linear PEI. The mixture was incubated for 5 to 10 min at room temperature before it was added onto subconfluent HEK 293T cell monolayer culture submerged in 5 ml of fresh culture medium. Transfected cells were detached from the plastic surface two days after transfection using 5 ml/dish phosphate-buffered saline containing 1 mM EDTA and were collected as pellets by centrifugation at 200g for 3 min. The pellets were stored at −20°C until use.
[3H]Histamine Binding Assay.
Radioligand binding assays were performed using homogenized transfected cells in 50 mM Tris-HCl binding buffer, pH 7.4 at room temperature, in a total assay volume of 200 μl. Saturation binding analysis was performed using different concentrations of [3H]histamine (18.10 Ci/mmol) in the absence and presence of 3 to 10 μM JNJ 7777120. For displacement studies, cell homogenates were typically coincubated at different concentrations of ligands in the presence of approximately 7 to 20 nM [3H]histamine, in a total volume of 200 μl. The reaction mixtures were incubated for 1 h at room temperature (22°C), harvested on 96-well glass fiber C plates (PerkinElmer Life and Analytical Sciences) that were pretreated with 0.3% PEI, followed by three washes with ice-cold 50 mM Tris-HCl buffer, pH 7.4 at 4°C. The radioactivity retained on the filters was measured by liquid scintillation counting. The equilibrium dissociation constant (KD) and inhibition constant (Ki) values were calculated by nonlinear regressions for a single binding site model using Prism 4.0 (GraphPad Software, Inc., San Diego, CA).
Residue Numbering and Nomenclature.
The Ballesteros-Weinstein residue numbering scheme (Ballesteros and Weinstein, 1995) is used throughout the article for GPCR transmembrane (TM) helices, whereas a recently proposed numbering scheme (de Graaf et al., 2008) was used to number the residues in the second extracellular loop (EL2). For explicitly numbering EL2 residues in specific receptors, the UniProt residue number is given in superscript after the EL2 number (e.g., Phe45.55169 in human H4R).
Construction H4R Models.
First, a preliminary high-throughput receptor model of only the seven TM helices of H4R was generated using the GPCRgen program (Bissantz et al., 2004) based on the high-resolution carazolol bound crystal structure template of the adrenergic β-2 receptor (Cherezov et al., 2007). The amino acid sequence alignments used for constructing the receptor models are shown in Supplemental Figure I. This preliminary H4R model was minimized with AMBER 10 (http://ambermd.org/) using the AMBER03 force field (Wang et al., 2004) to relax the structure and remove steric bumps. The minimizations were performed by 1000 steps of steepest descent followed by conjugate gradient until the root-mean-square gradient of the potential energy was lower than 0.05 kcal/mol · Å. A twin cut-off (12.0, 15.0 Å) was used to calculate nonbonded electrostatic interactions, and the nonbonded pair list was updated every 25 steps. Histamine was docked into this structure using “two-times speed-up” settings of GOLD ver. 4.0 (Verdonk et al., 2003). Experimentally driven receptor-ligand H-bond constraints were used to guide the docking process in the receptor between 1) the protonated amine nitrogen atom of histamine and one of the carboxylate oxygen atoms (OD1) of Asp3.32 and 2) the τ nitrogen of the imidazole group of histamine and one of the carboxylate oxygen atoms of Glu5.46. Fifteen histamine poses were generated. The histamine-H4R complex was minimized with AMBER 10 using the same settings as described above, including the same H-bond (hydrogen-acceptor distance and donor-hydrogen-acceptor angle) constraints restraints as used for docking with addition H-bond constraints between 3) the sulfur atom of Cys3.36 and one of the carboxylate oxygens (OD2) of Asp3.32, in line with an earlier experimentally supported histamine-bound model of H1R (Jongejan et al., 2005) and 4) the amide nitrogen atom of the Gln7.42 side chain and one of the carboxylate oxygen atoms (OD2) of Asp3.32 in line with an earlier histamine-bound H4R model (Jongejan et al., 2008). This minimized complex was refined by a second AMBER energy minimization without distance restraints. Histamine force-field parameters were derived using the Antechamber program (Wang et al., 2004), and partial charges for the substrates were derived using the AM1-BCC procedure in Antechamber. The EL2 was constructed using two subsequent Modeler 9 ver. 1 (Sali and Blundell, 1993) runs with an explicit disulfide bridge constraint between Cys3.25 and Cys45.50 and including the histamine binding pose in the TM template as “block” residue. In the first run, the β2 adrenergic receptor crystal structure (Protein Data Bank code 2rh1; Cherezov et al., 2007) was used to model the part upstream of EL2. Of the 15 generated models, the model with highest Modeler score and EL2 loop conformations properly accommodating the original histamine binding orientation in the original TM model were selected as input for a second Modeler run. In this second run, the EL2 segment downstream from Cys45.50 was constructed. One of 15 models was selected based on the criteria described before. The H4R-histamine H-bond constraints earlier used for energy minimization were transformed into explicit upper-bound (3.5 Å) distance constraints in the Modeler runs. After optimization of the EL2 conformation, extracellular loops 1 and 3, intracellular loops 1 and 2, and helix 8 were constructed based on the β2 adrenergic receptor crystal structure (Cherezov et al., 2007) using Modeler 9 ver. 1. Intracellular loop 3 and the N and C termini were not modeled. The amino acid sequence alignments used for constructing the receptor models are shown in Supplemental Figure I. The final receptor model was energy minimized with the initially minimized histamine docking pose as described before.
Clozapine and JNJ 7777120 were docked into this model using the following experimentally guided (Shin et al., 2002; Jongejan et al., 2008) H-bond constraints: 1) between the protonated piperidine nitrogen atom of the ligand and one of the carboxylate oxygen atoms (OD1) of Asp3.32 or 2) between the carboxamide (JNJ 7777120) or tricyclic ring (clozapine) nitrogen atom of the ligand and one of the carboxylate oxygen atoms of Glu5.46. In the JNJ 7777120-H4R complex, the 32 χ1 torsional angle of Cys3.36 was manually rotated from its g- to its t rotamer to mimic the inactive state of the receptor (Jongejan et al., 2005). The models of human H4R L5.39V, N4.57H, and N4.57H/S5.43L mutants were built by mutating the corresponding residues of the wild-type ligand bound H4R model using the “mutate” function of MOE 2008.10 (http://www.chemcomp.com). The resulting wild-type and mutant receptor-ligand models, including the docked ligands, were further minimized as described above.
Results
Expression of H4R Orthologs.
We comprehensively analyzed the ligand binding properties of the different H4R species variants. Transient transfection of HEK 293T cells with cDNAs of the different H4R orthologs resulted in an adequate expression of functional H4R proteins (Bmax values, 1–6.9 pmol/mg protein; Supplemental Figure II), as estimated by the binding of the agonist radioligand [3H]histamine with nanomolar affinities. The KD values of [3H]histamine for the human, monkey, pig, guinea pig, dog, mouse, and rat H4Rs are 9, 15, 11, 11, 75, 78, and 134 nM, respectively (Supplemental Figure II). These values indicate species differences up to 10-fold in binding the endogenous agonist and are in agreement with values reported previously (Oda et al., 2000, 2002, 2005; Liu et al., 2001b). It should be noticed that differences in expression host as well as method may lead to variability in measured pharmacological values. Interspecies ligand affinity ratios determined with individual experimental setups, however, are consistent. The KD value of [3H]histamine is 4-fold higher for dog H4R expressed in HEK 293T cells (current study) than for dog H4R expressed in COS-7 cells (Jiang et al., 2008), whereas KD values for human H4Rs expressed in HEK 293T (Oda et al., 2000) is 3-fold higher for human H4Rs expressed in SK-N-MC cells (Liu et al., 2001). Furthermore, the Ki values of other H4R ligands, such as 4-methylhistamine and thioperamide, for the dog H4R from this study in HEK 293T cells are approximately 5-fold higher than those reported previously in COS-7 cells (Jiang et al., 2008), whereas JNJ 7777120 affinity is in good agreement in both reports.
We showed that the binding of [3H]histamine is not affected by the presence of GTP or GTPγS (Supplemental Figure III), indicating that the binding of histamine is independent of the G-protein coupling state of the receptor, as proposed earlier by Schneider et al. (2009). This implies that the affinities of tested H4R ligands determined by [3H]histamine displacement are independent of G-protein coupling-state of the receptors. We also observed that all of these H4R orthologs are able to dose-dependently respond to histamine in a Gαqi5/nuclear factor of activated T cells-luciferase reporter gene assay performed according to a method described previously (Lim et al., 2008; Supplemental Figure IV). As described previously (Liu et al., 2001b; Lim et al., 2008), and in line with the binding studies (Table 1), histamine was less potent at rat and mouse H4Rs (Supplemental Table 2).
Affinity (pKi) of H4R ligands for different H4R species variants. The data are presented as mean ± S.E.M. of at least three independent experiments.
Ligand Binding Affinity for H4R Orthologs.
Histamine binds the different H4R orthologs, as described above, with affinities that vary up to 10-fold (Table 1 and Supplemental Figure II). As can be seen in Fig. 2 and Table 1, other H4R ligands interact with the different species orthologs with varying affinities. The H4R agonist 4-methylhistamine (Lim et al., 2005) consistently shows a slightly lower affinity than histamine for each of the orthologs, and the binding affinity of 4-methylhistamine shows the same trend as histamine for the various species variants (Table 1). The H4R affinity of VUF 8430, a potent human H4R agonist (Lim et al., 2006), does not completely follow this trend. Like histamine, VUF 8430 shows high affinity for human (pKi, 7.5) and monkey (pKi, 7.3) H4Rs, but for the H4Rs of the other species evaluated, the pKi value of VUF 8430 is between 5.9 (dog) and 6.8 (rat) (Table 2, Fig. 2).

Displacement of [3H]histamine binding by H4R ligands VUF 8430 (A), clozapine (B), JNJ 7777120 (C), and thioperamide (D) at the human, monkey, pig, and dog H4Rs. The error bars indicate the S.E.M. of results of at least three independent experiments.
Affinity (pKi) of H4R ligands at the human, pig, and dog H4Rs and selected human H4R mutants
Equilibrium dissociation constants (KD) and Bmax values for [3H]histamine (pmol/mg protein) and pKi of H4R ligands are presented as average ± S.E.M. of results of at least three independent experiments. The full list of investigated mutants is presented in Supplemental Table 1.
It is noteworthy that the affinity of the nonimidazole H4R agonist clozapine spans almost 3 log units across the tested H4R orthologs, with the order of increasing affinity (pKi values): dog, 4.5; pig, 5.2; mouse, 5.5; rat, 5.6; human, 6.4; monkey, 7.3; and guinea pig, 7.3 (Table 1, Fig. 3). This large difference in affinity is also shown by other tested nonimidazole H4R ligands, such as JNJ 7777120 (Fig. 2, Table 1) and its benzimidazole analog VUF 6002. Compared with JNJ 7777120, VUF 6002 consistently shows a 10-fold lower affinity for binding to the various orthologs, except for the guinea pig H4R, which binds JNJ 7777120 and VUF 6002 with equal affinity (Table 1).

Alignment of the partial sequences (from residue Asp3.49 to Phe5.47 according to the Ballesteros-Weinstein numbering system) of various species variants of the H4R. The residues that differ between the human and pig H4Rs are shaded, and the human H4R residues that are mutated into the pig/dog or monkey H4R counterparts are printed in bold and indicated by Ballesteros-Weinstein number. The FF-motif in EL2 is in italics. The domains of HPH and PHP chimeric receptors were swapped at the indicated ClaI and EcoRI restriction sites in the cDNAs of the human and pig H4Rs.
The H4R agonist/H3R inverse agonist clobenpropit shows equipotent affinity for H4Rs of human, monkey, mouse, and rat (pKi values of 7.5, 7.5, 7.3, and 7.3, respectively), lower affinity for H4Rs of pig and dog (pKi values of 6.6 and 6.4, respectively), and an higher affinity for the guinea pig H4R (pKi value of 8.2) (Table 1). The Ki values for the human, mouse, and rat H4Rs (stably expressed in HEK 293T cells) in this study are higher than those reported previously using H4Rs expressed in SK-N-MC cells (Liu et al., 2002b; Lim et al., 2005). Finally, we observed that the H4R inverse agonist thioperamide binds equipotently to all of the H4R orthologs (pKi values between 7.0 and 7.6), except for the dog H4R, which binds thioperamide with a lower pKi of 6.4 (Fig. 2, Table 1).
Residues involved in ligand binding affinity differences between human, rat, and mouse H4R have already been analyzed in earlier studies (Liu et al., 2001b; Lim et al., 2008). We therefore focused on the identification of key residues responsible for ligand binding affinity differences between human, pig, dog, and monkey H4R orthologs.
Chimeric Human-Pig H4R Approach.
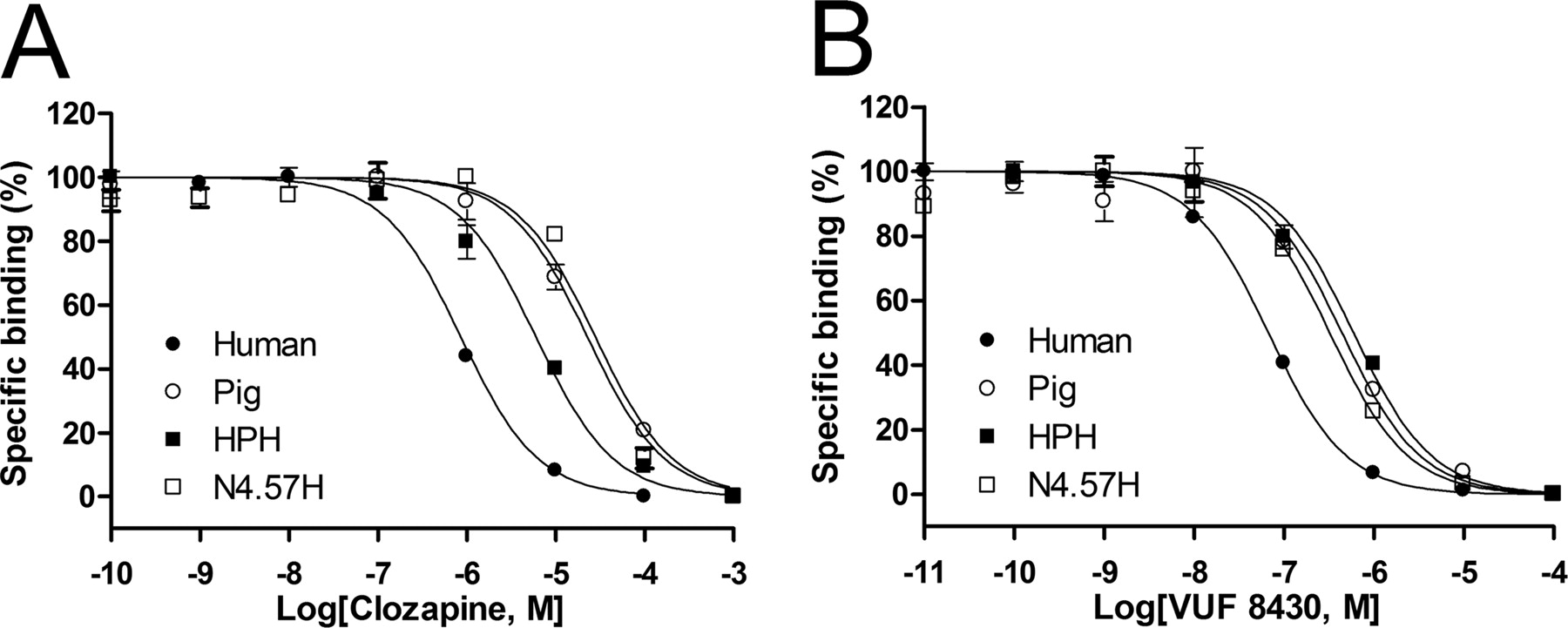
The human and pig H4Rs show equipotent affinity for histamine but different affinity for clozapine, JNJ 7777120, and VUF 8430 (Table 1). These four ligands were therefore used in further studies to probe human-pig H4R chimeras and pig H4R-mimicking site-directed mutants of human H4R. On the basis of our previous study on the pharmacological differences of the human and mouse H4Rs, we employed a chimeric receptor approach to investigate the differences in binding profiles between the pig and human H4Rs (Fig. 3). The chimeric HPH (after human-pig-human) receptor expressed in HEK 293T cells (Bmax = 2.9 pmol/mg protein) exhibits a KD value of 18 nM for [3H]histamine (Table 2). Although the affinity of histamine for the HPH chimeric receptor is conserved, HPH shows significantly lower affinity for clozapine, JNJ 7777120, and VUF 8430, with pKi values of 4.7, 6.1, and 6.3, respectively (Table 2, Fig. 4) compared with the human H4R. Expression of PHP (after pig-human-pig) chimeric receptor is significantly lower yet measurable (Bmax = 0.3 pmol/mg protein). The affinity of clozapine, JNJ 7777120, and VUF 8430 (pKi values of 6.8, 7.8, and 7.4, respectively) for the PHP chimera is significantly increased compared with pig H4R, mimicking the affinity profile of human H4R (Table 2, Fig. 4). These data clearly show that the middle H4R domain (Table 2) is playing a crucial role in the binding of clozapine, JNJ 7777120, and VUF 8430.

Displacement of [3H]histamine binding by clozapine (A) and VUF 8430 (B) at the human H4R, pig H4R, chimeric HPH H4R, and human H4R N4.57H mutant expressed in HEK 293T cells. The error bars indicate the S.E.M. of results of at least three independent experiments.
Site-Directed Mutagenesis of the Human H4R.
Following the results of the chimeric approach, we decided to continue with an SDM approach to further pinpoint the amino acid residues involved in the binding of clozapine, JNJ 7777120, and VUF 8430. The human and pig H4R species variants show a total of 16 divergent amino acid residues in the middle domain of the HPH and PHP chimeras (Fig. 3, shaded areas). Four divergent amino acids are located in the second intracellular loop or cytoplasmic half of TM4 (Fig. 3) and were omitted for further analysis, because this domain is not likely to be involved in ligand binding to bioaminergic GPCRs (Shi and Javitch, 2002). We also excluded from our analysis the highly divergent stretch of 4, 6, or 10 amino acid residues in the second extracellular loop (DEGSE in the human H4R and QGKQD in the pig H4R; Fig. 3), because our previous study did not implicate this region in ligand binding to human or mouse H4Rs (Lim et al., 2008). All other amino acid residues that differ between human and pig H4Rs were investigated for their involvement in ligand binding by constructing the human H4R mutants N4.57H, M4.60V, S45.42156A, F45.55169L, F45.55169L/S45.56170K, I5.38V, S5.43L, and L5.45F. After expression in HEK 293T cells, all mutant receptors still bound [3H]histamine with nanomolar affinity (6–51 nM; Supplemental Table 1) and were well expressed, except for the human H4R (hH4R) mutant S45.42156A, which showed high affinity for H4R but had very low expression (Supplemental Table 1). The M4.60V, S45.42156A, I5.38V, S5.43L, and L5.45F mutants of hH4R did not show the change in pharmacology observed in the HPH chimeric H4R (Supplemental Table 1). Only the S5.43L mutation resulted in a slight, but significant, 3-fold loss of the affinity of JNJ 7777120, but this mutation did not affect the affinities of histamine, clozapine, or VUF 8430 (Supplemental Table 1). In line with our previous work showing the importance of the FF motif in EL2 for the binding of clozapine (Lim et al., 2008), both the F16945.55L and the double mutant F16945.55L/S17045.56K show reduced affinity for clozapine (Supplemental Table 1). However, the binding of none of the other ligands was altered upon the F16945.55L and F16945.55L/S17045.56K mutations (Supplemental Table 1).
The role of position 45.55 in ligand binding to H4R has already been described in an earlier study (Lim et al., 2008). Table 2 presents three newly identified residues found to be responsible for ligand binding affinity differences between human, pig, dog, and monkey H4R orthologs (Asn/His4.56, Ser/Leu5.43, and Leu/Val5.39). The full list of investigated mutants is presented in Supplemental Table 1. The human, monkey, guinea pig, pig, rat, and mouse H4Rs contain an asparagine residue at position 4.57, whereas the pig and dog H4Rs possess a histidine residue at this position (Fig. 4). Although pig and human H4Rs bind [3H]histamine with high affinity (KD values of 9 and 11 nM, respectively; see Table 2), dog H4R and the N4.57H hH4R mutant bind [3H]histamine with low affinity (KD values of 75 and 51 nM, respectively). The N4.57H hH4R mutant mimics the pig and dog H4R and binds clozapine, VUF 8430, and JNJ 7777120 with significantly lower affinity than wild-type hH4R (Table 2, Fig. 4). The affinities of the agonists clozapine and VUF 8430 for this mutant are similar to those observed for the HPH chimeric H4R protein, but the affinity of the H4R antagonist JNJ 7777120 is only partially reduced by the N4.57H mutation (Table 2, Fig. 4). Apparently, other residues within the swapped region of the HPH chimeric receptor contribute to the difference in pharmacology between human and pig H4Rs as well. It is noteworthy that pig differs from human and dog H4R at position 5.43 (a serine instead of a leucine residue; Fig. 3), and we hypothesized that this residue might compensate for the negative effect of the N4.57H mutation on histamine binding while further decreasing binding affinity for JNJ 7777120. We therefore constructed the N4.57H/S5.42L hH4R double mutant, increasing the resemblance with the binding pocket of the pig H4R (Fig. 3). It is noteworthy that the double hH4R mutant N4.57H/S5.43L showed the predicted increase in affinity for [3H]histamine (Table 2). The double mutant does not show full conversion to the pharmacological profile of pig H4R, because the affinity for JNJ 7777120 is only slightly further decreased in the double mutant compared with the N4.57H and/or S5.43L single mutants (Table 2).
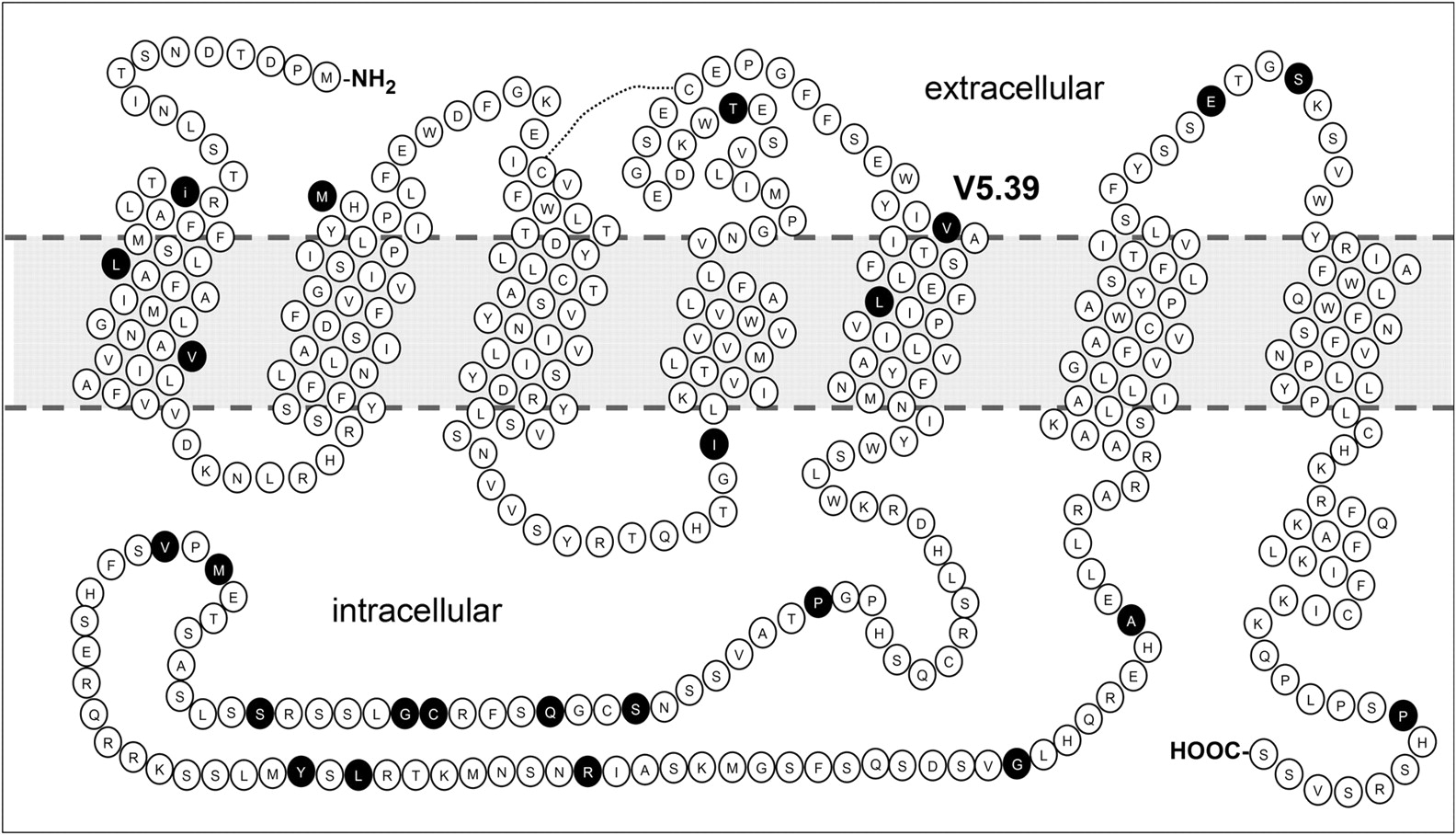
The monkey and human H4Rs have a high sequence homology (93%; Fig. 1), but they show significantly different affinity for JNJ 7777120 and clozapine (Table 1). Compared with the human H4R, the monkey H4R shows a 10-fold higher affinity for clozapine (pKi, 7.3 versus 6.4), but shows an almost 10-fold lower affinity for JNJ 7777120 (pKi, 7.5 versus 8.3). We exploited the small differences in protein sequence between the monkey and human H4Rs to study ligand-H4R interactions. In the extracellular domains, two residues within EL3 and one within EL2 differ (Fig. 5). Our previous study on the difference between human and mouse H4Rs indicated that these residues are not involved in ligand binding (Lim et al., 2008), and thus they were not included in our SDM approach. Within the transmembrane domains, only six amino acids differ between the human and the monkey receptor protein (Fig. 5). Four divergent amino acids are located in TM1 and TM2, which are usually not part of the main ligand binding pocket of bioaminergic GPCRs (Shi and Javitch, 2002). Two other residues are located in TM5; the human H4R has a Val residue at position 5.48, whereas a Leu residue is present in the monkey H4R and the other species orthologs, including the mouse and rat H4Rs (Jiang et al., 2008). Because JNJ 7777120 shows equipotent affinity for the human, mouse, and rat H4Rs, we argued that the difference of residue 5.48 is unlikely to be responsible for the difference in JNJ 7777120 affinity between monkey and human H4Rs. Residue 5.39 is valine in the monkey or leucine in the human, mouse, and rat H4Rs. We therefore selected this residue as the prime cause for the difference in affinity of JNJ 7777120 between the human and monkey H4Rs. The human H4R mutant L5.39V was constructed and expressed in HEK 293T cells. Compared with the wild-type human H4R, the H4R L5.39V mutant, like the monkey H4R, shows a low affinity for JNJ 7777120 (Table 2, Fig. 6). Moreover, clozapine shows an increase in affinity at the H4R L5.39V mutant and binds the mutant H4R similarly to the monkey H4R (Table 2, Fig. 6).

Snake plot of the monkey H4R. The residues indicated in black differ between the monkey and human H4R.
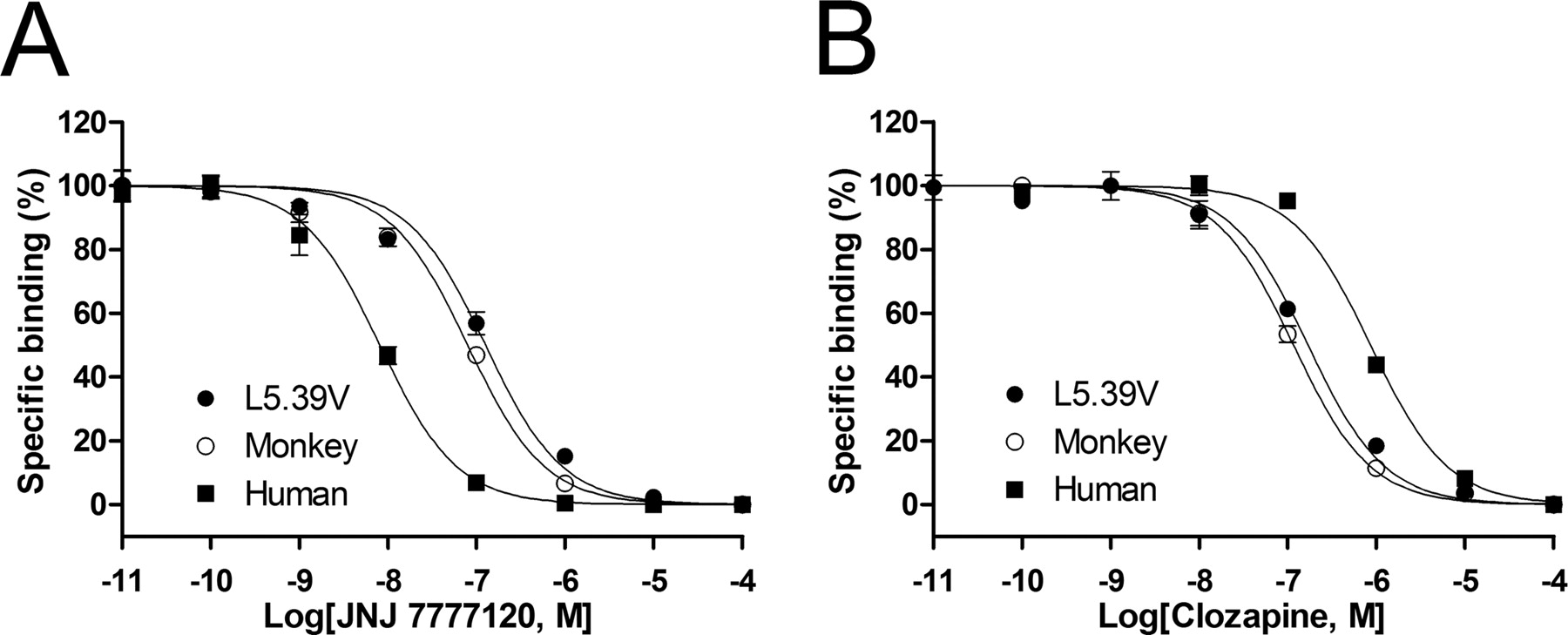
Displacement of [3H]histamine binding by JNJ 7777120 (A) and clozapine (B) at the human H4R, monkey H4R, and human H4R L5.39V mutant expressed in HEK 293T cells. The error bars indicate the S.E.M. of results of at least three independent experiments.
Discussion
Significant Differences in Ligand Binding Affinity between H4R Orthologs.
To understand the action of newly developed H4R ligands in translational preclinical studies in animal models of disease, we comprehensively characterized the binding of selected H4R ligands on heterologously expressed H4R species variants in HEK 293T cells, including those of human, monkey, pig, dog, mouse, rat, and guinea pig. Important results from our studies are the identification of substantial compound-specific pharmacology across the various H4R proteins, suggesting potential problems in the interpretation of in vivo results in animal models. The H4R proteins of human, monkey, pig, and guinea pig bind histamine with high affinity, whereas those of dog, mouse, and rat interact with the agonist with lower affinity (Table 1 and Supplemental Figure II). It is noteworthy that the high histamine affinity for the H4Rs expressed in HEK 293T cells are not affected by G-protein uncoupling reagents, such as GTP or GTPγS (Supplemental Figure III). Indeed, the H4R alone shows a high affinity for the agonist histamine in Sf9 cells, which is lacking G-proteins that are able to couple to H4R (Schneider et al., 2009). Moreover, neither coexpression nor fusion of Gαi2 changes the histamine affinity for H4R, which suggests the existence of a G-protein-independent high-affinity receptor state of H4R (Schneider et al., 2009).
4-Methylhistamine shows the same trend as histamine in binding affinity for H4R orthologs (Table 1) (Lim et al., 2005), albeit with slightly lower affinity. The recently discovered H4R agonist VUF 8430 (Lim et al., 2006), does not follow the trend of histamine affinity for the orthologs. The tricyclic H4R agonist clozapine shows large difference in affinity for the H4R orthologs. Compared with the affinity for the human H4R, a significant drop in affinity is observed for the pig, dog, mouse, and rat H4Rs (Table 1). Monkey and guinea pig H4Rs, on the contrary, show higher affinity for clozapine. It is noteworthy that JNJ 7777120 and VUF 6002, two H4R antagonists that have been used in several in vivo H4R studies, show significantly lower affinity for monkey, pig, dog, and guinea pig H4Rs (Table 1). The interspecies differences in ligand affinity have limited their use, and these H4R antagonists have to be used with caution for experiments in the indicated animals. In contrast, thioperamide shows an equipotent affinity for the species variants and consistently acts as an antagonist or inverse agonist at these species variants (H. D. Lim, unpublished observations). Therefore, despite its cross reactivity at the H3R, thioperamide might be used as an H4R antagonist in the species variants that show low affinity for JNJ 7777120.
Using chimeric human-pig-human and pig-human-pig H4R proteins and SDM studies, we have systematically identified residues at positions 45.55 (in EL2), 4.57, 5.39, and 5.43 as residues responsible for the observed species differences (Table 2 and Supplemental Table 1). Because the role of position 45.55 in ligand binding to H4R has already been described in an earlier study (Lim et al., 2008), we will discuss the role of the other three residues in more detail in the following paragraphs.
His4.57 Negatively Affects Ligand Binding in Pig and Dog H4Rs.
SDM studies have identified the residue at position 4.57 (Asn in human, mouse, rat, monkey, and guinea pig, His in dog and pig) as an amino acid responsible for differences in ligand affinity for H4R orthologs (Table 2). Although earlier SDM studies already showed the importance of Asn4.57 in histamine-induced H4R activation (Shin et al., 2002), current studies show that clozapine affinity is largely decreased in the human H4R N4.57H mutant (Table 2). The effect of this mutation on histamine, JNJ 777120, and VUF 8430 affinity is less dramatic but still significant (Table 2). It is noteworthy that the high affinity for histamine is recovered in the pig mimicking N4.57H/S5.43L double mutant. The subtle roles of N4.57H and S5.43L in histamine binding are rationalized by our H4R modeling studies as demonstrated in Fig. 7, A and B. The protonated amine group of histamine forms a complementary H-bond network with Asp3.32, Cys3.36, and Gln7.42, whereas the histamine imidazole group stacks between Tyr3.33 and Tyr6.51 and donates an H-bond to Glu5.46 (Fig. 7A). This binding mode is in line with earlier SDM studies indicating the essential role of Asp3.32 and Glu5.46 in histamine binding in H4R (Shin et al., 2002; Jongejan et al., 2008), and the experimentally supported role of the homologous Ser3.36 residue in H1R (Jongejan et al., 2005). In wild-type H4R, Asn4.57 donates H-bonds to the backbone carbonyl atom of Ala4.53 and the hydroxyl group of Thr3.37. An alternative H-bond network is formed in the pig and dog mimicking N4.57H mutant in which the histidine residue is able to donate an H-bond to the carboxylate group Glu5.46, which in its turn also accepts an H-bond from Thr3.37. This H-bond network reorients Glu5.46 more toward TM4. To maintain the essential H-bond with Glu5.46 (Jongejan et al., 2008), histamine has to reorient its imidazole ring deeper into the binding pocket. This alternative binding pose is stabilized by the leucine side chain in the S5.43L/N4.57H mutant (Fig. 7B), explaining the increased binding affinity for histamine in this pig-mimicking double mutant over the dog-mimicking N4.57H single mutant (Table 2). The full agonist clozapine forms an H-bond between its positively charged piperidine nitrogen atom and the carboxylate group of Asp3.32 in the pocket between TMs 2, 3, and 7 (subpocket i) and forces Cys3.36 into its g- conformation [which has been associated with the activated state of H1R (Jongejan et al., 2005)] by placing its chlorinated aromatic ring in the hydrophobic binding pocket between TMs 3, 4, 5, and 6 (subpocket ii) (Figs. 7C and 8A). The nonchlorinated aromatic ring stacks between Tyr3.33 and Tyr6.51, whereas the nitrogen atom in the tricyclic ring system donates an H-bond to Glu5.46 (Fig. 8A). This binding mode is in line with the selectivity profile of clozapine and olanzapine for bioaminergic receptor subtypes (Selent et al., 2008) and structure-activity relationship studies of clozapine analogs indicating the steric restriction around the tricyclic nitrogen atom and the importance of 7- and 8-substitution over 2- and 3-substitution (Smits et al., 2006). The reorientation of Glu5.46 in the pig and dog mimicking N4.57H mutant toward TM4 disrupts the H-bond with the NH group of clozapine because its rigid cyclic ring is tightly bound in subpocket ii (Fig. 7C), explaining the large decrease in clozapine binding affinity at the N4.57H mutant as well as at pig and dog H4Rs.

Binding modes of histamine (magenta carbon atoms, see Table 2 for two-dimensional representation) in the wild-type human H4R receptor model (A), histamine in the N4.57H/S5.43L double mutant (B), and clozapine in the N4.57H single mutant (C). The backbone of TM helices 4, 5, 6, and 7 are represented by yellow ribbons, and part of TM3 is shown as ribbon (the top of the helix is not shown for clarity). Important binding residues are depicted as ball-and-stick models with gray carbon atoms. The carbon atoms of mutated residues are colored green. Oxygen, nitrogen sulfur, chlorine, and hydrogen atoms are colored red, blue, orange, brown, and cyan, respectively. H-bonds described in the text are depicted by black dotted lines.

Binding modes of clozapine (magenta carbon atoms; see Table 2 for two-dimensional representation) (A) and JNJ 7777120 in wild-type/L5.39V mutant human H4R receptor models (B). Rendering and color coding is the same as in Fig. 7. The side-chain atoms of Leu5.39 and Val5.39 are depicted by gray and green ball and sticks models, whereas the CG2 methyl group of Val5.39 is additionally shown as semitransparent green van der Waals spheres. Numbers of different positions on the aromatic rings of clozapine and JNJ 7777120 are discussed in the text. The C3 methyl group of clozapine and the 5-chlorine atom of JNJ 7777120 are depicted by semitransparent magenta and brown van der Waals spheres, respectively.
Residue 5.39 Distinguishes the Human and Monkey H4Rs.
Despite the high homology between these two orthologs (93%), significant differences exist for the binding of clozapine and JNJ 7777120. Our SDM studies identified residue 5.39 as the cause of the pharmacological differences between the human and monkey H4R. In H1R, Lys5.39 is known to interact with histamine and zwitterionic H1R antagonists (Leurs et al., 1995; Gillard et al., 2002). In human H4R, the full agonist clozapine is positively affected by the L5.39V mutation. The valine residue in the monkey H4R mimicking L5.39V mutant is sterically limited because of a steric clash with the helical backbone (Lovell et al., 2000) and forms a complementary cap with its CG2 methyl group on top of the nonchlorinated aromatic ring of clozapine (Fig. 8A). The leucine residue in wild-type human H4R on the other hand, is more flexible and can orient its isobutyl side chain in a trans conformation (Lovell et al., 2000) pointing outwards of the binding pocket, explaining the gain of affinity in the L5.39V mutant. The antagonist JNJ 7777120 forms an ionic/H-bond between its positively charged piperazine nitrogen atom and Asp3.32 but stabilizes Cys3.36 in its inactive (Jongejan et al., 2005) trans conformation by accepting an H-bond with its piperazine carbonyl oxygen and donating an H-bond from its indole nitrogen to the carboxylate group of Glu5.46. The chlorinated aromatic ring is stacked between Tyr3.33 and Tyr6.51 and occupies a pocket between TMs 3, 5, 6, and EL2 (Fig. 8B). Previous SDM studies have shown the importance of Asp3.32 as well as Glu5.46 as critical ionic interaction point and H-bond acceptor, respectively (Jongejan et al., 2008) and are in line with the proposed JNJ 7777120 binding mode. Structure-activity relationships of JNJ 7777120 analogs, indicating the importance of the H-bond donor functionality of the indole nitrogen, the preference for 4- and 5-substitution over 6- and 7-substitution, and the toleration of polar groups at the 5-position of the aromatic ring (Jablonowski et al., 2003), support this binding pose instead of an orientation in which the chlorinated aromatic ring binds into the highly hydrophobic pocket close to Trp6.48 between TMs 3, 4, 5, and 6. The negative effect of the L5.39V mutation further supports the proposed binding mode. In the mutant, the CG2 methyl group of the sterically restricted valine residue bumps into the chlorine atom of JNJ 7777120, whereas the more flexible leucine residue in the wild-type can avoid this clash (Fig. 8B).
conclusions
In conclusion, we have pharmacologically characterized seven H4R species orthologs that are relevant in drug discovery (i.e., human, monkey, pig, dog, mouse, rat, and guinea pig H4R). We have described profound differences in the binding of an extensive set of reference H4R ligands, providing important information for a good understanding of the action of these ligands in animal models of disease. The current work demonstrates the usefulness of the differences in amino acid sequence between the various species variants (natural mutagenesis) to study H4R-ligand interactions. Domain swapping of the protein sequence of the human and pig H4Rs enabled us to identify the middle domain of the H4R as the cause of the observed species difference. SDM studies identified the residue at position 4.57 as an important determinant for the species difference in ligand affinity, whereas the difference between human and monkey H4Rs in ligand binding can be explained by a single mutation of position 5.39. Structural models of wild-type and mutant human H4R were used to explain the role of these critical residues in ligand binding. These results altogether improve our understanding of H4R-ligand interactions and provide valuable information for the construction and refinement of structural models of H4R-ligand complexes, which can eventually be used for structure-based H4R virtual screening and ligand design.
Acknowledgments
Saskia Nijmeijer is acknowledged for technical assistance with the functional assays.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the Top Institute Pharma [Project Number D1.105: the GPCR Forum] and European Cooperation in Science and Technology (COST) [Action BM0806].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.063040.
-
ABBREVIATIONS:
- H4R
- histamine H4 receptor
- H1R
- histamine H1 receptor
- H3R
- histamine H3 receptor
- GPCR
- G protein-coupled receptor
- HEK
- human embryonic kidney
- VUF 8430
- guanidinylethyl isothiourea
- JNJ 7777120
- 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methylpiperazine
- VUF 6002
- 1-[(5-chloro-1H-benzimidazol-2-yl)carbonyl]-4-methylpiperazine
- SDM
- site-directed mutagenesis
- DMEM
- Dulbecco's modified Eagle medium
- PEI
- polyethylenimine
- PCR
- polymerase chain reaction
- HPH
- human-pig-human
- TM
- transmembrane
- EL2
- second extracellular loop.
- Received December 9, 2009.
- Accepted January 26, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}