


Abstract
The μ-opioid receptor (MOR) contains four highly conserved cytoplasmic tyrosine residues that may serve to regulate receptor activity. For Xenopus laevis oocytes coexpressing the rat MOR and the heteromultimeric potassium channel, KIR3.1/3.2, pretreatment with insulin produced both a 40% suppression in the basal channel conductance and potentiation of response to the μ-opioid agonist [d-Ala2,methyl-Phe4,Gly5-ol]enkephalin (DAMGO) to 155% of matched, untreated control cells. Insulin-induced potentiation of the DAMGO response was concentration-dependent and reversed after 1 h. Insulin pretreatment increased the maximal effect of DAMGO, but did not change its EC50 value. Potentiation of the DAMGO response did not result from a recruitment of MOR to the cell surface, as measured by specific binding of the opioid peptide antagonist [3H]d-Phe(3H)-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2(cyclic) to whole-oocytes, but instead the potentiation was probably caused by an increase in intrinsic efficacy of G protein coupling. The involvement of tyrosine residues on the putative intracellular loops of the MOR was demonstrated with four point-mutated receptors, replacing tyrosine with phenylalanine to create MOR(Y96F), MOR(Y106F), MOR(Y166F), and MOR(Y336F). None of these mutations significantly altered the EC50 value for DAMGO compared with wild-type MOR, and insulin pretreatment still potentiated the effect of 1 μM DAMGO in oocytes containing either MOR(Y96F) or MOR(Y336F) to 137 ± 10 and 124 ± 8%, respectively. However, insulin did not significantly potentiate the DAMGO response with oocytes containing either MOR(Y106F) or MOR(Y166F), suggesting that these two sites were responsible for the insulin-induced opioid potentiation. The tyrosine-kinase inhibitors genistein (100 μM) or K-252a (20 μM) did not block the insulin-induced potentiation of the DAMGO response, but coincubation of insulin with either the MAP kinase inhibitor PD98,059 (20 μM) or phosphatase inhibitor orthovanadate (30 μM) completely blocked the potentiation. The results suggest the hypothesis that the potentiation was caused by dephosphorylation of the two tyrosines in MOR. To test this hypothesis, we measured the recovery rates after insulin treatment. As predicted, tyrosine kinase inhibition by K-252a significantly slowed the reversal and phosphatase inhibition by orthovanadate significantly accelerated the recovery. These findings support a rapid modulatory role for insulin on opioid signal transduction, possibly through the dephosphorylation of the MOR at tyrosines 106 and 166 by an insulin-activated MAP kinase/protein tyrosine phosphatase cascade. We conclude that tyrosine phosphorylation of the μ-opioid receptor regulates receptor-G protein coupling efficacy.
Phosphorylation of opioid receptors at cytoplasmic serines and threonines is a crucial mechanism for regulating opioid receptor signaling (Kovoor et al., 1997; Celver et al., 2000; Koch et al., 2000; Law et al., 2000; Xiang et al., 2000). The opioid receptor is also regulated by the insulin receptor tyrosine kinase cascade (Appleyard et al., 2000). However, although tyrosine phosphorylation is known to modulate the signaling of many growth factors and cytokines (Schlessinger, 2000), relatively little is known about the effect tyrosine phosphorylation has on G protein-coupled receptor function. This is of interest, because tyrosine residues are highly conserved throughout the seven-transmembrane domain (7-TM) receptor superfamily. For example, examination of the sequence alignment of many 7-TM receptors, including the human opioid, β2-AR, 5-hydroxytryptamine1A, and melatonin stimulating hormone receptors, shows a highly conserved aspartate-arginine-tyrosine (“DRY”) motif on the edge of the second cytoplasmic loop (Seibold et al., 1998). This trend of conserved tyrosines is seen throughout receptor subfamilies as well. Sequence alignment of the μ-, δ- and κ-opioid receptors shows that they share four conserved tyrosines in similar locations in cytoplasmic space (Thompson et al., 1993). Given the importance of phosphorylation in receptor control and the highly conserved distribution of cytoplasmic tyrosines throughout the 7-TM superfamily, it seems likely that tyrosine phosphorylation may also play a role in opioid receptor regulation.
Recent evidence suggests tyrosine phosphorylation state may influence receptor trafficking and signaling. Agonist-induced down-regulation of the rat μ-opioid receptor (MOR) was reduced by 50% after inhibition of tyrosine kinase, and abolished upon mutagenesis of the four intracellular tyrosine residues (Pak et al., 1999). Likewise, phosphorylation of tyrosine 318 in the DOR was recently shown to occur after agonist treatment and to be subsequently involved in receptor activation of MAP kinase and receptor internalization (Kramer et al., 2000a,b). Insulin-receptor tyrosine kinase was shown to phosphorylate tyrosine 141 in the β2AR, resulting in an increase in β2AR-stimulated adenylyl cyclase activity (Valiquette et al., 1995). Finally, an insulin-induced potentiation of KOR-induced potassium currents was blocked by both inhibition of tyrosine kinase or point-mutation of conserved tyrosines 87 or 157 in the KOR, suggesting tyrosine phosphorylation may modulate opioid receptor signaling (Appleyard et al., 2000). These findings suggest tyrosine phosphorylation state may provide an important general mechanism for regulation of 7-TM receptor signaling.
In the present study, the impact of MOR tyrosine phosphorylation state on the activation of the G protein-gated, inwardly rectifying potassium channel (KIR3) was analyzed using theXenopus laevis oocyte expression system. BecauseX. laevis oocytes endogenously express the insulin receptor tyrosine kinase cascade (Scavo et al., 1991) as it is found in brain (Bruning et al., 2000), this was a reasonable model system for our functional study. The link between insulin-induced potentiation of the MOR response and tyrosine phosphorylation state was identified using site-directed mutagenesis and demonstrated with selective inhibitors to probably result from a dephosphorylation of MOR triggered by insulin pretreatment.
Materials and Methods
Chemicals.
DAMGO was obtained from Peninsula Laboratories (Palo Alto, CA). [3H]CTAP was supplied by Multiple Peptide Systems (San Diego, CA) through the National Institute on Drug Abuse Intramural Drug Program. K-252a and staurosporine were purchased from Calbiochem (San Diego, CA). Sodium orthovanadate was obtained from Fisher Scientific (Seattle, WA). All other chemicals were obtained from Sigma (St. Louis, MO).
Complementary DNA Clones, Mutagenesis of MOR, and cRNA Synthesis.
The MOR cDNA was obtained from Dr. Lei Yu (GenBank accession number L13069). cDNA sequences for the inwardly rectifying potassium channel (KIR)3.1 (GenBank accession number U01071) and KIR3.2 (GenBank accession number U11859) were obtained from Drs. Cesar Lebarca and Henry Lester (California Institute of Technology, Pasadena, CA). KIR3.2(S146T) was made as described previously (Rogalski et al., 2000). The rat MOR cDNA was subcloned into the HindIII site of pBluescript (Stratagene, La Jolla, CA) as described elsewhere (Celver et al., 2000). Point mutations were made in wild-type MOR to produce MOR(Y96F), MOR(Y106F), MOR(Y166F), and MOR(Y336F), using techniques described previously (Befort et al., 1996). Briefly, mutations were introduced by polymerase chain reaction amplification using Taq DNA polymerase (Life Technologies, Grand Island, NY) with complimentary pairs of oligonucleotide primers incorporating the desired mutation used to generate the substitution of the MOR cDNA. The sense oligonucleotides used in site directed mutagenesis were: ATTGTAAGATTCACCAAAATGAAGACT (MORY96F), ACATCTTCATTTTCAACCTAGCTCT (MORY106F), ATGAGCGTCGACCGCTTCATTGCT (MORY166F), and AGCTGCCTTAATCCAGTTCTTTTCGCCTT (MORY336F). All MOR cDNA templates for cRNA synthesis were amplified from corresponding mutagenized clones using a sense oligonucleotide that introduced an SP6 transcriptional recognition site: AATCTAGCATTTAGGTGACACTATAGAATAGGGGCCATGGACAGCAGCAC, and an antisense oligonucleotide that introduced a 3′poly(A) tail: T(30)AGGG- CAATGGAGCAGTTTC. All mutations were confirmed by DNA sequencing. A mMESSAGE mMACHINE SP6 kit (Ambion, Austin TX) was used to generate capped cRNA from the PCR templates.
Oocyte Culture and Injection.
Defolliculated, stage IV oocytes were prepared as described previously (Snutch, 1988). Using a Drummond automatic microinjector, cRNA was injected into oocytes (50 nl/oocyte), and oocytes were incubated at 18°C for 2 to 3 days in normal ND96 oocyte buffer (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM HEPES, pH 7.5) solution supplemented with 2.5 mM sodium pyruvate and 50 μg/ml gentamycin. Each oocyte was injected with 1 ng of MOR cRNA and either 0.1 ng KIR3.1 and KIR3.2 wild-type cRNA or 1 ng KIR3.2(S146T) pore mutant channel cRNA.
Electrophysiology.
Oocytes were clamped at −80 mV with two electrodes filled with 3 M KCl having resistances of 0.5 to 2.0 MΩ, using an Axoclamp 2A unit and pCLAMP 6 software (Axon Instruments, Foster City, CA). Membrane current traces were recorded using a chart recorder. To facilitate recording inward K currents through the KIR3 channels, the normal oocyte saline buffer was modified to increase KCl concentration to 96 mM K+ (high potassium, or hK, buffer). Correspondingly, the concentration of NaCl was decreased to maintain osmolarity. Before recording, oocytes were pretreated with normal oocyte buffer or 0.08 to 8 μM insulin, pH 7.5, for 11 to 15 min as described previously.
Whole Oocyte [3H]CTAP Binding.
Oocytes were pretreated with ND96 buffer with or without 8 μM insulin, pH 7.5, for 11 to 15 min, then used in binding experiments. Each condition tested measured total binding with three intact oocytes per tube incubated for 30 min with 50 nM [3H]CTAP (Multiple Peptide Systems, San Diego, CA), in a final volume of 100 μl of ND96 buffer. Nonspecific binding was measured with the addition of 10 μM naloxone in parallel tubes. Incubations were terminated by filtration over Whatman GF/C 25 mm circular glass microfiber filter paper (VWR Scientific, San Francisco, CA), washing three times with 3 ml of cold ND96 buffer under vacuum pressure. Filters with intact oocytes were then placed in scintillation vials with 3 ml of Ecolite scintillation fluid (ICN, Costa Mesa, CA), and bound [3H]CTAP counts measured by β-scintillation counter. All experiments were performed in triplicate, with specific binding calculated from the mean total binding minus the mean nonspecific binding.
Statistical Analysis.
EC50 values from the dose-response curves were determined using NFIT software (Island Products, Galveston, TX). Confidence intervals (95%) were used for comparison of the independent means and were plotted under standard population assumptions using the formula: {[log(SUM EC50)] − 1.98[log(SEM)], [log(SUM EC50)]+1.98[log(SEM)]} (Pagano and Gauvreau, 1993). Student's unpaired t test was used for comparison of the independent means, with significance reported as two-tailedp values.
Results
Insulin Pretreatment Potentiates the μ-Opioid Receptor Activation of KIR3.
X. laevis oocytes were coinjected with cRNA for the rat μ-opioid receptor (MOR) and the inwardly rectifying potassium channel subunits (KIR)3.1 and KIR3.2 (Fig.1). Superfusion of oocytes with hK buffer increased the basal current, representing the influx of potassium through the KIR3.1/3.2 heteromultimer (IBasal) in two-electrode voltage clamp experiments. Activation of MOR by 1 μM DAMGO, a μ-selective agonist, caused an increased influx in the potassium current as previously shown (Chen and Yu, 1994; Kovoor et al., 1997). The response was blocked by the opioid antagonist naloxone (1 μM), demonstrating the opioid-induced current (IMOR). Pretreatment of the oocytes with 8 μM insulin for 11 to 15 min activated the endogenously expressed insulin receptor and potentiated the K current evoked by DAMGO (Fig. 1).
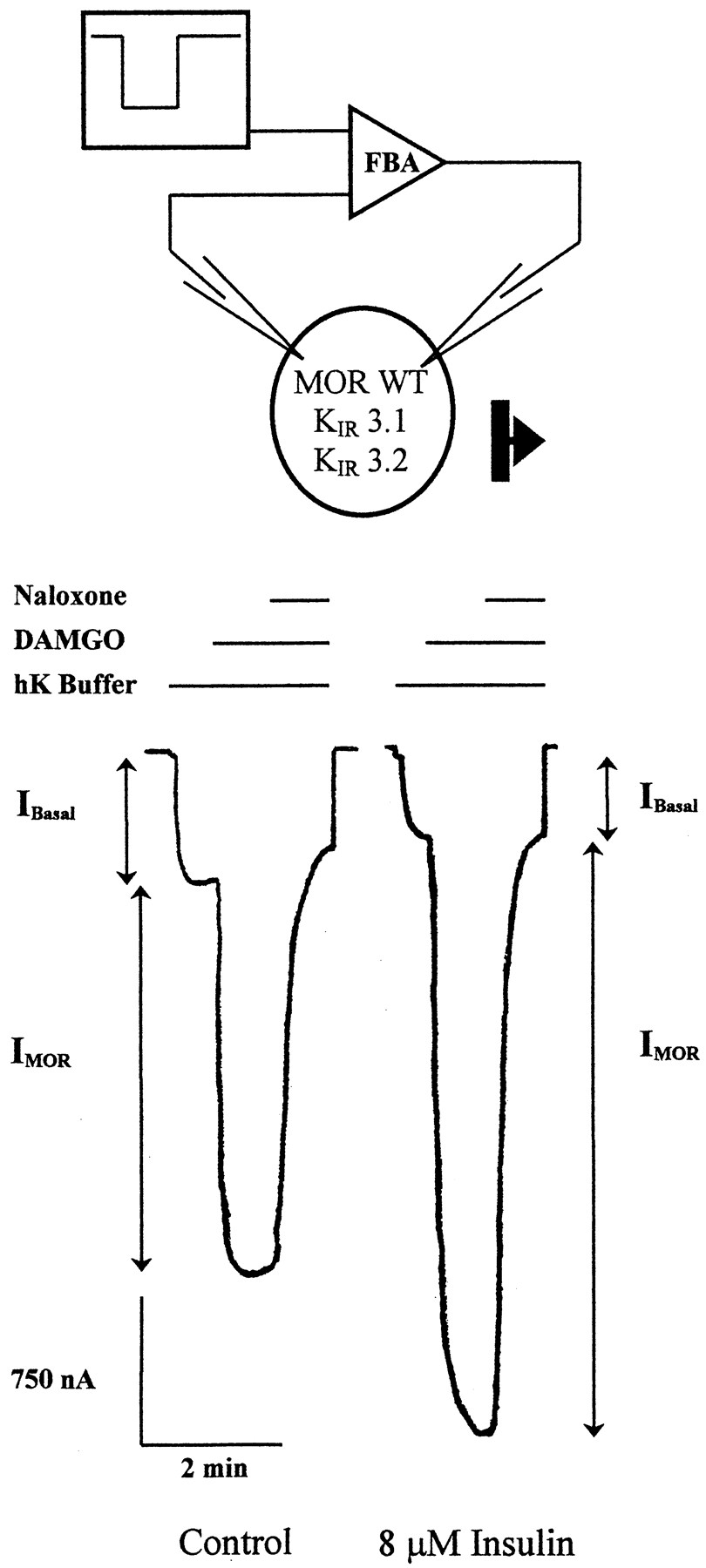
Insulin potentiates the MOR activation of KIR 3 channels. Oocytes were injected with 1 ng MOR cRNA and 0.1 ng each of KIR3.1 and KIR3.2 cRNA. Representative traces are shown from an untreated oocyte and an oocyte pretreated for 11 to 15 min with 8 μM insulin. In these experiments, the oocyte membrane potential was clamped at −80 mV while bathed in normal saline buffer. Basal K-channel currents (Ibasal) were generated by superfusion with high potassium saline buffer (hK buffer), increasing the driving force on K current flowing inward through K IR3 at −80 mV. After equilibration, oocytes were superfused with a 1 μM concentration of the μ-opioid agonist DAMGO, further increasing the inward current (IMOR). The increase in current was blocked by the opioid antagonist naloxone (1 μM).
Insulin Pretreatment Inhibits the IBasal Current through a Tyrosine Kinase-Dependent Mechanism.
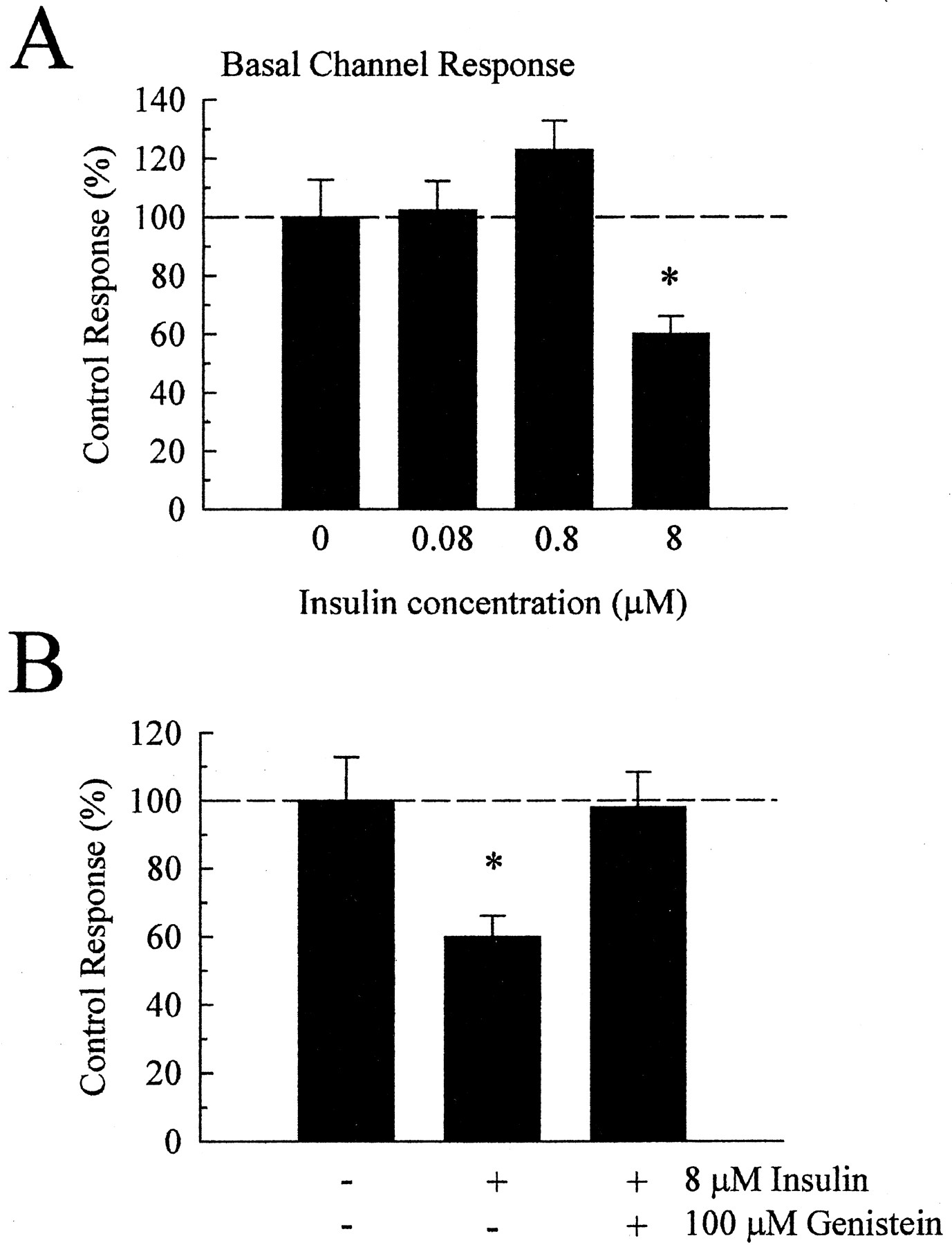
Insulin pretreatment (8 μM) also produced a significant inhibition of IBasal to 60% of untreated control cells (Fig.1). Basal current in hK was 1100 ± 140 nA in untreated oocytes (n = 24) and 660 ± 69 nA in insulin-pretreated oocytes (n = 28). The insulin-induced suppression of the basal current was statistically significant (p < 0.01) and concentration-dependent (Fig.2A). At a concentration shown to be effective in blocking the tyrosine kinase activity of the activated insulin receptor (100 μM; Akiyama et al., 1987; Wischmeyer et al., 1998), genistein completely blocked the inhibitory effect of insulin on the basal potassium currents (Fig. 2B). These results suggest that the insulin-induced inhibition of the KIR3.1/3.2 channel was mediated by tyrosine kinase activity, consistent with a previous report (Rogalski et al., 2000).

Insulin pretreatment produces a concentration-dependent inhibition of the basal channel response through tyrosine-kinase activity. Oocytes injected with cRNA for MOR and KIR3.1 and KIR3.2 were superfused with hK buffer and used once in two-electrode voltage clamp recordings as described. Multiple recordings were summarized and normalized against results from untreated oocytes of the same batch and recording day. A, pretreatment of oocytes with different concentrations of insulin produced a concentration-dependent inhibition of Ibasal, with maximal inhibition of current demonstrated after pretreatment with 8 μM insulin for 11 to 15 min. B, inhibition of Ibasalinduced by 8 μM insulin pretreatment was blocked by coincubation with the tyrosine kinase inhibitor genistein (100 μM). Data represent summarized recordings taken from 20 to 43 oocytes, drawn from three to five donors, for each bar. Differences were determined through comparison of control and insulin-pretreated conditions by Student'st test; *p < 0.01.
As both the basal channel current and the receptor induced responses were affected by insulin, we tested the effects of insulin on oocytes coexpressing MOR with a tyrosine kinase-insensitive, homomeric form of the G protein-gated, inwardly rectifying potassium channel, KIR3.2(S146T) (Vivadou et al., 1997; Rogalski et al., 2000). KIR3.2(S146T) is missing the critical tyrosine residues in the amino terminal domain of the channel where tyrosine kinase action inhibits conductance (Rogalski et al., 2000), and the S146T substitution in the pore domain of the channel facilitates expression of the homomeric form of the channel (Rogalski et al., 2000). Unlike the heteromeric KIR3 channel, the KIR3.2(S146T) channel was not sensitive to the insulin-induced inhibition of IBasal. The IBasal value of untreated oocytes was 253 ± 34 nA (n = 8), whereas the IBasal value after insulin pretreatment was 319 ± 43 nA (n = 8). In contrast, the DAMGO-evoked current in oocytes expressing the KIR3.2(S146T) channel was still potentiated by insulin pretreatment to 145% of control, with an increase in IMOR value in untreated oocytes from 490 ± 52 nA (n = 8) to 713 ± 142 nA in insulin pretreated oocytes (n = 8; p < 0.01). These results suggest that the insulin-induced potentiation of the DAMGO response was independent from insulin effects at the channel. Moreover, the genistein-sensitive inhibition of the basal current was probably caused by direct tyrosine phosphorylation of KIR3.
Insulin-Induced Potentiation of the MOR Current.
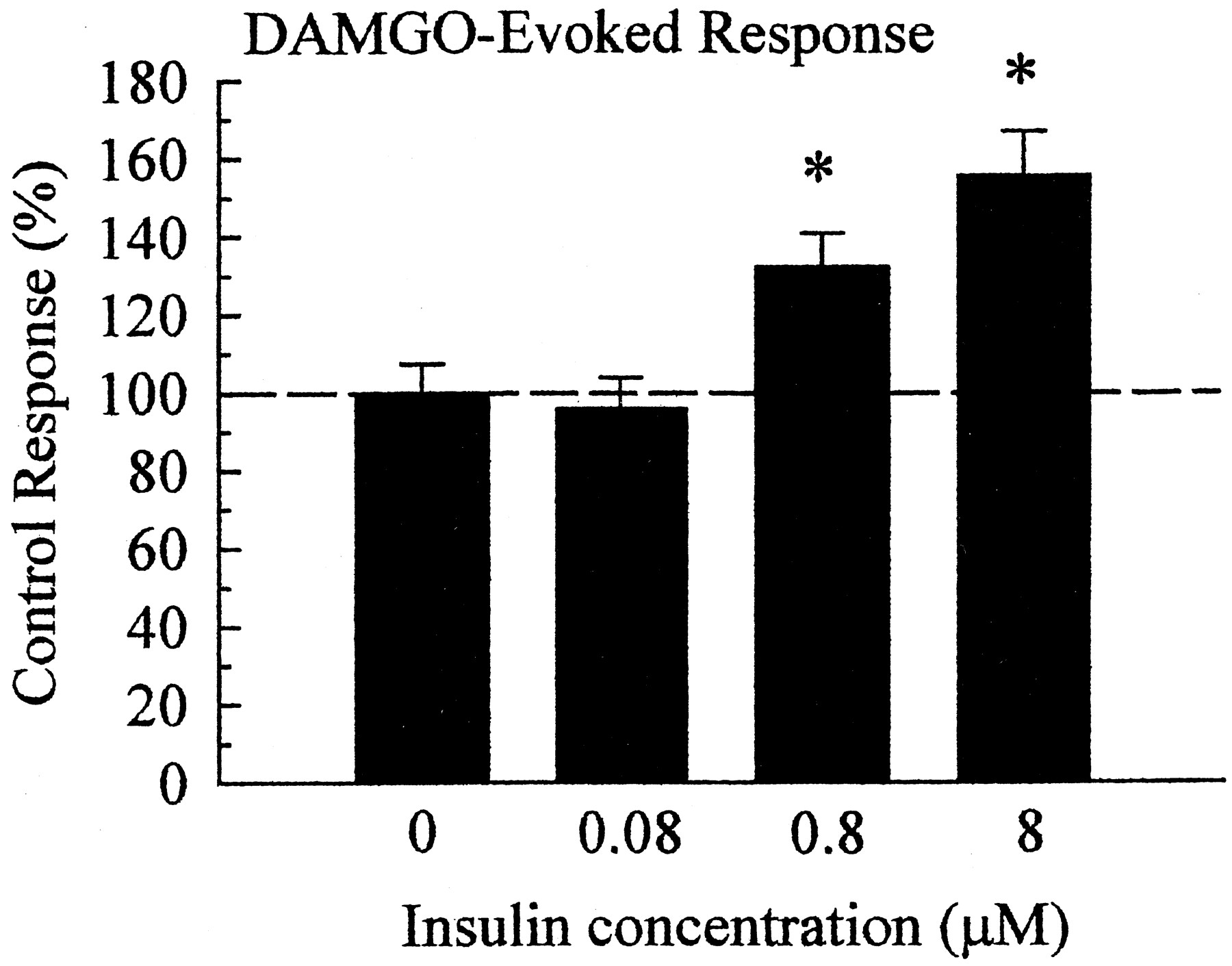
Insulin produced a concentration-dependent potentiation of the DAMGO response, reaching a statistically significant effect at 0.8 μM pretreatment, and producing a larger potentiation after 8 μM pretreatment (Fig.3). Insulin (8 μM) pretreatment significantly (p < 0.01) increased IMOR from 2900 ± 220 nA in untreated oocytes (n = 20) to 4520 ± 340 nA, or 155 ± 11.7%, after pretreatment (n = 33).

Insulin pretreatment induces a concentration-dependent potentiation of the DAMGO-induced current. Multiple recordings were summarized and normalized against results from untreated oocytes of the same batch and recording day. Pretreatment of oocytes with different concentrations of insulin produced a concentration-dependent potentiation of IMOR, with a maximal stable potentiation of current demonstrated after pretreatment with 8 μM insulin for 11 to 15 min. Pretreatment with 16 μM insulin resulted in unstable recordings, suggesting a maximal limit for insulin pretreatment. Data represent summarized recordings taken from 20 to 43 oocytes, drawn from three to five donors, for each bar. Differences were determined through comparison of control and insulin-pretreated conditions by Student's t test; *p< 0.01.
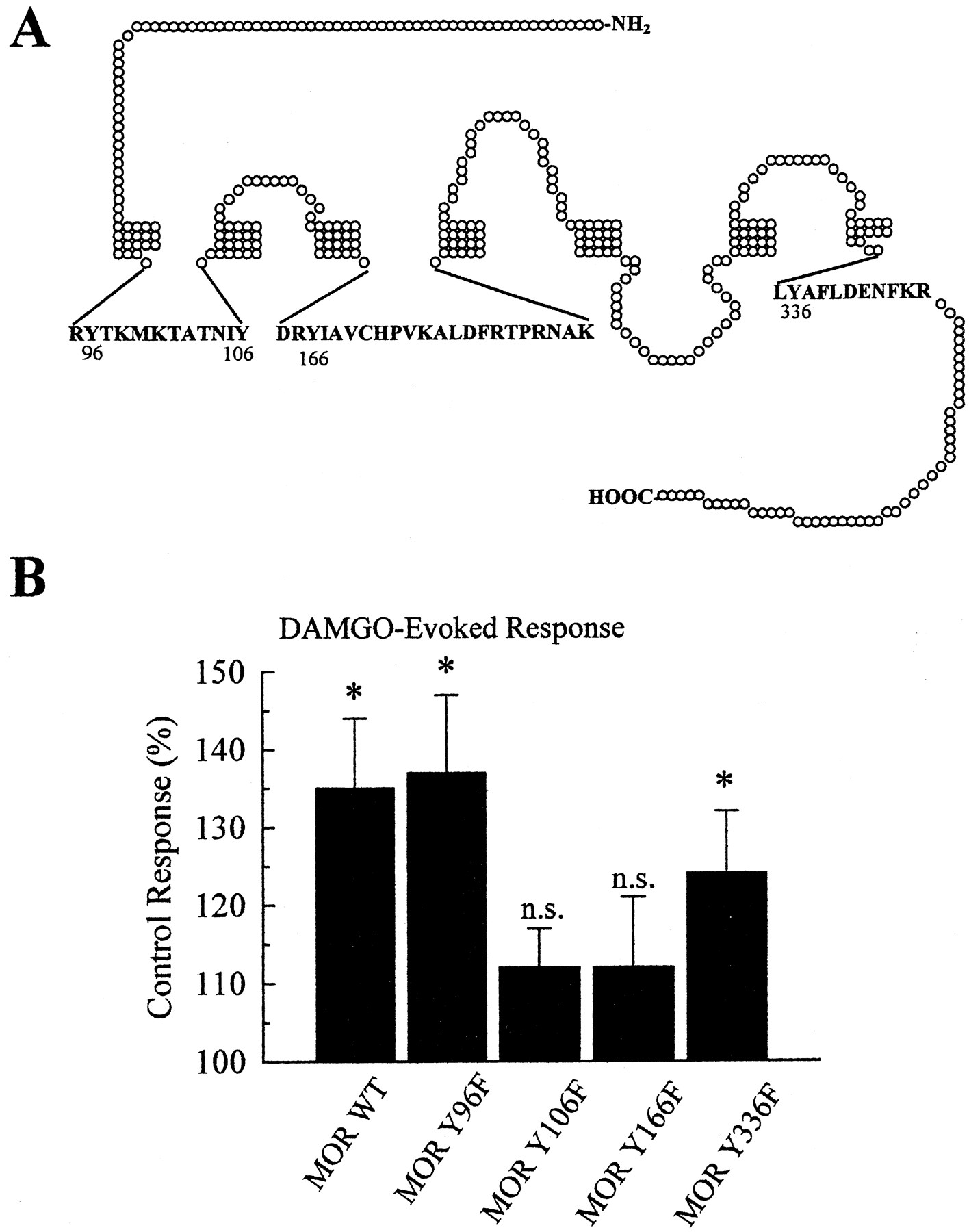
To determine the role of the four potential tyrosine phosphorylation sites in MOR, point mutants were constructed and tested (Fig.4). MOR(Y96F), MOR(Y106F), MOR(Y166F) and MOR(Y336F) were expressed in Xenopus oocytes with KIR3.1/3.2 as above. Mutation of cytoplasmic tyrosines to phenylalanines in MOR did not significantly (p > 0.1) alter the maximal response or the EC50 value of DAMGO compared with wild-type MOR values (Table 1), suggesting these residues did not affect DAMGO affinity for MOR. For this group of oocytes, insulin potentiated the response of MOR wild-type to 1 μM DAMGO to 135 ± 9% of those of the matching, untreated control cells. Similarly, oocytes expressing mutants MOR(Y96F) and MOR(Y336F) also displayed potentiation of the DAMGO response after insulin pretreatment, producing K currents of 137 ± 10% and 124 ± 8% compared with untreated control cells, respectively (Fig. 4B). In contrast, neither MOR(Y106F) nor MOR(Y166F) showed the insulin-induced potentiation of the DAMGO response. After insulin pretreatment, the response to DAMGO of MOR(Y106F) was 112 ± 5% (n= 30) of untreated control and MOR(Y166F) was 112 ± 9% (n = 30) of untreated control (Fig. 4B). Neither response was statistically different from the matched oocytes not pretreated by insulin (p > 0.05).

Site-directed mutagenesis of MOR intracellular tyrosine residues to phenylalanines produces no significant change in receptor response to DAMGO, but prevents insulin-induced potentiation of the DAMGO response. A, diagram of the MOR shows tyrosine residues in the first and second cytoplasmic loops, as well as on the edge of the cytoplasmic tail. Specific tyrosine residues Y96, Y106, Y166, and Y336 were individually mutated to phenylalanine. The accompanying table (Table 1) lists the EC50 values (and 95% confidence interval) and peak currents for the wild-type and mutated receptor response to DAMGO in two-electrode voltage clamp recordings. None of the mutant receptors produced peak currents or EC50 values that were significantly different from the wild-type values, suggesting that the Y-to-F mutations did not affect receptor expression or response to DAMGO. B, point-mutation of MOR cytoplasmic tyrosine residues 106 or 166 to phenylalanine prevented insulin-induced potentiation of the DAMGO response. Oocytes were injected with 1 ng MOR, MOR(Y96F), MOR(Y106F), MOR(Y166F), or MOR(Y336F) cRNA and 0.1 ng each of KIR3.1 and KIR3.2 cRNA. After 2 to 3 days expression, oocytes were used in two-electrode voltage clamp experiments as shown. The bar graph shows the response of oocytes to DAMGO after pretreatment with 8 μM insulin for 11 to 15 min, compared with matching, untreated controls. DAMGO-induced responses were significantly enhanced in insulin pretreated oocytes expressing the wild-type, Y96F, or Y336F MOR. However, no significant change in MOR response was seen after insulin pretreatment of oocytes expressing MOR(Y106F) or MOR(Y166F). Each bar represents the mean ± S.E.M. summarized from 24 to 31 oocytes from at least two different donors (*, significantly different from control, untreated matching oocytes, withp < 0.05 Student's t test; n.s.,p > 0.05 from control, untreated matching oocytes).
Peak DAMGO-evoked current responses and EC50 values for MOR wild-type and the four point mutants.
Insulin Pretreatment Increases Apparent Intrinsic Efficacy.
The basis of the insulin-induced potentiation was investigated by measuring the effect of insulin pretreatment on the DAMGO concentration-response curves (Fig. 5A). Pretreatment with insulin produced potentiation of the DAMGO response at each concentration tested. However, the EC50value for the insulin pretreated oocytes was not changed from the control value, with the untreated oocytes demonstrating an EC50 value (and 95% C.I). of 41.8 (19.3–90.8) nM, whereas the insulin-pretreated oocytes displayed an EC50 value of 44.5 (23.5–84.1) nM (p > 0.05).

Insulin-induced potentiation of the DAMGO response is caused by an increase in the intrinsic efficacy of the μ-opioid receptor. A, dose-response curve to DAMGO in oocytes expressing the wild-type MOR, with or without insulin pretreatment. Cumulative dose response curves were generated with two-electrode voltage clamp experiments, using oocytes first pretreated with either media (control, ●) or 8 μM insulin (▪) for 11 to 15 min. Each data point on the curves represents the mean ± S.E.M. of 9 to 16 replicates from at least two different donors. Insulin pretreatment significantly potentiated the effect of DAMGO at each concentration tested over the control dose-response curve (p < 0.05, Student'st test). B, opioid specific binding experiments with 50 nM [3H]CTAP and intact oocytes, with or without insulin pretreatment. Oocytes were first pretreated with either media (control) or 8 μM insulin for 11 to 15 min, then incubated in 50 nM [3H]CTAP for 30 min as described under Materials and Methods. Nonspecific binding was determined with the addition of 10 μM naloxone in parallel tubes. Binding was terminated by filtration over glass fiber filters. Oocytes not injected with MOR cRNA showed no specific opioid binding (leftmost bar), whereas oocytes expressing the MOR showed significantly increased opioid specific binding to [3H]CTAP (middle bar, p < 0.01, Student's t test). Insulin pretreatment of oocytes expressing MOR produced no change in the specific binding of [3H]CTAP (rightmost bar). The data suggest that the insulin-induced potentiation of the DAMGO response in 5A was not caused by an increase in receptor number but instead by an increase in intrinsic efficacy. Bars represent average specific binding ± S.E.M. from five experiments, each performed in triplicate.
This result suggested that the increased potency of DAMGO induced by insulin was not caused by an increase in the affinity of DAMGO for MOR, but instead was caused either by an increase in intrinsic efficacy of DAMGO at MOR or by up-regulation of MOR. The latter possibility was tested by pretreating oocytes expressing MOR and KIR3.1/3.2 with media or 8 μM insulin for 11 to 15 min, then performing whole-oocyte binding assays with 50 nM [3H]CTAP. As a peptide, this MOR-selective antagonist bound only the MOR expressed on the outer membrane of the oocyte, providing a measure of cell surface receptor number. Uninjected oocytes demonstrated no selective [3H]CTAP binding (Fig. 5B). Oocytes expressing the MOR showed saturable, naloxone-sensitive [3H]CTAP binding of 20.8 ± 2.6 fmol/tube. Notably, insulin pretreatment produced no change in [3H]CTAP binding, with a specific binding value of 21.2 ± 8.1 fmol/tube (n = 5 independent experiments, each performed in triplicate). Taken together, these data support the hypothesis that insulin-induced potentiation of the MOR response was not caused by a change in receptor number but instead by an increase in the intrinsic efficacy of DAMGO.
MAP Kinase and a Protein Tyrosine Phosphatase Mediate Insulin-Induced MOR Potentiation: Effects of Selective Inhibitors of Signal Transduction.
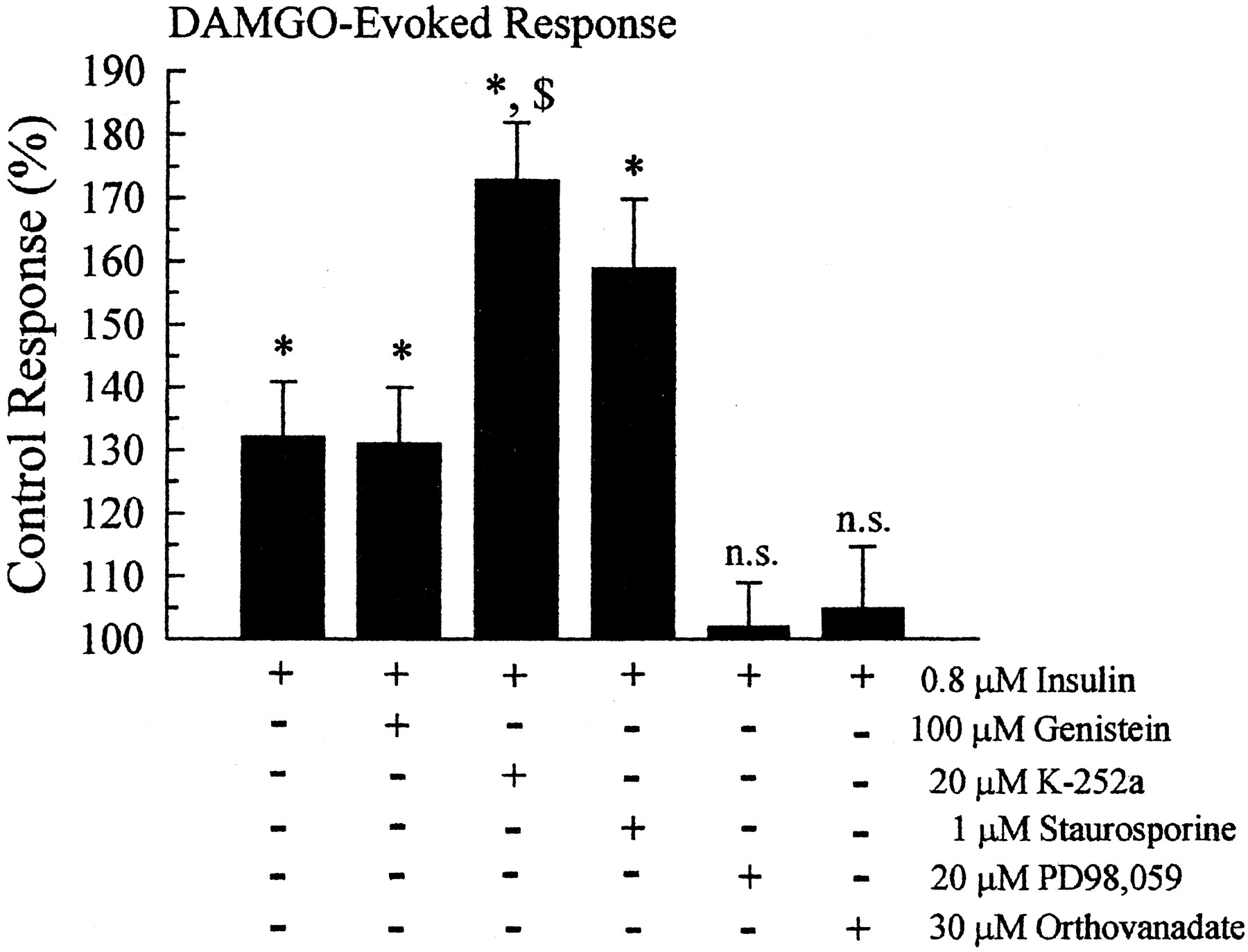
Signal transduction pathways acting on tyrosines 106 and 166 were investigated by coincubating oocytes with 0.8 μM insulin and a series of selective signal transduction inhibitors. Insulin alone produced a potentiation to 132 ± 8.8% of untreated MOR wild-type control cells (Fig.6). Surprisingly, pretreatment of oocytes for up to 1 h with tyrosine kinase inhibitors during insulin pretreatment failed to block the insulin-induced DAMGO potentiation. Genistein, at a 100 μM concentration shown to be effective at blocking insulin-induced inhibition of Kir3 basal currents in this same series of experiments (Fig. 2B), had no significant effect on insulin-induced DAMGO potentiation (Fig. 6). DAMGO-induced currents were 131 ± 9% of the untreated control values (n= 44; p < 0.05). The tyrosine kinase inhibitor K-252a (20 μM) not only failed to prevent insulin-induced potentiation of the DAMGO response, but enhanced the potentiation significantly above both the untreated and insulin-pretreated oocytes. After K-252a and insulin copretreatment, the DAMGO-evoked response was 173 ± 9.1% of the untreated oocytes (n = 24). The nonselective PKC and PKA inhibitor staurosporine (1 μM) also failed to block insulin-induced MOR potentiation; the DAMGO response was 159 ± 11% of the control response (n = 18). However, 30-min pretreatment of the oocytes with the MAP kinase inhibitor PD98,059 (20 μM) blocked the insulin-induced potentiation; the DAMGO response was 102 ± 7% of the untreated control currents (n = 19). Additionally, the protein tyrosine phosphatase inhibitor orthovanadate (30 μM) also blocked the insulin-induced potentiation; the DAMGO response was 105 ± 10% of the untreated control currents (n = 24). These results suggest the hypothesis that insulin did not potentiate the MOR response by increasing receptor tyrosine kinase activity as expected, but instead worked through the activation of the MAP kinase pathway, which in turn may have activated a protein tyrosine phosphatase.

Insulin-induced potentiation of the DAMGO response is mediated through the activation of MAP kinase and a protein tyrosine phosphatase. In parallel incubations with insulin, specific signal transduction inhibitors were added to study the signal transduction pathway mediating the effects of insulin on the MOR response. The tyrosine kinase inhibitors genistein and K-252a, as well as the PKA and PKC inhibitor staurosporine, did not prevent the potentiation of DAMGO induced by insulin. In contrast, the MAP kinase inhibitor PD98,059 and the protein tyrosine phosphatase inhibitor orthovanadate prevented insulin-induced potentiation, restoring the DAMGO response to control, untreated values. Bars represent the mean ± S.E.M. of 18 to 44 oocytes, replicates from at least two different donors. (n.s.= no significant difference from untreated controls, *, significantly different from untreated controls, p < 0.01 Student's t test; $, significantly different from insulin-pretreated oocytes, p < 0.01 Student'st test).
Manipulation of MOR Tyrosine Phosphorylation State Alters the Duration of Insulin-Induced MOR Potentiation.
The hypothesis that receptor potentiation was produced by dephosphorylation of tyrosines 106 and 166 was tested by measuring the rate of recovery after insulin pretreatment. After 11 to 15 min of pretreatment with 8 μM insulin, oocytes were incubated in insulin-free media for various times before testing the response to DAMGO. Under these conditions, the insulin-induced potentiation of the DAMGO response lasted 30 min after pretreatment, and returned to untreated control values by 1 h (Fig. 7). If insulin pretreatment produced MOR potentiation through tyrosine dephosphorylation, subsequent incubation of the insulin-pretreated oocyte with a tyrosine kinase inhibitor would be expected to extend the duration of MOR potentiation by blocking rephosphorylation of the MOR tyrosines. Likewise, subsequent incubation of the insulin-pretreated oocyte with a protein tyrosine phosphatase inhibitor would be expected to shorten the duration of MOR potentiation by facilitating rephosphorylation of the MOR tyrosines by the same kinases. In fact, incubation of insulin-pretreated oocytes with 20 μM K-252a extended the DAMGO potentiation 1.5 h beyond the normal duration, whereas incubation of insulin-pretreated oocytes with 30 μM orthovanadate hastened the recovery from DAMGO potentiation to within 15 min (Fig. 7). Together, this data supports the hypothesis that dephosphorylation of the MOR tyrosines after insulin pretreatment results in DAMGO potentiation. The data suggest that MOR phosphorylation at Y106 and Y166 by a constitutively active tyrosine kinase caused a reduction in the efficiency of DAMGO activation of MOR.

Manipulation of the tyrosine phosphorylation state on the MOR alters the duration of the insulin-induced potentiation of the DAMGO effect. DAMGO-induced currents returned to untreated baseline values 60 min after insulin pretreatment (●). In contrast, when oocytes were incubated with 20 μM K-252a added after insulin pretreatment, the duration of insulin-induced DAMGO potentiation persisted for 120 min (⋄). Additionally, when tyrosine phosphorylation was facilitated after insulin pretreatment by incubating the oocytes in the protein tyrosine phosphatase inhibitor orthovanadate (30 μM), the duration of insulin-induced DAMGO potentiation was shortened, with MOR-induced currents returning to untreated values just 15 min after pretreatment (▪). Data points represent mean ± S.E.M. of 10 to 22 oocytes from at least two different donors.

Diagram representing signal transduction pathways mediating insulin-induced potentiation of the MOR effect. Activation of a receptor tyrosine kinase (RTK) such as insulin leads to an increase in RTK activity, and the simultaneous activation of the MAP kinase (MAP K) cascade mediated by Sos activation of RAS protein. RAS phosphorylates and activates MEK, which in turn activates targets such as extracellular-signal regulated receptor kinase MAP kinase (ERK) through threonine and tyrosine phosphorylation. Either MEK or ERK kinases are thought to activate cytoplasmic PTP. PTP may then dephosphorylate conserved cytoplasmic tyrosine residues on 7-TM-spanning receptors, such as the opioid receptor. The tyrosine dephosphorylation may serve to increase intrinsic receptor efficacy, thereby potentiating receptor induced K currents mediated by the inwardly-rectifying potassium channel (KIR).
We previously reported that oocytes expressing the κ-opioid receptor (KOR) instead of MOR also showed insulin-induced potentiation but by a different mechanism (Appleyard et al., 2000). To compare results directly, pretreatment here with 8 μM insulin also produced a significant KOR potentiation of K currents to 149 ± 15% of those of the untreated control cells, which returned to baseline values after 1 h (107 ± 9%, n = 7 oocytes for each time point). However, when insulin-pretreated oocytes expressing KOR were subsequently incubated in 20 μM K-252a for 1 h before recording, the oocytes demonstrated U69,593-induced K currents of 106 ± 11%, representing a return to control values (n = 7). The failure of tyrosine kinase inhibitors to extend the duration of KOR potentiation supports the hypothesis that insulin-induced potentiation of the KOR was caused by tyrosine phosphorylation, as suggested earlier (Appleyard et al., 2000). Moreover, the finding suggests that κ- and μ-opioid receptors are both regulated by tyrosine phosphorylation, but with different consequences. The reason for this difference between MOR and KOR remains unclear, but may lie in the structural differences between the receptors themselves and their differential ability to interact with the insulin receptor.
Discussion
This study has two principal findings. First, insulin receptor activation reduced KIR3 channel current. Incubation with 8 μM insulin inhibited the channel, although an increase in tyrosine kinase activity, as coincubation with genistein blocked the effect. Secondly, pretreatment with 0.8 μM insulin potentiated the MOR-induced response. The potentiation of the opioid response required tyrosines 106 and 166 in the MOR, and coincubation with selective kinase and phosphatase inhibitors demonstrated that the potentiation required MAP kinase and protein tyrosine phosphatase activity. These results suggest that tyrosine phosphorylation state of both the receptor and effector may regulate opioid responses.
The inhibitory effects of insulin on the KIR3 channel are similar to the reported effects of tyrosine kinases on other potassium channels. Insulin induced an acute inhibition of KIR2.1 channels in both tsA-201 cells andX. laevis oocytes that was blocked by genistein (Wischmeyer et al., 1998). Similarly, constitutively active, nonreceptor tyrosine kinase v-Src increased tyrosine phosphorylation of the Kv1.3 channel protein and reduced channel activity 95% (Fadool et al., 1997). Insulin receptor tyrosine kinase also suppressed Kv1.3 currents and induced channel protein phosphorylation, and mutation of Kv1.3 tyrosines 111 to 113, 137 and 479 to phenylalanine abolished both channel phosphorylation and suppression induced by insulin (Fadool et al., 2000). Brain-derived natriuretic factor-activation of the receptor tyrosine kinase TrkB suppressed KIR3 channel current up to 70% after directly increasing the phosphorylation of KIR3 channel tyrosine residues (Rogalski et al., 2000). In a previous study, we reported insulin had no effect on KIR3 basal currents when coexpressed with the KOR (Appleyard et al., 2000). An explanation for this discrepancy is not clear; however we attribute the difference to the levels of expression and seasonal differences in insulin receptor expression. Because the homomeric KIR3.2 was insensitive to insulin treatment in this present study and insensitive to brain-derived natriuretic factor treatment (Rogalski et al., 2000), the results suggest that insulin also increased KIR3 tyrosine phosphorylation at sites in the amino terminal domain of the channel. However, this was not directly demonstrated in this study.
The receptor mutagenesis results suggest that the potentiation resulted from a change in the phosphorylation state of MOR. Insulin receptor kinase cascades have been demonstrated to phosphorylate tyrosines of other receptor proteins, such as the β2AR (Karoor et al., 1998). Moreover, evidence supports the role of tyrosine phosphorylation in regulating G-protein coupled receptors (Valiquette et al., 1995; Appleyard et al., 2000). Unfortunately, direct measures of phosphorylation of specific residues in MOR were not feasible. With approximately 7 fmol of MOR expressed per oocyte, we were unable to resolve the phosphopeptide fragments derived from immunoprecipitated receptor.
The present data suggest that the dephosphorylation rather than phosphorylation of the tyrosines 106 and 166 was responsible for the potentiation observed. At a concentration shown previously to be selective for protein tyrosine phosphatases (PTP) (Swarup et al., 1982), we observed that orthovanadate also blocked the insulin-induced DAMGO potentiation and accelerated the recovery after washout of insulin. In contrast, tyrosine kinase inhibition by K-252a delayed the recovery. The accelerated recovery presumably occurred by enabling endogenous tyrosine kinases to rephosphorylate MOR, whereas kinase inhibition presumably extended the duration of potentiation by blocking rephosphorylation. This suggests that the DAMGO potentiation was produced by an insulin-induced activation of protein tyrosine phosphatase.
The MAP kinase inhibitor PD98,059 effectively blocked DAMGO potentiation at a concentration previously reported to be selective for MEK 1/2 (Alessi et al., 1995). Prior reports document that the insulin receptor cascade activates MAP kinase through a Ras/Raf/MEK 1/2 pathway (Taha and Klip, 1999). The basis for the PD98,059 effect was not established in this study and could result from a direct or indirect action of the MAP kinase cascade on MOR. Direct connections between the MAP kinase cascade and the opioid receptor are becoming increasingly clear (Polakiewicz et al., 1998; Bohn et al., 2000; Schmidt et al., 2000; Trapaidze et al., 2000). Hypothetically, PD98,059 may directly inhibit the insulin effect by blocking the insulin-activated MEK 1/2 phosphorylation of MOR. MOR contains a conserved TNIY region (T103 to Y106) in the first cytoplasmic loop that is similar to the T-X-Y recognition motif for MEK 1/2 (Taha and Klip, 1999). However, a more parsimonious explanation is that activation of MAPK by the insulin receptor cascade subsequently increases tyrosine phosphatase activity. Consistent with this hypothesis, the MAP kinase cascade activated protein phosphatase-1 in rat skeletal muscle cells (Ragolia and Begun, 1998), and in yeast, MAP kinase Hog1p regulates tyrosine phosphatases PTP2p and PTP3p (Wurgler-Murphy et al., 1997; Keyse, 1998). Thus, the insulin-activated MAP kinase cascade might indirectly dephosphorylate MOR.
Relatively little is known about the role of phosphatases in regulating opioid activity. A non–calcium-dependent protein phosphatase reversed homologous desensitization of MOR-induced currents in whole cell and intracellular recordings from locus ceruleus neurons (Osborne and Williams, 1995), and PMA-induced desensitization of opioid receptor currents was reversed by calcineurin, a protein phosphatase 2B (Ueda et al., 1995). Additionally, inhibition of protein phosphatases 1 and 2A with okadaic acid in opioid naı̈ve tissue caused an opioid-induced formation of cAMP as occurs after chronic in vivo morphine exposure (Wang et al., 1996). However, less is known about the role of tyrosine phosphatases in the regulation of G protein-coupled receptor activity. A recent report demonstrated blockade of PTP caused a pronounced inhibition of Ca2+ currents specifically stimulated by β-AR receptor in guinea pig ventricular myocytes, supporting the present study by suggesting that basal tyrosine kinase activity may be capable of inhibiting β-AR responses (Sims et al., 2000). Interestingly, the conserved amino acid sequence surrounding tyrosine 166, VDRYIA in MOR, has similarities to the key amino acids conferring PTP substrate specificity, DADEpYLIPQQG (Zhang et al., 1993) and may allow PTP to bind the second cytoplasmic loop of MOR.
There are several possible cellular mechanisms that might account for the increased response of the dephosphorylated MOR to agonist activation. First, insulin pretreatment might increase the number of μ-opioid receptors on the membrane surface. However, this hypothesis was excluded because [3H]CTAP binding to intact oocytes revealed no change in MOR number. Secondly, insulin pretreatment might have altered the agonist affinity. This seems unlikely, because dose response curves measuring DAMGO-induced currents showed no change in DAMGO EC50 value. Therefore, the increase in response was probably caused by an increase in intrinsic efficacy. A molecular definition of intrinsic efficacy is not yet established but a plausible explanation would be an increase in the efficiency of G protein coupling. Phosphorylation of the cytoplasmic region of the receptor responsible for G protein binding would be expected to affect the interaction. A direct effect on G protein binding affinity is one possibility. An indirect effect of insulin would result if dephosphorylation of MOR(Y106) and (Y166) reduced the binding of a competing protein. Precedence for this hypothesis exists, as the “DRY” motif in α1AR andN-formyl peptide receptors were shown to be involved in the interaction with β-arrestin (Mhaouty-Kodja et al., 1999; Bennett et al., 2000). Consistent with this hypothesis, mutations of the aspartate within the DRY motif have produced constitutively active G protein-coupled receptors (Scheer et al., 1996; Huang et al., 2000;Rhee et al., 2000). Thus, the concept that changes in phosphorylation of this receptor domain controls the efficiency of G protein activation seems plausible.
The physiological relevance of these findings remains to be established. Opioid receptors are expressed along with insulin receptor kinases and other growth factors in many different regions, both centrally and peripherally. This coexpression could provide the basis for interactions between the two signaling systems. Recent studies demonstrated that intraventricular insulin reduced the antinociceptive effect of DAMGO in mice through a mechanism involving a tyrosine kinase activation of PKC (Kamei et al., 1998; Ohsawa et al., 1999). This limited evidence suggests insulin may modulate opioid-induced antinociception in vivo.
In conclusion, this study shows that insulin potentiates the coupling of MOR to KIR3. This potentiation occurred through activation of both a MAP kinase cascade and a protein tyrosine phosphatase, and required specific conserved tyrosine residues in the first and second intracellular loops of the MOR. These results suggest a novel mechanism by which modulation of the tyrosine phosphorylation state of opioid receptors regulates intrinsic efficacy.
Acknowledgments
We thank Dr. Sherri Rogalski (Department of Pharmacology, University of Washington, Seattle, WA) for the helpful gift of KIR3.2(S146T) mutant cRNA.
Footnotes
- Received January 29, 2001.
- Accepted March 5, 2001.
-
Send reprint requests to: Dr. Charles Chavkin, Department of Pharmacology, University of Washington, Box 357280, Seattle, WA 98185-7280. E-mail: cchavkin{at}u.washington.edu
-
This work was supported by U. S. Public Health Service Grants DA07278 and DA11672 from the National Institute of Drug Abuse.
Abbreviations
- 7-TM
- seven-transmembrane domain
- AR
- adrenergic receptor
- MOR
- rat μ-opioid receptor
- KIR3
- G protein-activated inwardly rectifying potassium channel
- DAMGO
- [d-Ala2,methyl-Phe4,Gly5-ol]enkephalin
- [3H]CTAP
- d-Phe(3H)-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2(cyclic)
- hK
- high potassium
- MAP
- mitogen-activated protein
- PD 98,059
- 2′-amino-3′methoxyflavone
- KOR
- κ-opioid receptor
- PTP
- protein tyrosine phosphatase
- MEK
- mitogen-activated protein kinase kinase
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}