


Abstract
Competition experiments with [3H]mepyramine showed that cetirizine and its enantiomers, levocetirizine and (S)-cetirizine, bound with high affinity and stereoselectivity to human H1 histamine receptors (K i values of 6, 3, and 100 nM, respectively). Cetirizine and levocetirizine were 600-fold more selective for H1 receptors compared with a panel of receptors and channels. Binding results indicated that the interaction between cetirizine, its enantiomers, and histamine is compatible with a competitive behavior, in contrast with the noncompetitive profile of cetirizine and levocetirizine observed in isolated organs. Binding kinetics provided a suitable explanation for this observation, because levocetirizine dissociated from H1 receptors with a half-time of 142 min; that of (S)-cetirizine was only 6 min, implying that the former could act as a pseudo-irreversible antagonist in functional studies. The carboxylic function of levocetirizine seemed responsible for its long dissociation time. Indeed, hydroxyl or methyl ester analogs dissociated more rapidly from H1 receptors, with half-times of 31 min and 7 min, respectively. The importance of the carboxylic function of levocetirizine for the interaction with the H1 receptor was further supported by the results from the mutation of Lys191 to Ala191. This mutation decreased the dissociation half-time of levocetirizine from 142 to 13 min and reduced its affinity from 3 to 12 nM, whereas the affinity and dissociation kinetics of hydroxyl and methyl ester analogs were hardly affected. The mutation of Thr194 reduced the binding stereoselectivity by selectively enhancing the affinity of the distomer.
The bioamine histamine produces a variety of physiological and pathophysiological effects through binding and activation of histamine receptors belonging to the superfamily of seven transmembrane G-protein-coupled receptors (Hill et al., 1997). Today, four human histamine receptor subtypes have been cloned: H1(Moguilevsky et al., 1994), H2 (Gantz et al., 1991), H3 (Lovenberg et al., 1999), and, more recently, H4 (Oda et al., 2000). H1 receptors induce smooth muscle contraction and increase vascular permeability and H1 antagonists constitute a medication of choice to alleviate the symptoms of allergies.
Cetirizine and levocetirizine are second-generation antihistamines. As opposed to first generation drugs, exemplified by hydroxyzine, chlorpheniramine, diphenhydramine, or ketotifen, second-generation drugs are nonsedating or less sedating, probably because of an improved H1 binding selectivity and reduced brain penetration (Timmerman, 1999). Structure-activity relationships and site-directed mutagenesis experiments performed with the guinea pig H1 receptor have provided data that have led to model pharmacophores of H1antagonists (ter Laak et al., 1995; Wieland et al., 1999). Since the cloning of the H1 receptor, several studies have been published on mutant receptors designed to better identify the binding pocket and the amino acids residues involved in the binding of histamine and histamine antagonists (Fig.1). This is of particular interest today in light of recent findings showing that most histamine H1 antagonists exhibit inverse agonist properties (Bakker et al., 2000). Asp107, located in the third transmembrane domain of the human receptor, is crucial for the affinity of histamine and histamine antagonists (Ohta et al., 1994); this amino acid is a hallmark of G-protein-coupled receptors, whose natural ligands are bioamines, and is responsible for forming an ionic bond with the protonated nitrogen of the neurotransmitter (Hibert et al., 1991). Asn198 (Leurs et al., 1994; Ohta et al., 1994; Moguilevsky et al., 1995) and Lys200(Leurs et al., 1995) are also involved in histamine binding to the H1 receptor in human and guinea pig, respectively. By analogy with the histamine H2receptor, Thr194 was expected to participate in the binding of histamine to human H1 receptors, but the mutation of this residue into Ala led to a receptor that kept its ability to bind histamine and histamine antagonists with affinities very similar to that of the wild-type receptor (Leurs et al., 1994;Ohta et al., 1994). However, we have shown that the mutation of Thr194 to Ala decreased the stereoselectivity of the enantiomers of cetirizine by increasing the affinity of the distomer (Moguilevsky et al., 1995). Finally, Lys200 , the guinea pig equivalent of human Lys191, was reported to interact with the carboxylic acid moiety of two second-generation antagonists, acrivastine and cetirizine (Wieland et al., 1999). In this report, we further explore the binding characteristics of cetirizine and its enantiomers to the wild-type human H1receptor in comparison with receptors bearing mutations at key amino acids (Lys191 and Thr194) known to be involved in ligand binding. Close structural analogs of cetirizine were also included in this study to explore the role of the carboxyl group in binding to the H1 receptor, under both equilibrium and nonequilibrium conditions.

Chemical structures of H1 antagonists.
Materials and Methods
Chemicals.
Cetirizine (Zyrtec; UCB Group, Brussels, Belgium), hydroxyzine, and their respective enantiomers [levocetirizine (Xyzal; UCB Group), (S)-cetirizine, (S)-hydroxyzine, and (R)-hydroxyzine (all as dihydrochloride salts)], (R)-ucb 29992, and (S)-ucb 29993 (as dimaleates) were synthesized at UCB SA Pharma Sector (Braine l'Alleud, Belgium). Fexofenadine was purchased from Ultrafine Chemicals (Manchester, UK). Histamine, (+)-chlorpheniramine, terfenadine, atropine, 2-chloroadenosine, chlorpromazine, ranitidine, pirenzepine, pargyline, and adenosine deaminase (EC 3.5.4.4. from bovine spleen) were from Sigma-Aldrich (Bornem, Belgium). WB-4101, (±)-isoproterenol, thioperamide, ritanserin, ketanserin, buspirone, (±)-propranolol, phentolamine, RX821002, Rα-methylhistamine, and butaclamol were purchased from Sigma/RBI (Natick, MA). Serotonin was purchased from Fluka (Bornem, Belgium). Pyridinyl-5-[3H]pyrilamine (27 Ci/mmol),l-N-methyl-[3H]scopolamine methyl chloride (83 Ci/mmol), [3H]RX821002 (59 Ci/mmol), [3H]SCH23390 (80 Ci/mmol) and wheat germ agglutinin-coated polyvinyltoluene SPA beads were purchased from Amersham Biosciences (Rosendaal, the Netherlands).N-α-[methyl-3H]methylhistamine (79 Ci/mmol), [propyl-2,3-ring-1,2,3-3H] 8-hydroxy-2-dipropylaminotetralin (154 Ci/mmol), [5,7-3H](−)CGP-12177 (45 Ci/mmol), [benzene ring-3H]spiperone (19 Ci/mmol), 8-[dipropyl-2,3-3H(N)]cyclopenthyl-1,3-dipropylxanthine (109 Ci/mmol), [ethylene-3H]ketanserin hydrochloride (77 Ci/mmol), [methyl-3H]tiotidine (84 Ci/mmol), and [7-methoxy-3H]prazosin (72 Ci/mmol) were purchased from DuPont de Nemours (Brussels, Belgium). α-Modified Eagle's minimal essential medium (α-MEM), Dulbecco's phosphate-buffered saline, penicillin, gentamicin, streptomycin, fetal calf serum, and l-glutamine were bought from BioWhittaker (Verviers, Belgium). All other reagents were of analytical grade and obtained from conventional commercial sources.
Cloning and Site-Directed Mutagenesis
Cloning and stable expression of human histamine H1 receptors in Chinese hamster ovary (CHO) cells were done in collaboration with Dr. A. Bollen (Department of Applied Genetics, Free University of Brussels, Brussels, Belgium) (Moguilevsky et al., 1994). To perform the mutagenesis, we used the human H1 receptor cDNA cloned into the plasmid pRc/RSV (pNIV 3604) as template for the synthesis of DNA with site-specific mutations using a polymerase chain reaction strategy. For the introduction of the mutations Thr194 to Ala and Lys191 to Ala in the TM5, a 1286-bpDraIII-XbaI fragment from pNIV 3604 was isolated before amplification of a 147-bp internal region flanked by the sitesXmnI and BglII. After sequencing to ensure that there were no Taq polymerase-induced mutations, theXmnI-BglII fragments carrying the mutations Thr194→Ala and Lys191→Ala were ligated with a 831-bpBglII-XbaI fragment, a 507-bpHindIII-XmnI and theHindIII-XbaI fragment of the eukaryotic vectors pRc/RSV and pRc/CMV, leading to the recombinant plasmids pNIV 3608 and pNIV 3626, respectively. CHO cells (American Type Culture Collection, Manassas, VA), grown in 5% CO2 at 37°C in α-MEM medium supplemented with 2 mM l-glutamine and 5% fetal calf serum, were transfected by electroporation using plasmids pNIV 3608 and pNIV 3626 (10 μg of DNA per 107 cells). Stably-transfected CHO cells were selected in medium containing Geneticin at 400 μg/ml. Resistant clones were isolated, subcloned, and expanded for subsequent [3H]mepyramine binding assays.
Cell Culture and Membrane Preparation
CHO cells were subcultured in α-MEM medium containing 2 mMl-glutamine, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 400 μg/ml Geneticin, and supplemented with 5% fetal calf serum. The cells were grown at 37°C in a humidified atmosphere of 5% CO2/95% air. Confluent cells were detached by a 10-min incubation in phosphate-buffered saline containing 1 mM EDTA. All subsequent operations were performed at 4°C. The cell suspension was centrifuged for 10 min at 500 g. The pellet was homogenized in 20 mM Tris-HCl, pH 7.4, 250 mM sucrose buffer, and frozen in liquid nitrogen. After thawing the homogenate was centrifuged at 30,000g for 15 min. The crude membrane pellet obtained was resuspended in the same buffer at a protein concentration of 6 to 8 mg/ml and stored in liquid nitrogen.
Equilibrium Binding Experiments
Equilibrium H1 binding assays were performed as described by Moguilevsky et al. (1994) and in Table1. For saturation binding isotherms, membranes (15 to 50 μg of proteins) from CHO cells expressing wild-type or mutant H1 receptors were incubated with increasing concentrations of [3H]mepyramine (from 0.2 to 20 nM). Binding experiments, at one drug concentration, were also carried out on a variety of other receptors or channels. Experimental conditions are listed in Table 1. Typically, after the incubation period, receptor-bound radioligand was separated from the free ligand by rapid vacuum filtration of the samples over GF/C glass fiber filters (Whatman, VEL, Belgium). Filters were presoaked in 0.1 to 0.3% polyethylenimine to reduce the nonspecific binding of the radioligand. Adsorbed samples were washed four times with 2 ml of ice-cold 50 mM Tris-HCl buffer, pH 7.4. The entire filtration procedure did not exceed 10 s/sample. Radioactivity trapped onto the filter was determined by liquid scintillation counting at 50 to 60% efficiency. For H1 SPA binding assay, 500 μg of wheat germ agglutinin-coated SPA beads were incubated with 50 μg of membranes in 200 μl of 50 mM Tris HCl buffer, pH 7.4, containing 2 mM MgCl2, 7.5 nM [3H]mepyramine and increasing concentration of drugs. The 96-well microplates were centrifuged (5 min at 1000g), sealed, and counted at various intervals of time in a scintillation counter.
Binding selectivity profile of cetirizine and its enantiomers
To determine whether the interactions between cetirizine or its enantiomers and histamine were competitive or allosteric, we used an experimental design based on the model described byLazareno and Birdsall (1995). Briefly, competition curves between histamine and [3H]mepyramine were carried out in the presence or absence of several concentrations of cetirizine or its enantiomers: each individual histamine binding curve was obtained in the presence of a single concentration of cetirizine or its enantiomers.
Kinetic Binding Experiments
Association.
Binding was initiated by the addition of membranes to the incubation buffer containing 3.5 nM [3H]mepyramine in the presence or absence of 10 μM cetirizine to define nonspecific binding. At increasing intervals of time thereafter, samples were filtered as described above.
Dissociation.
Membranes were added to the incubation buffer containing 3.5 nM [3H]mepyramine and binding was allowed to proceed for 60 min. At that time, radioligand dissociation was induced by the addition of cetirizine 10 μM. Sample aliquots were taken at increasing time intervals and filtered as explained above.
To determine the binding kinetics of unlabeled drugs to H1 receptors, we measured the association kinetics of [3H]mepyramine in the presence of a concentration of drug inhibiting by ±70% the specific binding of the radioligand at equilibrium.
Data Analysis
Data analysis was performed by computerized nonlinear curve fitting methods according to equations describing several binding models.
Competitive Interactions.
Analysis of equilibrium data according to competitive interactions between labeled and unlabeled ligands which obey to the law of mass action (Molinoff et al., 1981). IC50 values were corrected toK i values by applying the Cheng and Prusoff (1973) equation: IC50 =K i × [1 + ([L] /K D)], where IC50 is the concentration of unlabeled drug inhibiting by 50% the radioligand specific binding, [L] is the free radioligand concentration, and K D andK i are the equilibrium dissociation constants of the radioligand and unlabeled drug, respectively.
In the presence of a second unlabeled drug, the equation is extended to: IC50 = K i × [1 + ([L] / K D) + ([X] /K X)], where [X] andK X represent the concentration and equilibrium dissociation constant of the second unlabeled drug, respectively.
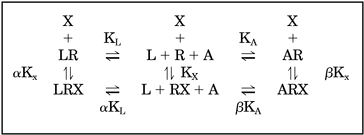
Allosteric Interactions.
Analysis of equilibrium data according to allosteric interactions of a molecule with labeled and unlabeled ligands (Lazareno and Birdsall, 1995),
Kinetic Constants.
Determination of the kinetic constants of unlabeled drugs according to a model described by Motulsky and Mahan (1984),
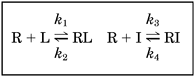
Statistics.
Partial F-tests were performed to compare two models (De Lean et al., 1982) and unpaired, two-tailed Studentt tests were used to compare pKi or kinetic constants.
Results
Equilibrium Binding Experiments.
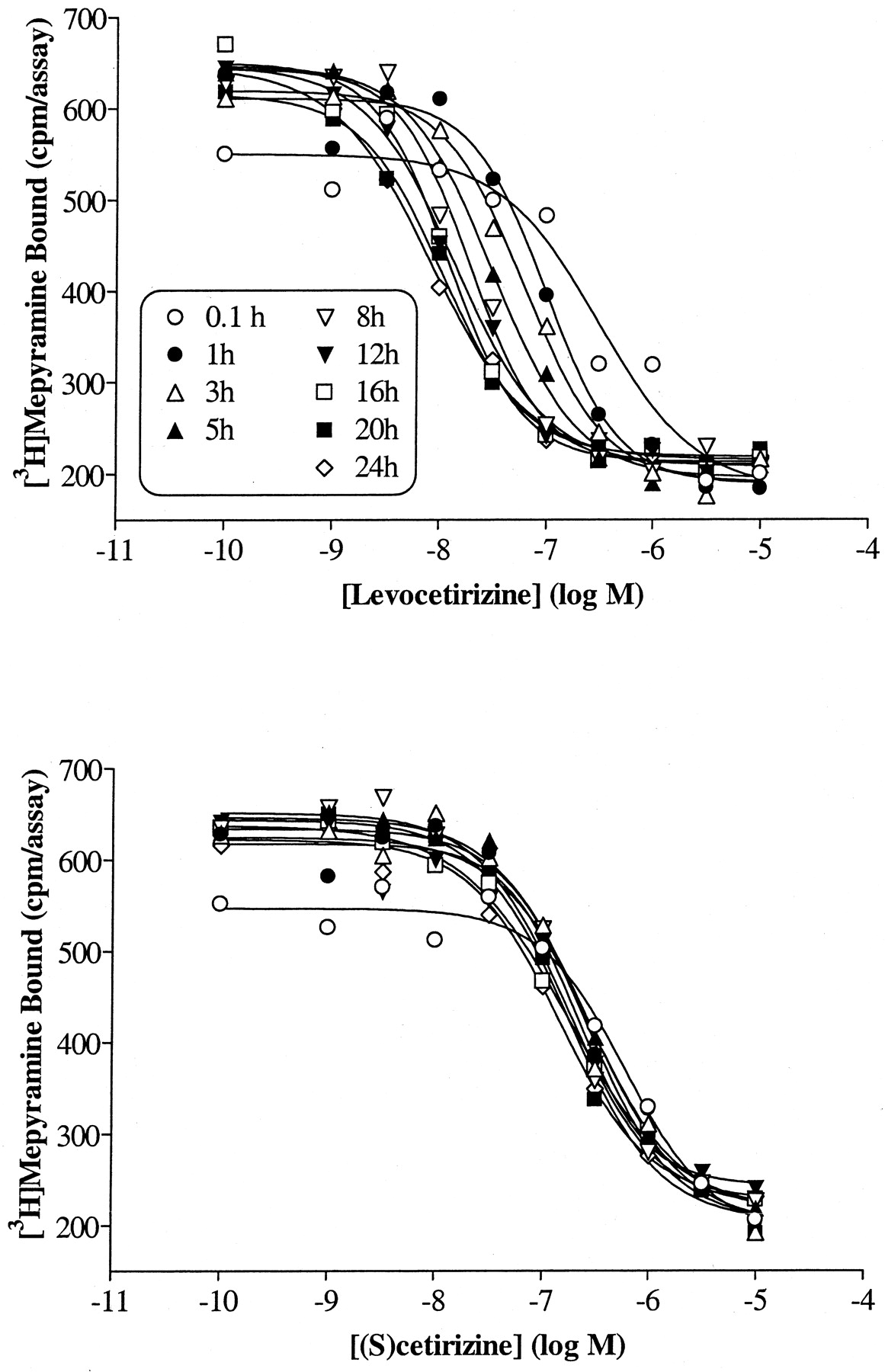
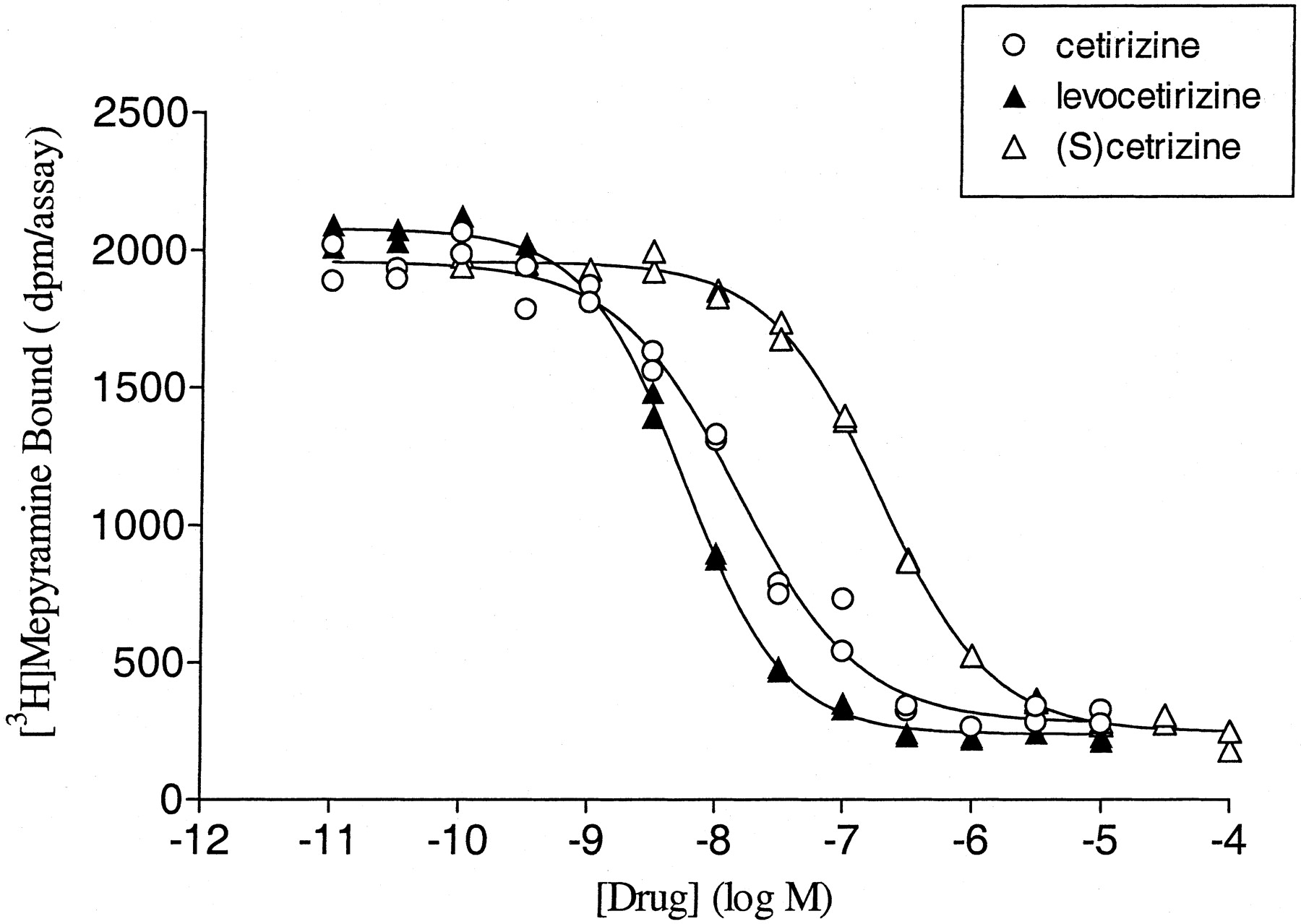
Preliminary experiments indicated that competition curves with cetirizine and levocetirizine shifted to the left (decreasing the IC50 value) with time. This is exemplified in Fig. 2, which depicts an SPA binding assay in which the IC50 values of levocetirizine are clearly decreasing with time whereas those of (S)-cetirizine are time-independent, suggesting different binding kinetics for the two enantiomers. Therefore, although the [3H]mepyramine equilibrates extremely rapidly, the incubation time was increased to 3 h to allow drugs with slow binding kinetics to reach steady state binding. As shown in Fig.3 and Table2, levocetirizine, the eutomer, has a 2-fold higher affinity than the racemic compound cetirizine (p < 0.01). The distomer, (S)-cetirizine, is about 30-fold less potent. The selectivity of cetirizine, levocetirizine, and (S)-cetirizine for H1 receptors, compared with a variety of GPCRs or channels that are known to bind first generation antihistamines, was evaluated (Table 1). No significant interactions were observed for any of the three compounds (tested at 10 μM), except for levocetirizine with the human α2C4 adrenergic receptor. The affinity of levocetirizine for these receptors (pK i = 5.8 ± 0.1;n = 2) was still 600 times less than its affinity for H1 receptors.

Effect of incubation time on the IC50 of slowly and rapidly equilibrating drugs. Competition binding curves for levocetirizine and (S)-cetirizine were obtained after varying incubation times using an SPA binding assay. Incubations were started by adding 500 μg of SPA beads precoated with H1receptors to samples in 96-well plates containing 7.5 nM [3H]mepyramine and increasing concentrations of unlabeled drugs. Results are representative of two experiments.

Affinity of cetirizine and its enantiomers for cloned human H1 histamine receptors. Membranes from CHO cells expressing cloned human H1 histamine receptors were incubated for 3 h at 37°C with increasing concentrations of compounds as described under Materials and Methods. The binding curves presented are representative of at least three experiments. Duplicates obtained for each concentration are visible on the graph.
Affinity of compounds for wild-type and mutant H1 receptors
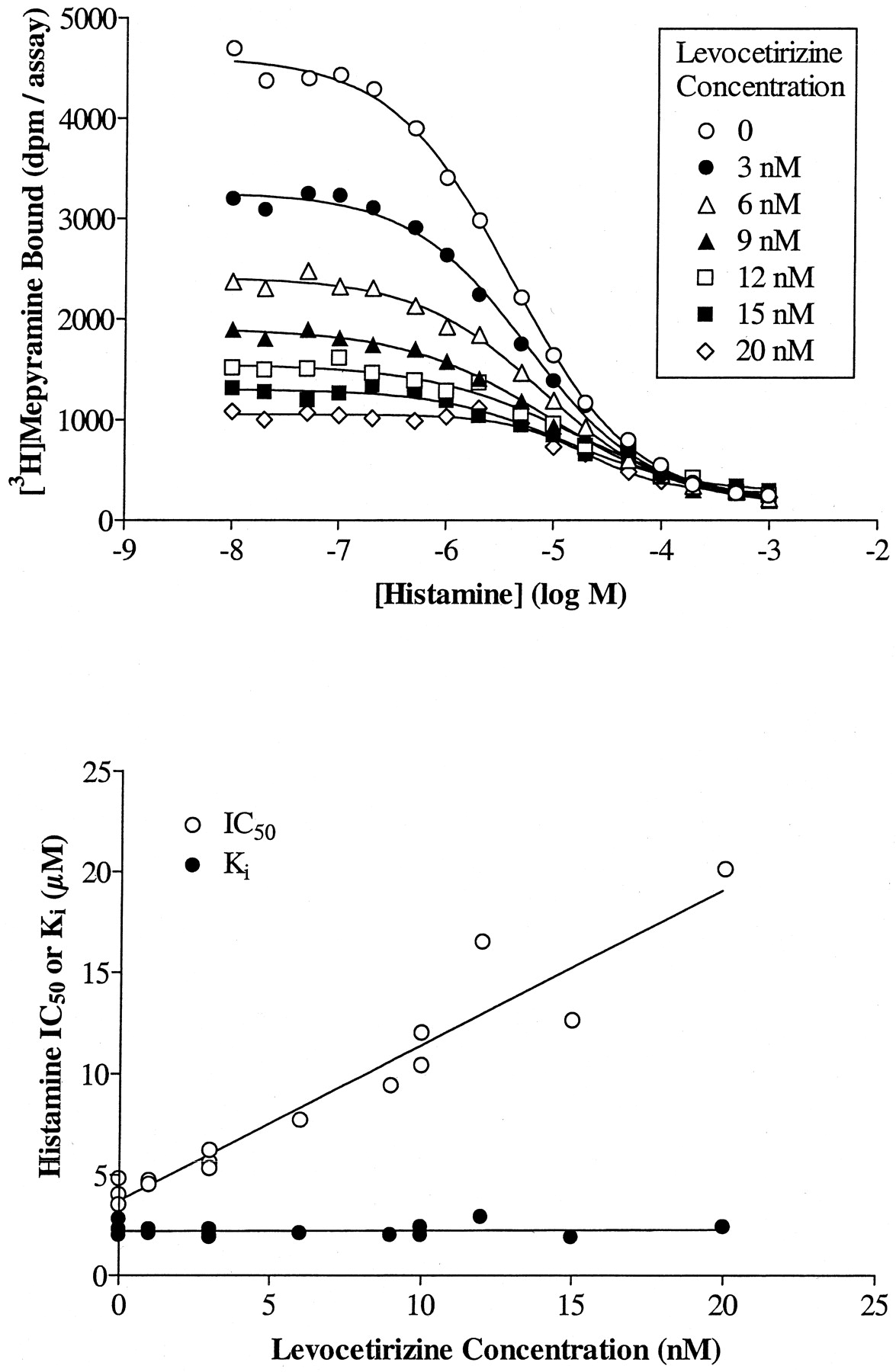
We further characterized the binding of cetirizine and its enantiomers to H1 receptors by verifying the competitive nature of their interactions, not only with respect to [3H]mepyramine but also to histamine. The IC50 of levocetirizine increased linearly with increasing concentrations of radioligand (Fig.4). TheK i values obtained by applying the Cheng and Prusoff equation were independent of the radioligand concentration and were identical to the value obtained by linear regression of the data (intercept of the ordinate) suggesting a competitive behavior of the compound. The results for cetirizine and (S)-cetirizine were similar (data not shown). Competition curves between [3H]mepyramine and histamine were performed in the presence or absence of given concentrations of cetirizine or its enantiomers. A representative set of curves is given for levocetirizine in Fig. 5, top. Histamine IC50 values increased linearly with increasing concentrations of levocetirizine, as would be expected for two compounds that interact competitively at a single binding site (Fig. 5, bottom). By analyzing the data with the allosteric/competitive ternary complex model (Lazareno and Birdsall, 1995), we found values for α (representing the interaction between cetirizine or its enantiomers with [3H]mepyramine) equal to 0 for all three compounds, whereas values for β (representing the interaction between cetirizine or its enantiomers with histamine) were equal to 0.07 (n = 1), 0.12 ± 0.06 (n = 3), and 0.07 (n = 1) for cetirizine, levocetirizine, and (S)-cetirizine, respectively, suggesting strong negative allosteric interactions, very close to a competitive behavior. Partial F-tests performed to compare the competitive model (with the allosteric constants set to 0) and the allosteric model indicated that the data were not better fitted with the allosteric model (p > 0.15 except for one of three experiments performed with levocetirizine where p < 0.05).

Cetirizine and its enantiomers interact competitively with [3H]mepyramine. The concept is exemplified in this figure for levocetirizine. Similar results were obtained for cetirizine and (S)-cetirizine. Top, competition binding curves between levocetirizine and [3H]mepyramine were obtained at increasing concentrations of radioligand. The curves presented are representative of two experiments performed in duplicate. Bottom, the IC50 values calculated by nonlinear regression analysis of all binding curves were plotted against the concentrations of radioligand. K i values were calculated by transforming the IC50 values according to the Cheng and Prusoff equation as described under Materials and Methods.
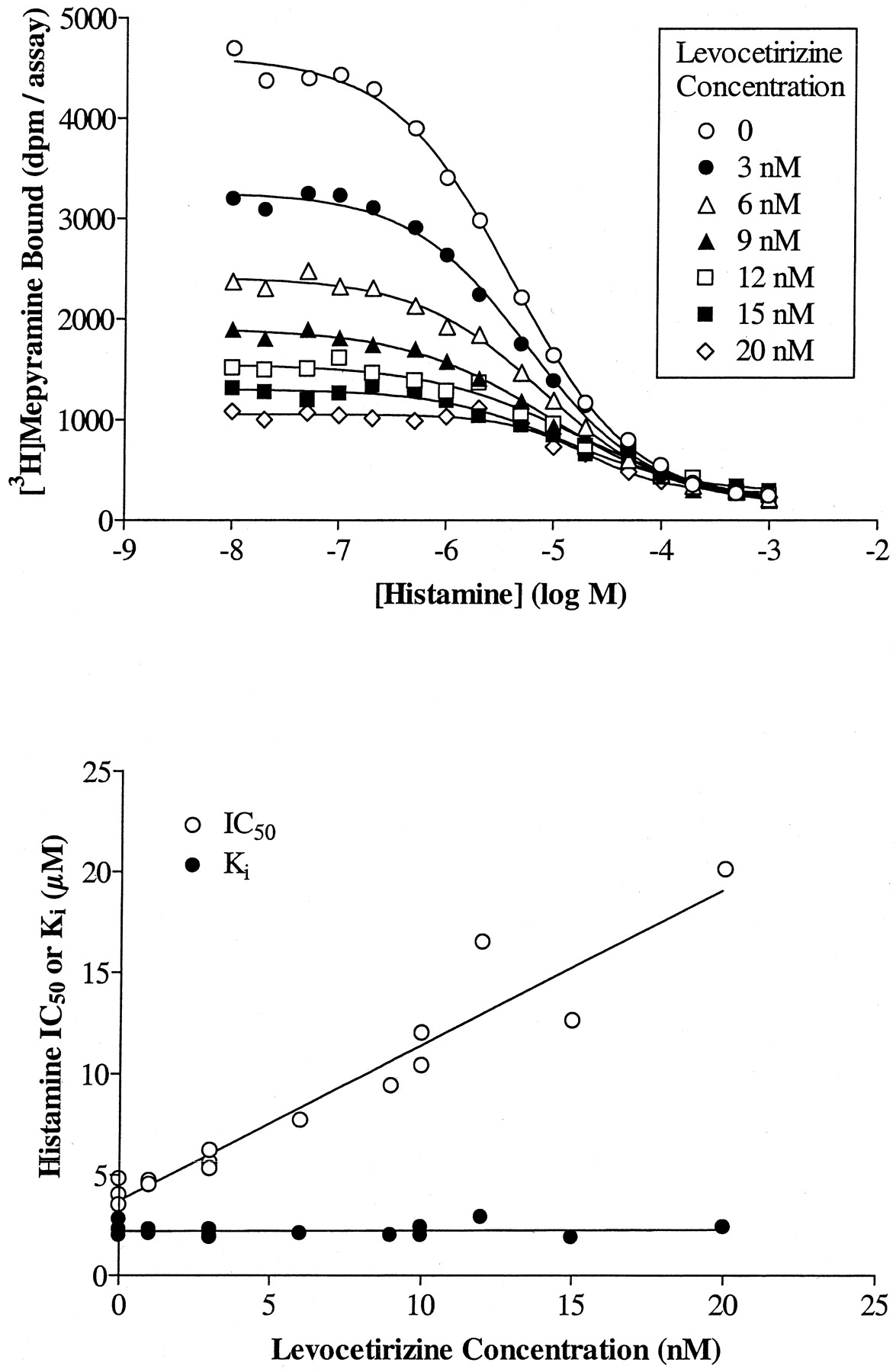
Cetirizine and its enantiomers interact competitively with histamine. The concept is exemplified in this figure for levocetirizine. Similar results were obtained for cetirizine and (S)-cetirizine. Top, competition binding curves between histamine and [3H]mepyramine in the presence of increasing concentrations of levocetirizine. The curves presented are representative of two experiments. Bottom, the IC50 values calculated for histamine by nonlinear regression analysis of the binding curves were plotted against the concentrations of levocetirizine. K i for histamine were calculated by transforming the IC50 values according to the modified Cheng and Prusoff equation as described under Materials and Methods.
Kinetic Binding Experiments.
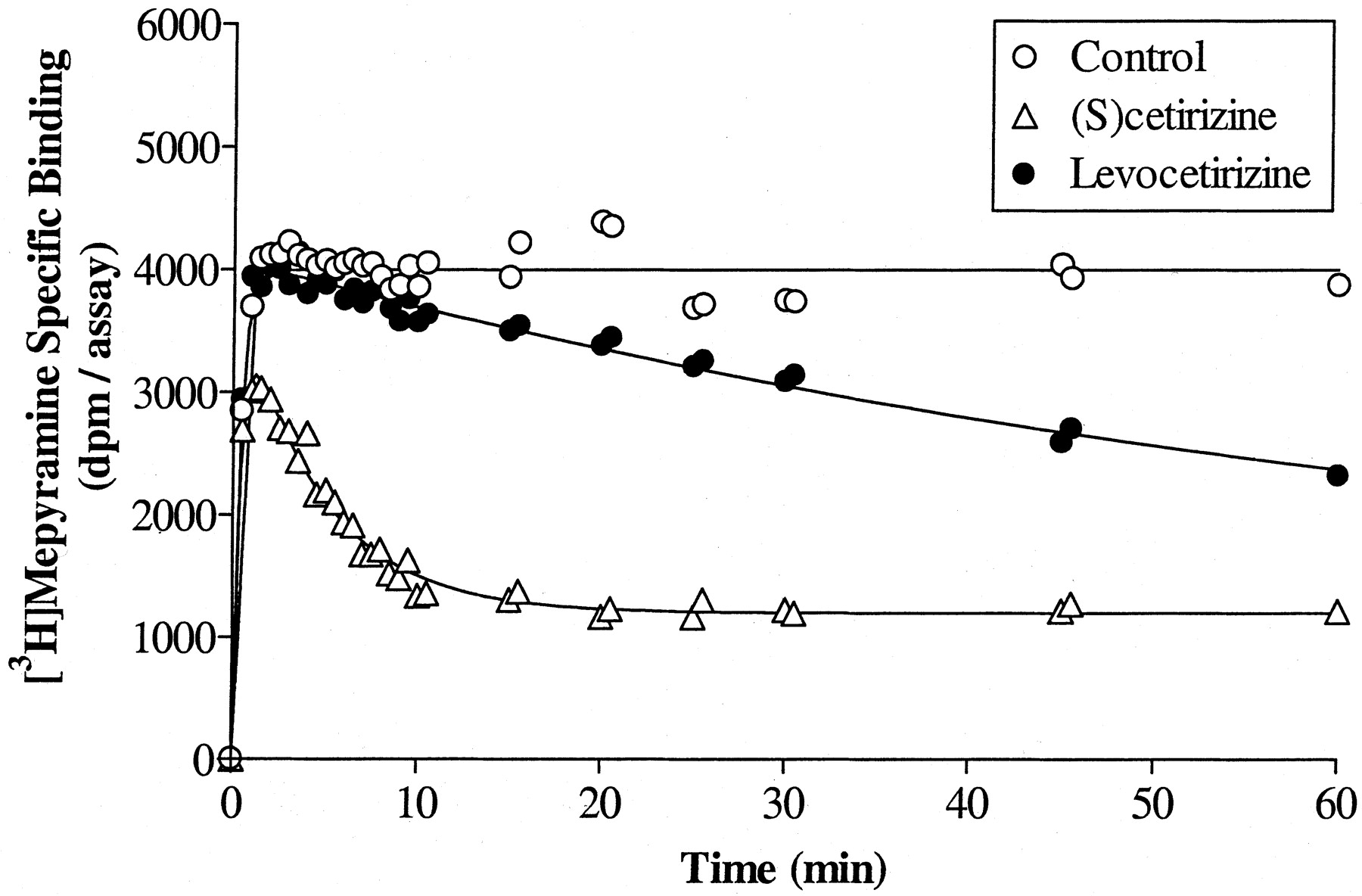
Association kinetics of [3H]mepyramine were performed in the absence and presence of a single concentration of unlabeled drug producing ± 70% inhibition of the radioligand specific binding at equilibrium. Data were analyzed according to the model of Motulsky and Mahan (1984)describing the kinetics of competitive radioligand binding as predicted by the law of mass action. The kinetic constants of [3H]mepyramine and the amount of receptors in the membrane preparations were determined independently and were kept constant to analyze the data in the presence of the unlabeled drug. It is clear from Fig. 6 that (S)-cetirizine reaches equilibrium faster than levocetirizine at equiactive concentrations. Indeed, the analysis shows that although levocetirizine and (S)-cetirizine have quite similar association constants, they differ strikingly when considering the dissociation constants; levocetirizine has a half-time of dissociation longer than 2 h compared with only 6 min for (S)-cetirizine (Table 3). It is noteworthy to point out that the pK i values calculated from the ratio of the two kinetic constantsk −1/k +1(8.7 ± 0.1 and 7.1 ± 0.1, respectively) agree perfectly with those observed experimentally at equilibrium (8.5 ± 0.1 and 7.1 ± 0.1, respectively). We also looked at the binding kinetics of close analogs of cetirizine, whose only structural differences reside in the carboxyl group being replaced either by an hydroxyl group or by a methyl ester (Fig. 1). Although all these pairs of enantiomers have quite similar affinities for the H1 receptor (Table 2), they differ strikingly from a kinetic point of view (Table3). Indeed, replacement of the carboxyl moiety leads to a sharp increase in the association rate (from 10- to 30-fold) but also, concomitantly, to an increase in the dissociation rate (from 4- to 20-fold).

Determination of the binding kinetic constants of levocetirizine and (S)-cetirizine. [3H]Mepyramine association kinetics were performed in the presence or absence of a single concentration of levocetirizine or (S)-cetirizine (chosen to inhibit approximately 75% of [3H]mepyramine specific binding at equilibrium). Data were analyzed according to the model proposed by Motulsky and Mahan (1984) as described under Materials and Methods. The curves are the actual fitting and are representative of three experiments.
Binding kinetic constants of antagonists to wild-type and mutant H1 receptors
Mutagenesis Experiments.
Two mutants were of particular interest: the Thr194 to Ala mutation, which we showed was important for the stereoselectivity of cetirizine enantiomers (Moguilevsky et al., 1995). Here we extend our previous observations on Thr194 by including other pairs of enantiomers and by studying the effects of this mutation on the kinetic constants of the molecules. The second mutant is Lys191 to Ala, which might theoretically be important for the recognition of the carboxyl group of cetirizine and its enantiomers. The pK i and kinetic constants are reported in Table 2 and 3, respectively.
Mutation Thr194→Ala.
The results are compiled in Table 2 and 3. [3H]Mepyramine bound with higher affinity to this mutant than to the wild-type receptor. The mutation provoked an 8- to 13-fold increase in the affinity of the distomers [i.e., (S)-cetirizine, (S)-hydroxyzine, and (S)-ucb 29993]. The increase in affinity of the corresponding eutomers was limited to 1.5- to 4-fold. As a consequence, the binding stereoselectivity of the enantiomers was reduced with eudismic indexes decreasing from 25 to 3 for levocetirizine and (S)-cetirizine, from 32 to 8 for (R)-hydroxyzine and (S)-hydroxyzine and from 13 to 6 for (R)-ucb 29992 and (S)-ucb 29993. Histamine and loratadine, on the contrary, had 3- to 5-fold lower affinity for the mutant receptor. On a kinetic level, levocetirizine and (S)-cetirizine experienced both an increase in theirk +1 but only (S)-cetirizine had a concomitant decrease in itsk −1. The same observation applies for the couple (R)- and (S)-hydroxyzine. As for (R)-ucb 29992 and (S)-ucb 29993, there is a similar trend, although it is not statistically significant because of the rather large variations in the kinetic constants calculated for compounds having very fast kinetics.
Mutation Lys191→Ala.
The results are compiled in Table 2 and 3. The binding of [3H]mepyramine was not significantly affected by this mutation whereas the affinity of histamine was decreased by 20-fold. At equilibrium, the affinity of levocetirizine and (S)-cetirizine for the mutant receptor was decreased by a factor of 4 to 6, whereas the affinity of the hydroxyl or methyl ester analogs was hardly changed. Terfenadine and fexofenadine (the carboxyl derivative of terfenadine) also experienced a slight decrease in affinity (about 2 to 4 fold), whereas that of loratadine remained unchanged. On a kinetic level, the association rates of all compounds increased by 2- to 5-fold, except for (R)-ucb 29992, for which no significant changes were observed. By contrast, the dissociation rates for levocetirizine and (S)-cetirizine were increased by 10- and 6-fold, respectively compared with only 2-fold for the hydroxyl analog (R)-hydroxyzine and with no change for the ester analog (R)-ucb 29992. Both terfenadine and fexofenadine showed a similar 3-fold increase in their dissociation rates whereas a 3-fold decrease was observed for loratadine.
Discussion
Cetirizine is a second generation antihistamine drug, displaying high affinity and selectivity for cloned human H1histamine receptors. As we showed previously, cetirizine and its enantiomers levocetirizine and (S)-cetirizine bind stereoselectively to this receptor with a eudismic ratio of 30 in favor of levocetirizine (Moguilevsky et al., 1994, 1995). Here, we characterize in more detail the molecular interactions of these three compounds with the human H1 receptor. First, it seems clear from the results that the two enantiomers bind to the receptors with quite different kinetics; although they have quite similar association constants, their dissociation rates are different, with levocetirizine dissociating from the receptors with a half-time of 142 min compared with only 6 min for (S)-cetirizine. The difference in dissociation rates between these compounds accounts for most of the difference in their affinities. The dissociation half-time found for levocetirizine agrees well with the 130 min measured for cetirizine on guinea pig H1 receptors using another method (Leysen et al., 1991). One practical consideration about long dissociation kinetics is the time needed to reach equilibrium in binding or other in vitro experiments. Short incubation times will lead to underestimation of the affinity of slowly equilibrating drugs, as exemplified in the SPA binding assay. With time, levocetirizine competition curves shifted to the left along the concentration axis giving decreasing IC50 values from 300 nM at 10 min to 10 nM after 8 h incubation, whereas IC50 values for (S)-cetirizine decreased only from 500 nM to 250 nM in the same interval of time, as expected for a compound that dissociates much faster and thus reaches equilibrium more quickly. As a consequence, the stereoselectivity ratio for such compounds will depend on the incubation time, going from approximately 1.5 after 6 min to 25 after 8 h.
Cetirizine has been reported as acting as a noncompetitive antagonist when inhibiting in vitro histamine-induced contractions of human bronchus (Advenier et al., 1991) and guinea pig trachea (Kahler and Du Plooy, 1994). These observations can now be easily explained by the slow dissociation kinetic of levocetirizine (which, as the eutomer, is the active component of cetirizine). Indeed, slowly dissociating drugs may virtually act as irreversible antagonists and produce what is now called insurmountable antagonism in functional studies; i.e., the maximal tissue response produced by the agonist will be depressed at high antagonist concentration (Kenakin, 1993; see Jenkinson et al., 1995, for nomenclature). Insurmountable antagonism related to slow dissociation kinetics has also been reported for AT2 antagonists (Olins et al., 1995). The extent of insurmountable antagonism caused by slowly dissociating drugs will also depend on the receptor reserve present in the tissue under study as illustrated with cetirizine and levocetirizine in guinea pig trachea and ileum (Christophe et al., 2000). Although interactions with L-type calcium channels can also cause insurmountable antagonism in the same experimental settings, we have shown that cetirizine and levocetirizine, up to 10 μM, did not interact with these channels. We also have shown in this study that cetirizine and both enantiomers interact competitively with histamine at the receptor level. Indeed, the three compounds increased, in a dose-dependent manner, the IC50 of histamine in competition binding assays as expected for competitive antagonists (Fig. 5). When we analyzed the data according to an allosteric model (Lazareno and Birdsall, 1995), best fits were obtained with allosteric constants close to or equal to 0, indicative of strong negative allosterism or competitive antagonism. Negative allosterism implies that in the presence of the antagonist, the agonist will still be able to bind to the receptor, albeit with a lower affinity. In this regard, competitive antagonism can be viewed as an extreme case of allosteric antagonism in which the agonist affinity is reduced to 0, making competitive and strongly negative allosteric antagonists quite difficult to distinguish (Ehlert, 1988).
The carboxyl group of cetirizine or levocetirizine, which is ionized at physiological pH (Pagliara et al., 1998), although not important for the affinity of the compounds, is responsible for the long dissociation time. Its replacement by a hydroxyl group or a methyl ester group hardly modifies the affinity but increases both the dissociation and association kinetic constants at the H1 receptor. The dissociation half-time decreases from 142 min for levocetirizine to 31 min for (R)-hydroxyzine (the hydroxyl analog) and 7 min for (R)-ucb 29992 (the methyl ester analog). A comparable effect was observed with the corresponding distomers.
The mutation of Lys200 into Ala in the guinea pig H1 receptor was first reported to lead to a decrease in histamine affinity without much change in antagonist affinity (Leurs et al., 1995). However, a second study by the same group showed that with antagonists bearing a carboxyl group, their affinity falls by 8- to 50-fold (Wieland et al., 1999). The human counterpart of the guinea pig Lys200 is located in position 191 of the fifth transmembrane domain. We mutated Lys191 into Ala and observed, as previously published for the guinea pig, a 20-fold lower affinity of histamine for this receptor compared with the wild-type receptor. More interesting, however, was the observation that cetirizine and its enantiomers also had a reduction in affinity between 3- and 5-fold, whereas the affinity of their structural analogs lacking the carboxyl group was unchanged. When looking at the kinetic constants, the picture is even clearer; if the mutation of Lys191 to Ala slightly (by 2-fold) increases the association constants, it had a much more pronounced effect on the dissociation rate, which was increased by a factor of 10, decreasing the dissociation half-life from 142 min to 13 min for levocetirizine. The hydroxyl analog [(R)-hydroxyzine] is much less sensitive to this mutation and its dissociation half-life was shortened by only 50%, whereas the mutation has no effect at all on the binding kinetics of the methyl ester analog [(R)-ucb 29992]. These results advocate for a strong interaction between the carboxyl moiety of cetirizine or its enantiomers and Lys191 of the human H1 receptors and indicate that this interaction is the key to the slow dissociation rates of these compounds. The lesser effect of the mutation on the hydroxyl analog is in line with the weaker energy of the hydrogen bond that can still occur between the primary amine of the lysine and the hydroxyl group of the compound compared with the ionic bond expected with the carboxyl group. However, these results obtained on Lys191 with levocetirizine and by Wieland et al. (1999) with cetirizine and acrivastine cannot be extended to all second-generation antihistamines bearing a carboxyl group. Indeed, fexofenadine, the carboxyl analog of terfenadine, and terfenadine are equally sensitive to the mutation of Lys191. A possible explanation might be the distance between the protonated nitrogen, believed to interact strongly with Asp107 (or Asp116 in the guinea pig) in the third transmembrane region and the carboxyl function. This distance is far greater in fexofenadine compared with cetirizine and places the carboxyl function of the former out of reach of any interaction with Lys191. The hydroxyl group present in both terfenadine and fexofenadine, however, could be at the right distance to make an hydrogen bond with Lys191. Alternatively, a rather hydrophobic environment (Moguilevsky et al., 1994) could stabilize a π cationic bond between the benzyl ring of terfenadine and fexofenadine and the nitrogen of Lys191.
While we were studying the influence of Thr194 on the binding of histamine, for which we observed a 5-fold decrease in affinity for the mutant as reported by others (Leurs et al., 1994; Ohta et al., 1994), we also found, surprisingly, that the mutation of Thr194 into Ala decreased the stereoselectivity of cetirizine enantiomers (Moguilevsky et al., 1995). Although the mutation increases the affinity of both enantiomers, it is more pronounced for (S)-cetirizine, leading to an 8-fold decrease in stereoselectivity. The other enantiomeric pairs were also sensitive to this mutation but the stereoselectivity decrease was limited to 4-fold for (R)- and (S)-hydroxyzine and to 2-fold for (R)-ucb 29992 and (S)-ucb 29993. Because the chiral centers are identical in the three pairs of compounds, we could speculate that the interactions taking place with other amino acids, like Lys191, are influencing the way the compounds are hindered by Thr194.
In conclusion, we have shown in this study that cetirizine and levocetirizine are high-affinity, selective H1antagonists (more than 600-fold compared with a variety of other G-protein-coupled receptors and channels) interacting competitively with histamine. The eutomer levocetirizine has 2-fold higher affinity for H1 receptors compared with cetirizine, the racemic compound. Its high affinity is related to slow dissociation kinetics, partly because of an interaction between the carboxylic moiety and Lys191 of the human H1 receptor. This slow dissociation rate also helps explain the insurmountable antagonism observed in certain functional assays. Finally, the 30-fold binding stereoselectivity of the enantiomers is, to some extent, the consequence of a hindrance caused by Thr194.
Acknowledgments
We thank Dr. Florence Moureau and Dr. Luc Queré for their helpful discussions and Mrs. F. Varsalona for her skillful technical assistance in constructing the mutant receptors.
Footnotes
- Received August 28, 2001.
- Accepted November 13, 2001.
Abbreviations
- [3H]RX821002
- 1,4-[6,7(n)-3H]benzodiazoxan-2-methoxy-2-yl)-2-imidazoline hydrochloride
- [3H]mepyramine
- pyridinyl-5-[3H]pyrilamine
- SCH23390 R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine
- SPA, scintillation proximity assay
- α-MEM
- α-Modified Eagle's minimal essential medium
- CHO
- Chinese hamster ovary
- [3H]NMS
- l-N-methyl-[3H]scopolamine methyl chloride
- [3H]Nα-methylhistamine
- N-α-[methyl-3H]methylhistamine
- [3H]8-OH-DPAT
- [propyl-2,3-ring-1,2,3-3H]8-hydroxy-dipropylaminotetralin
- [3H]CGP-12177
- [5,7-3H](−)CGP-12177
- [3H]spiperone
- [benzene ring-3H]spiperone
- [3H]DPCPX
- 8-[dipropyl-2,3-3H(N)]cyclopenthyl-1,3-dipropylxanthine
- [3H]ketanserin
- [ethylene-3H]ketanserin hydrochloride
- [3H]tiotidine
- [methyl-3H]tiotidine
- [3H]prazosin
- [7-methoxy-3H]prazosin
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}