


Abstract
The effects of phosphorylation of the tyrosine residue in the highly conserved DRY motif expressed in the putative second cytoplasmic loop of the μ-opioid receptor were assessed after expression in human embryonic kidney (HEK) 293 cells. Tyrosine kinase activation by epidermal growth factor (EGF) or hydrogen peroxide treatment effectively increased phosphorylation of the tyrosine-166 in the μ-opioid receptor (MOR-Tyr166p) as measured by a novel phosphoselective antibody. We were surprised to find that the increase in MOR-Tyr166p immunoreactivity (ir) required coactivation by the opioid agonist [d-Ala2,methyl-Phe4,Gly5-ol]enkephalin (DAMGO), as demonstrated by both Western blot imaging of membrane proteins and confocal microscopy of transfected cells; MOR-Tyr166p-ir did not significantly increase after either DAMGO, EGF, or H2O2 treatment alone. The increase in MOR-Tyr166p-ir was blocked by pretreatment with the opioid antagonist naloxone or the Src kinase inhibitor 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine. Consistent with these data, mutation of the tyrosine-166 to phenylalanine blocked the increased immunoreactivity, and untransfected HEK293 cells did not increase MOR-Tyr166p-ir after treatment. DAMGO increased guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding to membranes from cells expressing wild-type MOR or MOR-Y166F receptors in a dose-dependent manner. Pretreatment of the wild-type MOR-expressing cells with the combination of DAMGO and EGF completely blocked subsequent DAMGO stimulation of [35S]GTPγS binding membranes, whereas [35S]GTPγS binding to membranes from cells expressing mutated MOR(Y166F) was only partially inhibited. These results suggest that G-protein activation as measured by [35S]GTPγS binding can be regulated by DAMGO and EGF by convergent mechanisms and support the hypothesis that tyrosine phosphorylation within the DRY motif may reduce μ-opioid receptor–G-protein coupling efficiency.
The μ-opioid receptor (MOR; OPRM1) belongs to the class A (rhodopsin family) Gi/o-coupled family of G-protein-coupled receptors (GPCRs) and functions to reduce neuronal excitability primarily by increasing potassium conductance and inhibiting voltage-gated calcium channels (Law et al., 2000; Williams et al., 2001). The opioid system is usually described within the context of drug abuse and analgesic drug action; however, the normal physiological role of the opioid system is to regulate pain sensitivity, endocrine functioning, gut motility, and smooth muscle tone in response to physiological stressors (López et al., 1999; Drolet et al., 2001). The regulation of μ-opioid signaling is a dynamic and complex process (Law et al., 2000). A primary desensitization mechanism involving G-protein receptor kinase (GRK) and β-arrestin-dependent internalization through cytoplasmic serine/threonine phosphorylation has been well described previously (Celver et al., 2001, 2004; Williams et al., 2001). In addition, MOR contains four highly conserved cytoplasmic tyrosine residues (Thompson et al., 1993), and tyrosine kinase-mediated mechanisms regulating MOR signaling have also been described previously (McLaughlin and Chavkin, 2001; Zhang et al., 2009). Tyrosine phosphorylation may influence MOR trafficking and signaling (Pak et al., 1999), consistent with the effects of tyrosine phosphorylation on internalization and signaling of the δ- and κ-opioid receptors (Kramer et al., 2000; Appleyard et al., 2000). A recent study by Law and colleagues showed that tyrosine phosphorylation of MOR at Tyr166 and Tyr336 controlled the switch from inhibition to stimulation of adenylyl cyclase after prolonged agonist application (Zhang et al., 2009). A prior receptor mutagenesis study from our group also showed that tyrosine phosphorylation of MOR regulates agonist coupling efficiency after heterologous gene expression of MOR in Xenopus laevis oocytes (McLaughlin and Chavkin, 2001). The latter study demonstrated that the increase in Kir3-mediated potassium conductance evoked by μ agonist stimulation could be strongly suppressed by MOR tyrosine phosphorylation; this could be blocked by the mutation of Tyr166 and Tyr106 to phenylalanines, whereas mutation of Tyr96 or Tyr336 had no effect on signaling (McLaughlin and Chavkin, 2001). Inhibition of the high basal level of tyrosine kinase activity and stimulation of tyrosine phosphatases in these cells robustly increased μ-opioid activation of Kir3 induced by wild-type MOR but not MOR(Y106F)- or MOR(Y166F)-expressing cells (McLaughlin and Chavkin, 2001). These results suggested that tyrosine phosphorylation of MOR at the 106 or 166 sites could reduce coupling efficiency, but the underlying mechanism and relevance to signal transduction in mammalian cells was not evident.
Tyrosine 166 is part of the highly conserved DRY motif among class A GPCRs (Johnston and Siderovski, 2007; Rovati et al., 2007). The DRY motif, located at the boundary of transmembrane 3 and intracellular loop 2, is believed to be important for regulating the conformational states of the GPCR and G-protein activation. Molecular modeling of class A GPCRs suggests that in the inactive conformation, the arginine residue (Asp3.50) forms a double salt bridge with its neighboring aspartate (Arg3.49) and a charged residue on helix 6 (Rovati et al., 2007). Computational methods predict that agonist-induced conformational changes involve breaking the ionic lock between Asp3.50 and a glutamic acid on helix 6 (Glu6.30) (Bhattacharya et al., 2008a,b). Mutation of the aspartic acid in many class A GPCRs, including Asp(3.49) in MOR, leads to constitutive, agonist-independent activation of the receptor (Li et al., 2001b). Other research has shown that in some GPCRs, nonconservative mutations lead to a loss of G-protein coupling (Rovati et al., 2007).
To better understand the role of phosphorylation of the tyrosine in the DRY motif, we generated a phosphoselective antibody for the μ-opioid receptor at tyrosine 166 and measured the effects of receptor phosphorylation in the DRY motif on G-protein coupling. We found that in human embryonic kidney (HEK) 293 cells expressing GFP-tagged μ-opioid receptors, prior receptor activation by agonist is required for receptor phosphorylation by tyrosine kinases. Phosphorylation of MOR-Tyr166, measured as an increase in immunoreactivity, was found to be dependent on the activation of Src, and that phosphorylation reduced agonist-induced G-protein activation.
Materials and Methods
Chemicals.
Mouse epidermal growth factor (EGF) and PP2 were from Calbiochem (San Diego, CA). Hydrogen peroxide (H2O2) was from VWR International (West Chester, PA). [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) was from Peninsula Laboratories (Palo Alto, CA). Naloxone was from Sigma-Aldrich (St. Louis, MO). Drugs were dissolved in distilled H2O except for PP2, which was dissolved in dimethyl sulfoxide, and the final concentration of dimethyl sulfoxide in the assays did not exceed 0.1%.
HEK293 Cell Culture.
HEK293 cells were cultured in a 1:1 mixture of Dulbecco's modified Eagle's media and Ham's F-12 (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Sigma-Aldrich), l-glutamine, and penicillin/streptomycin at 37°C and 95% O2/5% CO2.
Mutagenesis of MOR and Transfection of HEK293 Cells.
Mutation of the μ-opioid receptor of tyrosine 166 to a phenylalanine was done as described previously (McLaughlin and Chavkin, 2001). Stable transfections of the GFP-tagged μ-opioid receptor (MOR-GFP) and MOR(Y166F)-GFP constructs into HEK293 cells were obtained as described previously (Celver et al., 2004).
Antibody Generation and Affinity Purification.
A polypeptide containing residues 158 to 177 (KTMSVDRpYIAVCHPVKALD) of the μ-opioid receptor phosphorylated at tyrosine-166 was generated by PeptidoGenic Research and Co., Inc. (Livermore, CA). Polyclonal antibody generation and affinity purification using 2 to 5 μmol MOR-Tyr166p peptide was done as described previously for other phosphopeptide antibodies (Ippolito et al., 2005). Sera aliquots (4 ml) were added to the peptide-conjugated Sepharose beads and were incubated for 72 h at 4°C with gentle rocking. Unbound protein was washed off the beads using 20 ml of Tris (50 mM, pH 7.4) containing NaCl (150 mM) and NaN3 (0.02%). Antibody was eluted with 5 ml of MgCl2 (5 M) in Tris/NaCl and concentrated using a Centriprep-30 (Millipore Corporation, Billerica, MA). Antibody aliquots were stored at −20°C in 30% glycerol. Protein concentrations were determined by the by bicinchoninic acid assay (Pierce, Rockford, IL).
ELISA.
Ninety-six-well plates were incubated for 4 h at room temperature with a 10 μg/ml solution of a peptide containing residues 159 to 177 of the μ-opioid receptor or with a peptide containing the μ-opioid receptor phosphorylated at the tyrosine residue at the 166 site. The wells were then incubated for 2 h at room temperature in a blocking solution [3% bovine serum albumin in phosphate-buffered saline (PBS)]. Wells were washed with PBS/0.05% Tween 20 and then incubated with increasing concentrations of purified MOR-Tyr166p antibody in triplicate overnight at 4°C. Wells were then incubated in IgG/alkaline-phosphatase-conjugated secondary antibody (Promega, Madison, WI) diluted 1:3000 in 1× PBS/0.05% Tween 20 solution. The alkaline phosphatase was detected by 1 mg/ml p-nitrophenyl phosphate in 50 mM sodium carbonate buffer, pH 9.8, with 1 M MgCl2·NaOH (2.5 M) was the added to terminate the reaction, and the absorbance was measured via spectrophotometer at 410 nm. The absorbance readings were compared with the antibody dilutions (triplicates were averaged) to establish the serum antibody titer.
Immunocytochemistry.
HEK293 cells expressing either MOR-GFP or MOR(Y166F)-GFP were plated on poly(d-lysine)-coated coverslips (BD Biosciences, San Jose, CA) at 60% confluence and cultured for 24 to 48 h as described above. Cells were washed with sterile PBS (Invitrogen) and serum-starved overnight. After drug treatment, cells were washed once with sterile PBS and then fixed with 4% paraformaldehyde for 20 min at room temperature. After three 5-min washes in PBS, cells were incubated in blocking buffer (PBS containing 0.3% gelatin and 0.025% Triton X-100) for 1 h at room temperature. Cells were then incubated with rabbit anti-MOR-Tyr166p (15 μg/ml) and mouse anti-Src-pTyr418 (1:300; Calbiochem) diluted in blocking buffer for 72 h at 4°C. After primary antibody incubation, cells were washed four times for 10 min each time in PBS, pH 7.4, and then incubated in Alexa Fluor 555 anti-rabbit (1:500; Invitrogen) and Alexa Fluor 633 anti-mouse (1:500; Invitrogen) diluted in blocking buffer for 2 h at room temperature. After three 5-min washes with PBS and two 5-min washes in 0.1 M phosphate buffer, slides were allowed to air-dry for 10 min and then were mounted onto coverslips with Vectashield (Vector Laboratories, Burlingame, CA). In other experiments, the same protocol was followed except that cells were incubated with mouse anti-Src-pTyr418 (1:300; Calbiochem) and then incubated in Alexa Fluor 555 anti-mouse (1:500; Invitrogen). Slides were viewed by Leica confocal microscopy (Leica Microsystems, Inc., Deerfield, IL). Pixel intensity was quantified using Metamorph Imaging System software (Universal Imaging Corporation, Downington, PA), in which the membrane and cell body (excluding the nucleus) were outlined for >30 cells per field, and the average pixel intensity of the defined region was determined.
Membrane Preparation.
Untransfected HEK293 cells or cells expressing either MOR-GFP or MOR(Y166F)-GFP were plated on 100-mm culture plates at 50% confluence and harvested when they reached 80% confluence. Cells were cultured as described above for 24 to 48 h, washed once with sterile PBS, and then serum-starved overnight. DAMGO-stimulated [35S]GTPγS binding and Western blotting were assayed in HEK293 membrane homogenates as described previously (Carroll et al., 2005). In brief, cells were treated with vehicle, DAMGO (1 μM), EGF (50 ng/ml), or DAMGO + EGF at specified time points. After treatment, cells were washed twice with PBS. Cells were scraped into 2-ml membrane buffer containing 300 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 1× protease, and 1× phosphatase inhibitor cocktails (Calbiochem). Cells were then homogenized with a Polytron homogenizer (Kinematica, Littau-Lucerne, Switzerland) for 2 × 10-s pulses. Homogenates were centrifuged at 30,000g at 4°C for 20 min. Supernatant was discarded, and pellet was washed in membrane buffer, rehomogenized, and recentrifuged one more time before freezing the pellet at −80°C until use. In Western blot experiments, the remaining membrane pellet was resuspended in a buffer containing 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 10% glycerol, and 1% n-dodecyl-β-d-maltoside with 1× protease and 1× phosphatase cocktail inhibitor sets (Calbiochem) for 2 h on ice and then stored at −80°C.
Western Blot.
Membrane samples were thawed, and protein concentration was determined by bicinchoninic assay protein assay (Thermo Fisher Scientific, Waltham, MA). Protein (15–20 μg) was resolved by SDS-polyacrylamide gel electrophoresis on 4 to 12% Bis-Tris NuPAGE (Invitrogen) gels and transferred to nitrocellulose. Blots were blocked in 5% nonfat milk in Tris-buffered saline (TBS), pH 7.4, for 1.5 h at room temperature and then incubated in primary antibody (rabbit anti-MOR-Tyr166p; 15 μg/ml) diluted in blocking buffer overnight at room temperature. Blots were then washed four times for 10 min each in TBS containing 1% Tween 20 (TBST) and then incubated in IR-Dye 800-conjugated affinity-purified anti-rabbit IgG (Rockland Immunochemicals, Gilbertsville, PA) at a dilution of 1:7500 in a 1:1 mixture of 5% milk/TBS and LI-COR blocking buffer (LI-COR Biosciences, Lincoln, NE) for 1 h at room temperature. After incubation, cells underwent three 5-min washes in TBST and two 5-min washes in TBS and analyzed using the Odyssey infrared imaging system (LI-COR Biosciences). Blots were then reprobed with mouse anti-β-actin (AbCam, Cambridge, MA) 1:5000 diluted in 5% nonfat milk in TBS for 2 h at room temperature and underwent three 5-min washes with TBST. Blots were then incubated in Alexa Fluor 680 anti-mouse (Invitrogen) at a dilution of 1:7500 in a 1:1 mixture of 5% nonfat milk/TBS and LI-COR blocking buffer for 1 h at room temperature, washed three times for 5 min in TBST and twice for 5 min in TBS. Immunoblots were scanned using the Odyssey Infrared imaging system, and band intensities were measured as described previously (Bruchas et al., 2006).
[35S]GTPγS Binding.
For the GTPγS assay, membranes containing 20 mg of protein were incubated with DAMGO (0.1–1000 nM) in 50 mM binding buffer containing 50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 0.1% bovine serum albumin, and 1 mM dithiothreitol, pH 7.4 at 30°C for 1 h in the presence of 0.1 nM [35S]GTPγS and 10 μM GDP. Nonspecific binding was defined by adding 10 μM unlabeled GTPγS. Bound radioligand was separated from free by rapid filtration using a Brandel cell harvester (Brandel Inc., Gaithersburg, MD) onto GF/B filters (Whatman, Clifton, NJ). Filters were washed three times in a buffer containing 50 mM Tris, pH 7.4, with 0.1% bovine serum albumin. Bound radioligand was measured using Econoscint XR (National Diagnostics, Atlanta, GA) scintillation fluid and were counted using a Packard Tri-Carb 2200 CA liquid scintillation analyzer (PerkinElmer Life and Analytical Sciences, Waltham, MA). Triplicate determinations were normalized to the percentage of vehicle binding. Concentration-response curves were plotted using nonlinear regression analysis (Prism 4.0; GraphPad Software Inc., San Diego, CA).
Results
A phosphopeptide containing residues 159 to 177 of the MOR sequence phosphorylated at tyrosine 166 was used to generate an affinity-purified polyclonal antibody (Fig. 1A). The resulting reagent demonstrated a high degree of selectivity for the phosphopeptide compared with the unphosphorylated version in the ELISA (Fig. 1B). Using membrane proteins isolated from HEK293 cells stably expressing an MOR-GFP, we found that basal MOR-Tyr166p-ir was low, as visualized by Western blot (Fig. 2A). Treatment of the cells with hydrogen peroxide (H2O2, 4.5 mM) for 15 min did not increase MOR-Tyr166p-ir (Fig. 2A, lane 2). This result was surprising because H2O2 is a known activator of tyrosine kinases and an inhibitor of tyrosine phosphatases that leads to an overall increase in tyrosine phosphorylation within the cell (Takano et al., 2002; Thakali et al., 2007). Likewise, treatment with the MOR-specific agonist DAMGO (1 μM) for 30 min alone did not increase MOR-Tyr166p-ir (Fig. 2A, lane 3). In contrast, 30-min treatment with 1 μM DAMGO followed by subsequent cotreatment with 4.5 mM H2O2 for 15 min caused a significant increase in MOR-Tyr166p-ir (Fig. 2A, lane 4). The apparent molecular mass of the immunoreactive band was approximately 82 kDa (Supplemental Fig. 1), which is consistent with prior determinations of MOR mobility in Western blots (Petraschka et al., 2007). The MOR-Tyr166p-ir protein band showed the same mobility as the band detected by anti-mouse GFP, and peroxide treatment did not affect MOR-GFP protein expression. Quantification of replicate images indicate that the MOR-Tyr166p-ir band intensity significantly increased 3.38 ± 0.56-fold (n = 5) after combined DAMGO-H2O2 treatment but was not significantly affected by treatment with either DAMGO or H2O2 alone (Fig. 2A, left).

MOR-Tyr166p antibody is phosphoselective and specific for the μ-opioid receptor. A, cartoon of the MOR phosphorylated at tyrosine 166. B, ELISA showing that the MOR-Tyr166p affinity-purified antibody is specific for the phosphorylated MOR-Tyr166 peptide (●) over the unphosphorylated peptide (○).

Phosphorylation of the μ-opioid receptor at tyrosine 166 requires prior activation of the receptor. HEK293 cells were treated either with the vehicle, hydrogen peroxide (H2O2, 4.5 mM, 15 min), DAMGO (1 μM, 30 min), or cotreated with DAMGO and H2O2. A, a representative Western blot of HEK293 cells stably expressing MOR-GFP and quantification of band intensities, n = 4 to 7. One-way ANOVA, p < 0.001, with Tukey's multiple comparison test. ∗∗, p < 0.01 compared with the H2O2-treated group or the DAMGO-treated group. B, a representative Western blot of HEK293 cells stably expressing MOR(Y166F)-GFP and quantification of band intensities, n = 5. One-way ANOVA, not significant. C, a representative Western blot of untransfected HEK293 cells and quantification of band intensities, n = 5. One-way ANOVA, not significant. Band intensities were normalized to actin and shown as -fold change over the vehicle-treated band.
Specificity of the MOR-Tyr166p antibody was assessed using HEK293 cells transfected with the MOR-Y166F-GFP, having tyrosine-166 mutated to phenylalanine (McLaughlin and Chavkin, 2001). Although these cells were shown to express equivalent amounts of MOR, treatment with DAMGO and H2O2 alone or in combination failed to significantly increase MOR-Tyr166p-ir (Fig. 2B). Likewise, untransfected HEK293 cells did not show MOR-Tyr166p-ir (Fig. 2C). These results suggest that the tyrosine-166 residue is occluded in the basal state and that agonist activation by DAMGO unmasks the phosphorylation site.
We confirmed these results using confocal microscopy of HEK293 cells stably expressing MOR-GFP. Treatment with 1 μM DAMGO for 30 min alone induced receptor internalization (green), as shown by the comparison between the cell surface labeling (Fig. 3A) and cytosolic localization of the green fluorescence in Fig. 3B. Treatment with DAMGO alone did not increase MOR-Tyr166p-ir (red) (Fig. 3B). In contrast, cotreatment with 1 μM DAMGO and H2O2 caused both internalization of MOR-GFP and a significant increase in MOR-Tyr166p-ir (Fig. 3C). HEK293 cells stably expressing MOR(Y166F)-GFP show equivalent plasma membrane localization of GFP (Fig. 3D) and robust internalization after DAMGO treatment (Fig. 3E). Previous reports have shown that mutation of the aspartate residue, a part of the DRY triad, resulted in agonist-independent internalization of MOR (Li et al., 2001a,b). However, mutation of the tyrosine-166 residue did not promote agonist-independent internalization of MOR(Y166F)-GFP (Fig. 3D). Treatment with 1 μM DAMGO alone or with 4.5 mM H2O2 did not increase MOR-Tyr166p-ir in MOR(Y166F)-GFP expressing cells (Fig. 3, E and F). Quantification of replicate images confirmed that MOR-Tyr166p-ir was significantly increased by combined DAMGO and H2O2 treatment of cells expressing MOR-GFP but not cells expressing MOR(Y166F)-GFP (Fig. 3G). These results further support the conclusion that the antibody generated is able to detect phosphorylation of tyrosine-166 and that prior activation of the μ-opioid receptor is required for this phosphorylation event to occur.

Cotreatment of HEK293 cells expressing MOR-GFP with DAMGO and H2O2 results in increased MOR-Tyr166p IR that colocalizes with the receptor. A to C, confocal images of HEK293 cells stably expressing MOR-GFP. Cells were treated with either the vehicle (A), DAMGO (B), or DAMGO + H2O2 (C). D to F, confocal images of HEK293 cells stably expressing MOR(Y166F)-GFP. Cells were treated with either the vehicle (D), DAMGO (E), or DAMGO + H2O2 (F). Scale bar, 10 μm. Green represents the GFP-tagged receptor, and red represents MOR-Tyr166p IR. G, quantification of MOR-Tyr166p pixel intensity. Data are shown as a -fold change over the vehicle control for each group, n = 4 to 6. One-way ANOVA, p < 0.0001, with a Tukey's multiple comparison post hoc test. ∗∗∗, p < 0.0001 compared with the vehicle control. †††, p < 0.0001 compared with the DAMGO-treated group.
Peroxide treatment is a fairly nonselective tyrosine kinase activator and activates several different tyrosine kinase cascades (Klann and Thiels, 1999). HEK293 cells endogenously express the EGF receptor tyrosine kinase system (Kramer et al., 2002), and other groups have shown that EGF receptor and MOR interact (Belcheva et al., 2003; Chen et al., 2008). We next asked whether activation of the EGF receptor could also increase MOR-Tyr166p-ir. Treatment of HEK293 cells stably expressing MOR-GFP with 50 ng/ml EGF alone for 5 min did not lead to tyrosine phosphorylation of the receptor (Fig. 4B, row 2). However, cotreatment of the cells with DAMGO (1 μM, 30 min) and EGF (50 ng/ml, 5 min) did cause a robust increase in MOR-Tyr166p-ir that seemed to colocalize with the green fluorescence of the receptor (Fig. 4C, rows 2 and 4). Quantification of replicate images confirmed that EGF alone did not significantly increase MOR-Tyr166p-ir, whereas cotreatment with DAMGO and EGF did increase staining by 1.72 ± 0.12-fold (n = 6) (Fig. 4D).

Cotreatment of HEK293 cells expressing MOR-GFP with DAMGO and EGF results in increased MOR-Tyr166p IR. A, HEK293 cells stably expressing MOR-GFP treated with a vehicle control. B, HEK293 cells stably expressing MOR-GFP treated with EGF (50 ng/ml, 5 min). C, HEK293 cells stably expressing MOR-GFP cotreated with DAMGO (1 μM, 30 min) and EGF (50 ng/ml, 5 min). Green represents the GFP-tagged receptor and red represents MOR-Tyr166p IR. Scale bar, 10 μm. D, quantification of MOR-Tyr166p pixel intensity. Data are shown as a -fold change over the vehicle control group, n = 6. One-way ANOVA, p < 0.0001, with Tukey's multiple comparison post hoc test. ∗∗∗, p < 0.001 compared with the vehicle control. †††, p < 0.001 compared with the EGF-treated group.
These results support the hypothesis that agonist-induced activation of MOR by DAMGO leads to a conformational change that allows access of the receptor tyrosine kinase-initiated cascade to the tyrosine 166 residue; however, the identity of the kinase directly responsible for the increase in MOR(Y166p)-ir was not established. Recent studies have suggested that Src can be activated by agonist-stimulated MOR and that this can result in Y166p (Zhang et al., 2009); however, they concluded that Tyr336 phosphorylation is functionally important and Tyr166 somehow regulates the phosphorylation state of Tyr336 but that Tyr166 was not phosphorylated by Src. To assess the role of Src, we measured pSrc(Tyr418)-ir, which probes for the activated state of Src, in HEK293 cells that express either MOR-GFP or MOR(Y166F)-GFP. We found that treatment with DAMGO, EGF, or cotreatment with DAMGO and EGF led to a significant increase staining intensity (Fig. 5) in both cell lines. It is noteworthy that pretreatment of either cell line with the μ-opioid receptor antagonist naloxone prevented increases in pSrc(Tyr418)-ir seen after cotreatment with DAMGO and EGF (Fig. 5, A, column 5, B, and C). Likewise, cotreatment with DAMGO and H2O2 leads to an increase in MOR-Tyr166p-ir that was blocked by pretreatment with either naloxone (1 μM) or the Src inhibitor, PP2 (5 μM) (Fig. 6A). Quantification of replicate experiments demonstrated that both naloxone and PP2 significantly blocked the increase in MOR-Tyr166p-ir caused by cotreatment with DAMGO and H2O2 (Fig. 6B). These results suggest that activation of Src may be responsible for the increased MOR-Tyr166p-ir.

Treatment of HEK293 cells expressing MOR-GFP or MOR(Y166F)-GFP with EGF, DAMGO, or cotreatment with EGF and DAMGO results in increased pSrc(Tyr418)-ir. A, confocal images taken of HEK293 cells expressing MOR-GFP (top row) or MOR(Y166F)-GFP (bottom row) treated with a vehicle, EGF (50 ng/ml, 5 min), DAMGO (1 μM, 30 min), cotreated with DAMGO and EGF, or pretreated with naloxone (1 μM, 30 min) and then cotreated with DAMGO and EGF. Green represents the GFP-tagged receptor, and red represents pSrc(Tyr418)-ir. Scale bar, 10 μm. B, quantification of pSrc(Tyr418) pixel intensity in cells expressing MOR-GFP. Data are shown as a -fold change over the vehicle control group, n = 5 to 8. One-way ANOVA, p < 0.01, with a one-sample t test. ∗, p < 0.05, ∗∗, p < 0.01 compared with the vehicle control. ††, p < 0.01 compared with the naloxone-pretreated group. C, quantification of pSrc(Tyr418) pixel intensity in cells expressing MOR(Y166F)-GFP. Data are shown as a -fold change over the vehicle control group, n = 4 to 5. One-way ANOVA, p < 0.01, with a one-sample t test. ∗, p < 0.05, ∗∗, p < 0.01 compared with the vehicle control.
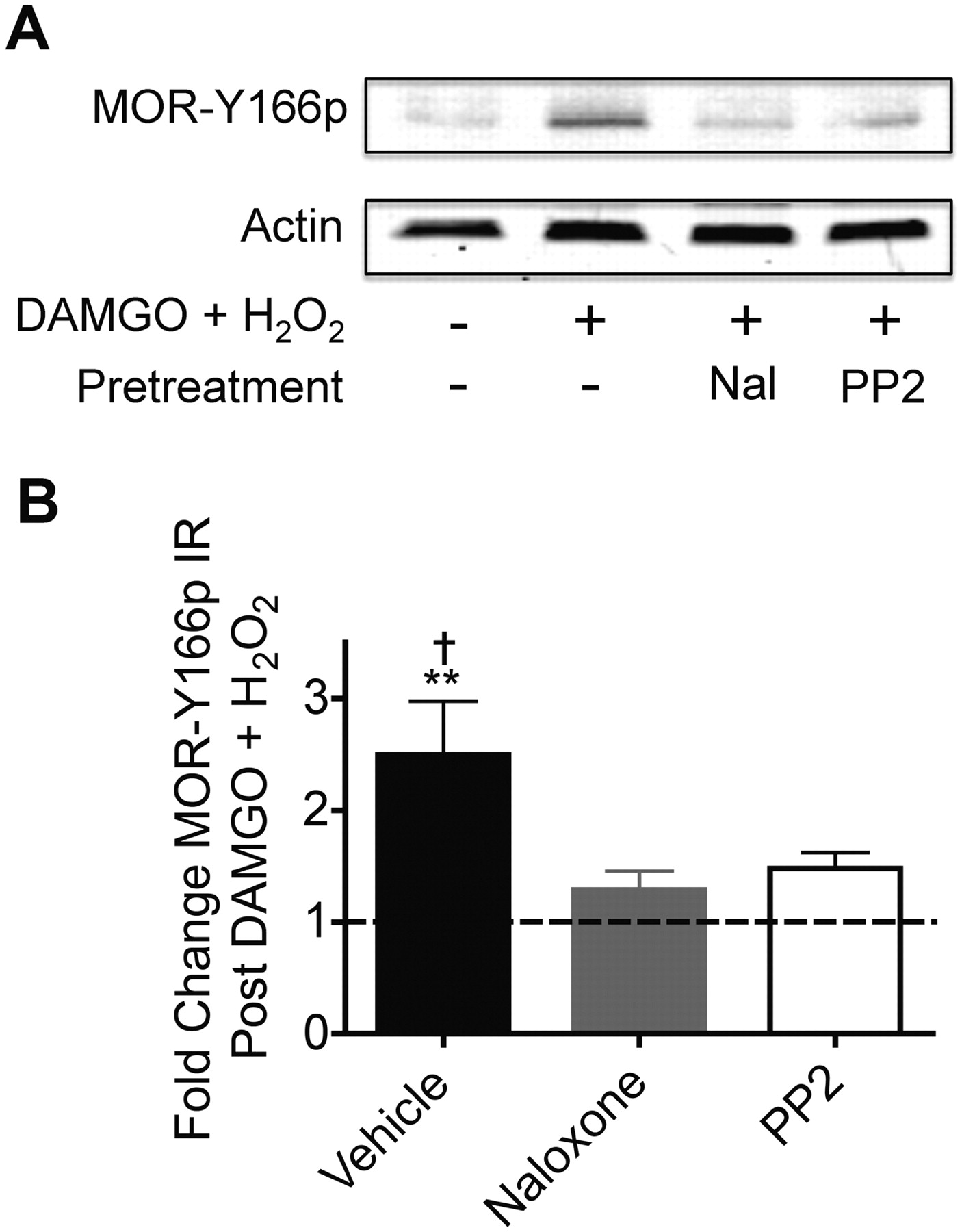
Tyrosine phosphorylation of the μ-opioid receptor is dependent on receptor activation and activation of the nonreceptor tyrosine kinase Src. A, a representative Western blot of MOR-Tyr166p and actin in HEK293 cells stably expressing MOR-GFP. Cells were pretreated with the vehicle control naloxone (1 μM, 30 min) or PP2 (5 μM, 30 min) and subsequently cotreated with DAMGO (1 μM, 30 min) and H2O2 (4.5 mM, 15 min). B, quantification of band intensities. All data points were normalized to actin and are shown as a -fold change over the untreated control group, n = 4 to 5. One-way ANOVA, p < 0.01 with a Newman-Keuls multiple comparison test. ∗∗, p < 0.01 compared with the untreated control. †, p < 0.05 compared with the naloxone and PP2-pretreated groups.
As discussed previously, the DRY motif has been suggested to have a significant role in agonist-dependent activation of G proteins (Rovati et al., 2007). To assess the effects of tyrosine-166 phosphorylation on G-protein activation by MOR, we used the [35S]GTPγS binding assay to measure changes in DAMGO efficacy (Harrison and Traynor, 2003). Membranes isolated from MOR-GFP-expressing HEK293 cells show a DAMGO concentration-dependent increase in [35S]GTPγS binding that was not evident in untransfected cells (Fig. 7A). Pretreatment of the HEK293 cells with DAMGO (1 μM) for 30 min before membrane isolation significantly reduced the maximal increase in [35S]GTPγS binding caused by DAMGO without affecting the EC50 of DAMGO (Fig. 7A and Table 1). This observation is consistent with prior reports of DAMGO-induced receptor desensitization (Celver et al., 2001; Virk and Williams, 2008) and with images shown in Fig. 3B of DAMGO-induced receptor internalization. Treatment of the cells with EGF alone also reduced the maximal effect of DAMGO-induced G-protein activation (Fig. 7A and Table 1). EGF treatment did not induce MOR-GFP internalization, but prior studies suggest that activation of receptor tyrosine kinases can reduce opioid receptor signaling (Chen et al., 2008). Cotreatment of the cells with DAMGO and EGF led to a near complete block of DAMGO-induced G-protein activation (Fig. 7A and Table 1).

Phosphorylation of MOR-Tyr166 by cotreatment with DAMGO and EGF show reduced G-protein activation in response to DAMGO. Dose-response relationship of DAMGO-induced G-protein activation on membranes from untransfected HEK293 cells (□) or cells stably expressing MOR-GFP (A) or MOR(Y166F)-GFP (B). Cells were pretreated with a vehicle (■), DAMGO (▴), EGF (○), or cotreated with DAMGO and EGF (●) and then isolated as described under Materials and Methods. [35S]GTPγS binding was then measured after the membranes had incubated with various concentrations of DAMGO.
Summary of the effects of DAMGO, EGF, or DAMGO + EGF on DAMGO-stimulated GTPγS binding on wild-type or mutant Y166F MOR
Pharmacological data for DAMGO-stimulated GTPγS binding was collected on membranes taken from HEK cells stably transfected with either wild-type MOR (MOR) or the MOR mutant Y166F (Y166F). GTPγS binding was measured and analyzed as described under Materials and Methods. Results represent the means ± S.E.M. of four to eight independent experiments performed in triplicate. Data were analyzed by one-way ANOVA followed by Tukey's post hoc test. EC50 values were determined from nonlinear regression analysis (Prism version 4.0).
Cells expressing MOR(Y166F)-GFP also showed a DAMGO concentration-dependent increase in [35S]GTPγS binding (Fig. 7B). Pretreatment with EGF did not significantly affect either the EC50 or Emax of DAMGO stimulated [35S]GTPγS binding to membranes from MOR(Y166F)-GFP-expressing cells (Fig. 7B and Table 1). As expected, pretreatment of the cells with DAMGO still caused a significant decrease in subsequent DAMGO-stimulated G-protein activation (Fig. 7B and Table 1). Both MOR-GFP and MOR(Y166F)-expressing cells show equivalent receptor internalization (Fig. 3), and prior results showed that Y166F mutation did not affect MOR receptor activation of G-protein-gated inwardly rectifying potassium currents (McLaughlin and Chavkin, 2001). Cotreatment of MOR(Y166F)-GFP expressing cells with both EGF and DAMGO did not produce a significantly greater reduction in Emax than did pretreatment with DAMGO alone (Fig. 7B and Table 1). This result suggests that tyrosine phosphorylation of residue 166 in the DRY motif strongly reduces G-protein coupling efficiency of the μ-opioid receptor.
Discussion
In this study, we investigated the mechanism and effects of phosphorylation of MOR-tyrosine 166, a residue previously found to be important in regulating receptor efficacy (McLaughlin and Chavkin, 2001). To elucidate the mechanism of receptor phosphorylation, we generated and characterized an affinity-purified antipeptide antibody selective for tyrosine-166 phosphorylation: MOR-Tyr166p. We found that increases in MOR-Tyr166p-ir were evident only if the receptor had been activated by DAMGO before treatment with either H2O2 or EGF, known activators of tyrosine kinase cascades. Furthermore, we found that phosphorylation of the tyrosine residue resulted in a decrease in agonist-mediated efficacy as measured using the GTPγS binding assay.
Tyrosine residue 166 is part of the highly conserved DRY motif found among class A GPCRs. The DRY motif, located in the second intracellular loop, is important in regulating conformation states of GPCRs and is important for agonist-induced G-protein activation (Rovati et al., 2007). A wide body of research has used mutational analyses to understand the role of this triad sequence. For example, mutation of the aspartic acid (Asp3.49) of the β2-adrenergic receptor results in structural instability of the receptor and constitutive activation (Rasmussen et al., 1999). Likewise, mutation of the aspartic acid (Asp3.49) of the MOR results in constitutive activation and receptor internalization in the absence of agonist stimulation (Li et al., 2001a,b). A charge-conserving mutation of the arginine residue (Arg3.50) of the α1β-adrenergic receptor led to constitutive activity of the receptor and an increase in binding affinity (Scheer et al., 2000), whereas mutation of this arginine residue in the α2α-adrenergic receptor led to a decrease in binding affinity (Chung et al., 2002). Although much research has investigated the role of the aspartate and arginine residues in the DRY motif, previously, there was little understanding of the role of the tyrosine.
We generated a mutant MOR in which tyrosine residue 166 was changed to a phenylalanine and found no evidence that this caused constitutive activity. There was no internalization of the receptor in the absence of agonist, and the receptor seemed to internalize normally in the presence of DAMGO. In addition, in the GTPγS binding assay, the mutant receptor was not different from the wild-type MOR in DAMGO-induced G-protein activation, EC50 values, or desensitization profile. These results are also consistent with the finding that MOR(Y166F) and the wild-type receptor show comparable activation of G-protein-gated inwardly rectifying potassium (Kir3) currents in response to DAMGO (McLaughlin and Chavkin, 2001). The tyrosine and phenylalanine amino acids are structurally similar, and this conservative mutation is advantageous when focusing on the effects of phosphorylation; however, future studies using alternative substitutions at this site might induce constitutive activity similar to that produced by mutation of the adjacent arginine (Rovati et al., 2007).
The conclusion that the affinity-purified antibody generated for this study was phosphoselective was based on a series of control experiments. First, the affinity-purified MOR-Tyr166p antibody had a higher affinity for the phosphopeptide than the nonphosphopeptide in the ELISA. In addition, cotreatment of HEK293 cells that express MOR(Y166F) or untransfected cells with DAMGO and H2O2 showed no increase in immunoreactivity as measured by either Western blot or confocal microscopy. Pretreatment with the MOR antagonist naloxone before cotreatment with DAMGO and H2O2 prevented the increase in MOR-Tyr166p-ir. The apparent molecular weight of the MOR-Tyr166p-ir was the same as that identified by anti-GFP-ir staining of MOR-GFP expressed by HEK293 cells. Further development of this tool will enable us to extend our research from transfected cells to in vivo models.
Increases in immunoreactivity were not seen in cells that had been treated with DAMGO alone but required coactivation of tyrosine kinases, by treatment with either H2O2 or EGF. This is probably due to a difference in the conformational state of the activated receptor and dissociation of the G-protein that would otherwise occlude the DRY motif. Alternatively, tyrosine phosphorylation of MOR may require β-arrestin recruitment, which has been demonstrated to be necessary for Src activation (Luttrell et al., 1999; Walwyn et al., 2007).
Although the C-terminal tail and third loop have been implicated in G-protein activation (Johnston and Siderovski, 2007), it is also believed that agonists can act by changing the conformation of the GPCR second loop to activate the switch on the G-protein, remove the GDP block required for G-protein activation, or stabilize nucleotide exchange (Rovati et al., 2007; Wacker et al., 2008). It is therefore possible that the prior treatment with DAMGO opens the second loop's DRY motif for Src phosphorylation, causing subsequent inhibition of G-protein activation. Computational research of agonist-induced conformational states of rhodopsin predicts a disruption of the ionic lock between Arg3.50 and Glu6.30 that results in an increase in the distance between Arg3.50 and Tyr3.51 (Bhattacharya et al., 2008b). Indeed, research on the ligand-stabilized conformational states of the β2-adrenergic receptor has shown that agonist-induced changes include conformational changes of transmembrane helices 3, 5, and 6 and breaking of the ionic lock between Arg3.50 and Glu6.30. It is noteworthy that comparing agonists, it was found that although all agonists tested were able to induce transmembrane conformational changes, there was a difference in the disruption of the ionic lock. Treatment with the full agonist norepinephrine was able to induce the disruption of the ionic lock, whereas treatment with catechol, a weak agonist, was not able to break the ionic lock (Bhattacharya et al., 2008a). In our experiments, robust phosphorylation of MOR-Tyr166 was seen when we cotreated the cells with DAMGO, a full agonist, and H2O2 or EGF. In the future, it would be interesting to compare this response with cotreatment with a weak or partial agonist.
We found that cotreatment of cells with DAMGO and EGF resulted in a significant increase in activation of the nonreceptor tyrosine kinase, Src. We also found that pharmacological inhibition of Src blocked DAMGO, and H2O2 induced increases in MOR-Tyr166p-ir. It is known that activation of both the EGFR and MOR increase Src activity (Walwyn et al., 2007). However, treatment with DAMGO or EGF alone was not able to increase MOR-Tyr166p-ir, despite the ability of both agonists to lead to a significant increase in Src activation. These results suggest that activation of both pathways results in a robust increase in Src activity and that this combined stimulus is needed for phosphorylation of MOR-Tyr166 to occur. Previous reports have also shown that in some cases, activation of both GPCR and receptor tyrosine kinase pathways are needed for full stimulation of an effector (Wetzker and Böhmer, 2003). For example, activation of mitogen-activated protein kinase by the bradykinin (B2) receptor requires both protein kinase C activation and transactivation of the EGFR (Adomeit et al., 1999). In addition, MOR-mediated activation of extracellular signal-regulated kinase by DAMGO in cortical astrocytes also requires transactivation of the EGFR (Belcheva et al., 2003). Our results may represent another example of multiple input-coordinated signal transduction.
To better understand the functional implications of phosphorylation of MOR-Tyr166, we used the GTPγS binding assay. Studies have shown the DRY motif to be important for regulating G-protein coupling and activation, and previous studies have shown this tyrosine phosphorylation event to be important for regulating receptor efficacy (McLaughlin and Chavkin, 2001; Rovati et al., 2007). First, we found that pretreating the membranes with DAMGO resulted in a significant decrease in subsequent DAMGO-induced G-protein activation. It is well known that agonist treatment of GPCRs results in desensitization and internalization of the receptor and a reduction in further G-protein activation. We found that cotreatment of membranes with DAMGO and EGF, a stimulus that results in tyrosine phosphorylation at residue 166, resulted in a complete obstruction of GTPγS binding with an Emax that was significantly less than the vehicle control and the DAMGO-treated group. This result suggests that phosphorylation of MOR-Tyr166 results in a decrease in agonist efficacy, and it is consistent with the hypothesis that dephosphorylation of this residue results in an increase in agonist efficacy (McLaughlin and Chavkin, 2001).
It is interesting that we found that treatment of membranes with EGF alone resulted in a decrease in agonist-mediated G-protein activation that was comparable with the DAMGO-treated group. This result is consistent with research showing that activation of receptor tyrosine kinase cascades transregulate GPCRs (Delcourt et al., 2007). It has been shown that activation of the EGFR in HEK293 cells results in phosphorylation and activation of GRK2. Activation of GRK2 then promotes internalization of opioid receptors and phosphorylation of DOR at the GRK phosphorylation site (Chen et al., 2008). Our results complement these studies by suggesting that activation of EGFR leads to a reduction in G-protein coupling. It is notable that the decrease in G-protein binding after EGF treatment was not seen in the mutant receptor, MOR(Y166F).
In summary, we generated and characterized an antibody that can selectively detect phosphorylation of tyrosine 166 of the μ-opioid receptor. Our data suggest that agonist activation of the receptor in combination with activation of tyrosine kinase cascades results in an increase in MOR-Tyr166p-ir. Phosphorylation of tyrosine 166 was dependent on Src activation and resulted in a block of agonist-mediated G-protein activation. This mechanism of regulation of opioid receptor signaling by coincident activation of opioid and tyrosine kinase cascades may provide a novel explanation for the reduction in opioid sensitivity during chronic stress.
Acknowledgments
We thank Hernan Navarro (Research Triangle Institute), Nephi Stella, and Faith Reyes (University of Washington) for procedural advice.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant DA11672].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.060558
-
ABBREVIATIONS:
- MOR
- μ-opioid receptor
- GPCR
- G-protein-coupled receptor
- HEK
- human embryonic kidney
- GFP
- green fluorescent protein
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- EGF
- epidermal growth factor
- GRK
- G-protein receptor kinase
- EGFR
- epidermal growth factor receptor
- PP2
- 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- ir
- immunoreactivity
- ELISA
- enzyme-linked immunosorbent assay
- PBS
- phosphate-buffered saline
- TBS
- Tris-buffered saline
- TBST
- Tris-buffered saline/Tween 20
- ANOVA
- analysis of variance
- MOR-GFP
- green fluorescent protein-tagged μ-opioid receptor
- MOR-Tyr166p
- phosphorylation of the tyrosine-166 in the μ-opioid receptor
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate.
- Received August 24, 2009.
- Accepted December 2, 2009.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics
References
- Adomeit et al., 1999.↵
- Appleyard et al., 2000.↵
- Belcheva et al., 2003.↵
- Bhattacharya et al., 2008a.↵
- Bhattacharya et al., 2008b.↵
- Bruchas et al., 2006.↵
- Carroll et al., 2005.↵
- Celver et al., 2004.↵
- Celver et al., 2001.↵
- Chen et al., 2008.↵
- Chung et al., 2002.↵
- Delcourt et al., 2007.↵
- Drolet et al., 2001.↵
- Harrison and Traynor, 2003.↵
- Ippolito et al., 2005.↵
- Johnston and Siderovski, 2007.↵
- Klann and Thiels, 1999.↵
- Kramer et al., 2000.↵
- Kramer et al., 2002.↵
- Law et al., 2000.↵
- Li et al., 2001a.↵
- Li et al., 2001b.↵
- López et al., 1999.↵
- Luttrell et al., 1999.↵
- McLaughlin and Chavkin, 2001.↵
- Pak et al., 1999.↵
- Petraschka et al., 2007.↵
- Rasmussen et al., 1999.↵
- Rovati et al., 2007.↵
- Scheer et al., 2000.↵
- Takano et al., 2002.↵
- Thakali et al., 2007.↵
- Thompson et al., 1993.↵
- Virk and Williams, 2008.↵
- Wacker et al., 2008.↵
- Walwyn et al., 2007.↵
- Wetzker and Böhmer, 2003.↵
- Williams et al., 2001.↵
- Zhang et al., 2009.↵




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}