


Abstract
β-Adrenergic receptor stimulation regulates the activity of several different cardiac ion channels through an adenylate cyclase/cAMP/protein kinase A-dependent mechanism. Previous work has suggested that basal tyrosine kinase activity attenuates the β-adrenergic responsiveness of these cardiac ion channels, supporting the idea that tyrosine phosphorylation exerts an inhibitory effect at some point in the common signaling pathway. To determine which element in the β-adrenergic pathway is regulated by tyrosine kinase activity, we studied the effects of various protein tyrosine phosphatase (PTP) inhibitors on the cAMP-dependent regulation of the L-type Ca2+ current in guinea pig ventricular myocytes. Three such compounds, sodium orthovanadate, peroxovanadate, and bpV(phen), had no effect on the basal Ca2+ current, yet each caused a pronounced inhibition of the Ca2+ current stimulated by the β-adrenergic receptor agonist isoproterenol. These observations are consistent with the idea that basal tyrosine kinase activity is capable of inhibiting β-adrenergic responses. However, these PTP inhibitors had no effect on cAMP-dependent stimulation of the Ca2+ current via activation of adenylate cyclase with forskolin or activation of H2-histaminergic receptors with histamine. These results are consistent with the idea that inhibition of PTP activity produces an inhibitory effect involving a tyrosine kinase-dependent mechanism acting selectively at the level of the β-adrenergic receptor. This signaling mechanism does not seem to be linked to tyrosine kinase activity associated with insulin and insulin-like growth factor receptors, because acute exposure to agonists of these receptors did not inhibit isoproterenol regulation of the Ca2+ current.
In the heart, binding of catecholamines to β1-ARs can regulate the activity of a variety of ion channels (Hartzell, 1988). This is caused by activation of a common signaling pathway that involves Gs-dependent stimulation of adenylate cyclase, cAMP-dependent activation of PKA, and serine/threonine-dependent protein phosphorylation. The cardiac L-type Ca2+, delayed rectifier K+, and the CFTR Cl− channels are all regulated by this signaling pathway.
Phosphorylation of tyrosine residues by PTKs represents another means of modulating target proteins. Although the role of protein tyrosine phosphorylation in the regulation of cell growth and differentiation has been extensively characterized, only more recently has attention been focused on the acute modulation of ion channel activity by PTKs. Activation of G protein-coupled receptors as well as growth factor receptors have been found to regulate the activity of a variety of ion channels through PTK-dependent signaling pathways (Huang et al., 1993;Chik et al., 1997; Bowlby et al., 1997; Hilborn et al., 1998; Hu et al., 1998). PTK activity independent of receptor stimulation has also been implicated in the regulation of L-type Ca2+channel activity in a variety of preparations, including cardiac myocytes. Evidence for this includes the ability of PTK inhibitors to attenuate and PTP inhibitors to enhance basal channel activity (Cataldi et al., 1996; Wijetunge et al., 1998; Hu et al., 1998; Wang and Lipsius, 1998; Ogura et al., 1999). This suggests that basal PTK activity exerts a stimulatory effect on L-type Ca2+ channel activity, although it is not clear whether such effects are caused by direct phosphorylation of the channel protein.
In addition to possibly having a direct stimulatory effect, basal PTK activity also seems to exert an inhibitory effect on L-type Ca2+ channel activity in cardiac myocytes that is caused by antagonism of β-adrenergic responses. Previous work from our laboratory has demonstrated that the PTK inhibitor genistein enhances the β-adrenergic sensitivity of not only L-type Ca2+ channels, but also delayed rectifier K+ channels and CFTR Cl−channels (Hool et al., 1998). Such results suggest that basal PTK activity exerts an indirect inhibitory effect on the β-adrenergic signaling pathway regulating these cardiac ion channels, rather than a direct effect on the channels themselves. However, the point in the β-adrenergic-signaling pathway at which PTK activity may exert an inhibitory effect is unknown. The goal of the present study was to verify the role of basal PTK activity in regulating the β-adrenergic responsiveness of cardiac ion channels and to determine where in the signaling pathway inhibitory modulation is mediated.
Materials and Methods
Cell Isolation.
Single ventricular myocytes were isolated from adult Hartley guinea pigs (250–350 g) by a modification of a method described previously (Hool et al., 1998). Briefly, hearts were excised from animals of either sex that had been anesthetized by an intraperitoneal injection of pentobarbital (150 mg/kg). Coronary arteries were perfused via the aorta with a PSS containing: 140 mM NaCl, 5.4 mM KCl, 1.5 mM CaCl2, 2.5 mM MgCl2, 11 mM glucose, and 5.5 mM HEPES, pH 7.4. The heart was first perfused with this Ca2+-containing PSS for 5 min. The solution was then switched to a nominally Ca2+-free PSS for another 5 min, after which enough collagenase B (Boehringer Mannheim) was added to achieve a final concentration of 0.27 to 0.33 mg/ml. After 30 min of digestion at 36°C, the ventricles were removed and placed in a Kraft-Brühe solution containing 110 mM potassium glutamate, 10 mM KH2PO4, 25 mM KCl, 2 mM MgSO4, 20 mM taurine, 5 mM creatine, 0.5 mM EGTA, 20 mM glucose, and 5 mM HEPES, pH 7.4. The tissue was then minced, and single myocytes were obtained by filtering through 100-μm nylon mesh. Cells were allowed to settle, the supernatant was aspirated, and the pellet was resuspended in Ca2+-containing PSS. Experiments were performed on the day of cell isolation only. The methods used in this procedure are in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University.
Data Acquisition and Analysis.
The β-adrenergically regulated L-type Ca2+ and CFTR Cl− currents were studied using the conventional whole cell configuration of the patch clamp technique (Hamill et al., 1981). Microelectrodes were pulled from borosilicate glass capillary tubing (Corning 7052; Garner Glass, Claremont, CA) and had resistances between 0.5 and 1.5 MΩ when filled with an intracellular solution containing130 mM CsCl, 20 mM tetraethylammonium chloride, 5 mM MgATP, 5 mM EGTA, 0.1 mM Tris-GTP, and 5 mM HEPES, pH 7.2. Cells were bathed in a K+-free control extracellular solution containing 140 mM NaCl, 5.4 mM CsCl, 2.5 mM CaCl2, 0.5 mM MgCl2, 11 mM glucose, and 5.5 mM HEPES, pH 7.4. Macroscopic currents were recorded using an Axopatch 200 voltage-clamp amplifier (Axon Instruments, Foster City, CA) and an IBM-compatible computer with a Digidata 1200 interface and pCLAMP software (Axon Instruments).
Isolated myocytes were placed in a 0.5-ml chamber on the stage of an inverted microscope where they were superfused with either control or drug containing solutions at a rate of 1 to 2 ml/min. All experiments were performed at room temperature. The L-type Ca2+ current was isolated by blocking K+ channels with Cs and tetraethylammonium, inactivating Na+ channels with a voltage clamp prepulse step to −30 mV, and eliminating the driving force for Cl− currents by measuring the Ca2+ current near the adjusted Cl− equilibrium potential. The time course of changes in the size of the Ca2+ current was monitored by measuring the absolute magnitude of the peak inward current recorded during 100-ms voltage-clamp steps to 0 mV applied after a 40-ms prepulse to −30 mV from a holding potential of −80 mV once every 5 s.
When recording the CFTR Cl− current, CsCl in the intracellular solution was replaced with Cs-glutamate, and the L-type Ca2+ current was blocked by adding 1 μM nisoldipine to all extracellular solutions. The time course of changes in Cl− conductance was monitored by applying 100 ms voltage steps to +50 mV from a holding potential of −30 mV once every 3 s. The Cl− current was defined as the agonist-induced difference current obtained by subtracting currents recorded in the absence of drugs from those recorded in the presence of drugs.
Responses to Iso and histamine were measured at steady state. Responses to PTP inhibitors, insulin, and IGF-1 were measured after exposure to drugs for 5 min. Results are reported as the mean ± S.E. of at least three independent experiments. Statistical comparisons between two groups of experimental data were performed using the paired Student's two-tailed t test where indicated.
Drugs and Reagents.
Sodium orthovanadate (vanadate) was added during the preparation of extracellular solutions before adjusting the pH to 7.4. Iso, histamine, insulin, IGF-1, and bpV(phen) (Calbiochem, San Diego, CA) were prepared as aqueous stock solutions and later diluted in extracellular solution to achieve the desired final concentration. Ascorbic acid (50 μM) was added to extracellular solutions to maintain the stability of Iso. Nisoldipine (a gift from Miles Laboratories, West Haven, CT) and forskolin were prepared as stock solutions in polyethylene glycol (Mr 400). The final concentration of polyethylene glycol never exceeded 0.1%, a concentration that by itself had no effect on basal currents. Insulin stock solutions (1 mM) were prepared in 2.5 mM HCl and diluted to a working concentration of 100 nM in extracellular solution. PVN was prepared by combining 10 mM H2O2 and 10 mM Na3VO4 in an aqueous solution containing 50 mM HEPES, pH 7.4. This mixture was allowed to stand at room temperature for 15 min, after which time, excess H2O2 was eliminated by incubating the mixture in the presence of 200 μg/ml catalase for 15 min before further dilution in extracellular solution. The resulting stock solution contained a mixture of peroxovanadium complexes (Posner et al., 1994). The final concentration of PVN used in our experiments is based on the concentration of vanadate used in preparing the stock solution. To control for potential nonspecific effects of catalase or any remaining H2O2, it was first verified that solutions containing H2O2 and catalase alone had no effect on agonist responses. All solutions containing Iso and PVN were stored in light resistant containers. All drugs were obtained from Sigma/Research Biochemicals International, except where noted.
Results
Effect of Vanadate on L-type Ca2+ Channel Regulation.
The β-adrenergic agonist Iso, acting through the cAMP/PKA signaling pathway, is a potent activator of L-type Ca2+ channel activity in cardiac ventricular myocytes (Kameyama et al., 1985). Previous studies from our laboratory have shown that the PTK inhibitor genistein increases the sensitivity of Ca2+ channels to β-adrenergic stimulation (Hool et al., 1998). To further characterize potential PTK-dependent modulation of cardiac signaling mechanisms, we examined the effect of the PTP inhibitor sodium orthovanadate (vanadate) on Iso-stimulated Ca2+ channel activity in guinea pig ventricular myocytes. To determine whether vanadate exhibits effects on L-type Ca2+ channels that are independent of β-adrenergic stimulation, we also monitored its effect on basal channel activity. As demonstrated in Fig.1, exposure to 1 mM vanadate alone consistently had no effect on basal Ca2+ channel activity. The same result was obtained independent of whether myocytes were exposed to extracellular solution containing 1 mM vanadate at the beginning (data not shown) or end of an experiment. After exposure to vanadate for 5 min, the magnitude of the current was 92 ± 4.4% (n = 10) of that observed before exposure to vanadate. The small decrease is consistent with current rundown.

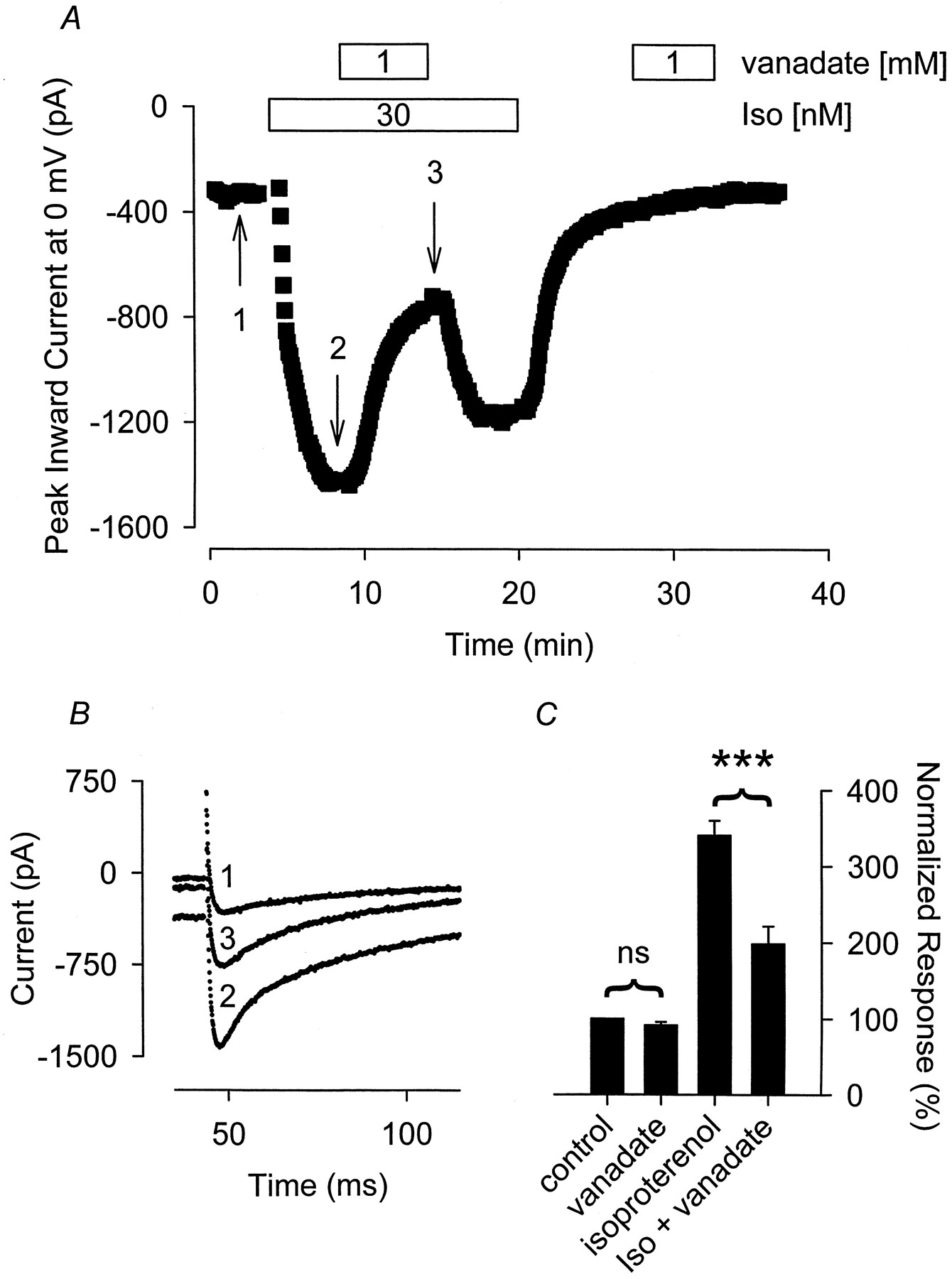
The tyrosine phosphatase inhibitor sodium orthovanadate (vanadate) selectively antagonizes the Iso-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 30 nM Iso alone, Iso plus 1 mM vanadate and vanadate alone. B, current traces recorded at time points indicated in A. Note, changes in prepulse current at −30 mV reflect parallel regulation of the time-independent CFTR Cl− current present in this cell. C, cumulative responses (n = 10–13) normalized to the magnitude of the Ca2+ current recorded under control conditions. ***, the magnitude of the current measured in the presence of Iso plus vanadate was significantly smaller than the magnitude of the current measured in the presence of Iso alone (P < .001). ns, the magnitude of the current measured in the presence of vanadate alone was not significantly different from the magnitude of the current measured in the absence of vanadate.
To assess the potential role of tyrosine phosphatase inhibition on the β-adrenergic responsiveness of the L-type Ca2+channel, we monitored the effect that 1 mM vanadate had on the Ca2+ current stimulated by 30 nM Iso. This concentration of Iso has been demonstrated to produce submaximal stimulation of the L-type Ca2+ current in guinea pig ventricular myocytes (Kameyama et al., 1985). In our hands, exposure to 30 nM Iso increased the Ca2+ current an average of 218 ± 14% (n = 23) over baseline. Superfusion of myocytes with 1 mM vanadate in the presence of 30 nM Iso produced a pronounced inhibition of the peak Ca2+current recorded with Iso alone (Fig. 1). In these cells, addition of 1 mM vanadate inhibited the Iso response by 61 ± 7.6% (n = 13, P < .001). The inhibitory effect of vanadate was at least partially reversible. After washout of vanadate, the Iso response was restored to 63 ± 13% (n = 7) of that observed before vanadate exposure.
The fact that vanadate was able to inhibit the L-type Ca2+ current in the presence, but not the absence of β-adrenergic stimulation, is consistent with the idea that vanadate acts at some point in the β-adrenergic signaling pathway, but not at the level of the channel itself. To assess where in the signaling cascade vanadate might be inhibiting β-adrenergic regulation of the Ca2+ current, we examined its effect on the Ca2+ current activated by direct stimulation of adenylate cyclase with forskolin. Exposure of myocytes to 3 μM forskolin increased the peak inward Ca2+ current by an average value of 260 ± 36% (n = 10) over control values. However, subsequent exposure of cells to 1 mM vanadate in the continued presence of forskolin consistently had no effect Ca2+ channel activity. The magnitude of the Ca2+ current measured in the presence of forskolin plus vanadate was 98 ± 2.4% (n = 5, P > .05) of that measured in the presence of forskolin before exposure to vanadate. The lack of effect of vanadate on the forskolin stimulated L-type Ca2+ current demonstrates that vanadate does not exert an inhibitory effect directly on the channel, even when it has been activated by PKA-dependent phosphorylation. It also suggests that vanadate is exerting its inhibitory effect upstream of adenylate cyclase in the β-adrenergic signaling pathway.
To further elucidate the likely point at which vanadate exerts its inhibitory action, we monitored the effect of this PTP inhibitor on the Ca2+ current stimulated by histamine. Histamine regulates cardiac L-type Ca2+ channel activity by interacting with H2 histaminergic receptors, which are coupled to adenylate cyclase via the same stimulatory G protein linking β-ARs to adenylate cyclase (Hescheler et al., 1987). Exposure to 300 nM histamine increased the Ca2+current an average of 219 ± 20% (n = 17) over control. Although the magnitude of this response was comparable with that produced by 30 nM Iso, exposure to 1 mM vanadate in the continued presence of histamine had no effect on the histamine stimulated Ca2+ current. The magnitude of the Ca2+ current measured in the presence of histamine plus vanadate was 105 ± 2.6% (n = 8,P > .05) of that measured in the presence of histamine before exposure to vanadate (Fig. 2). Because the only difference between the signaling pathways mediating Iso and histamine responses is the receptor through which they act, these data strongly suggest that the PTP inhibitor vanadate exerts its inhibitory effect at the level of the β-AR.

The tyrosine phosphatase inhibitor sodium orthovanadate (vanadate) has no effect on the hist-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 300 nM histamine alone and histamine plus 1 mM vanadate. B, representative current traces recorded at the time points indicated in A. Note, changes in prepulse current at −30 mV reflect parallel regulation of the time-independent CFTR Cl− current present in this cell. C, cumulative responses (n = 8) normalized to the magnitude of the peak Ca2+ current recorded under control conditions. ns, the magnitude of the current measured in the presence of vanadate plus histamine was not significantly different from the magnitude of the current measured in the presence of histamine alone.
Effect of PVN on L-type Ca2+ and CFTR Cl−Channel Regulation.
Although the ability of vanadate to inhibit Iso stimulation of the Ca2+ current is consistent with our previous observation suggesting that basal PTK activity exerts an inhibitory effect on β-adrenergic regulation of cardiac ion channels, vanadate is known to exert a number of other effects in addition to PTP inhibition. Furthermore, others have reported that vanadate has no effect on the Ca2+ current activated by Iso (see Wang and Lipsius, 1998). Therefore, one concern is that the apparent inability of vanadate to inhibit forskolin and histamine responses might simply be caused by the variability of its inhibitory action, rather than a true absence of inhibitory response. To address this point, we evaluated the effects of another more potent PTP inhibitor, PVN (Bevan et al., 1995).
As demonstrated in Fig. 3, application of 100 μM PVN alone had no significant effect on basal Ca2+ channel activity. The magnitude of the Ca2+ current recorded in the presence of 100 μM PVN was 89 ± 3.0% (n = 5) of that measured before exposure to PVN. Again, the slight decrease could be explained by current rundown, which was evident even in the absence of PVN. To determine whether inhibition of PTP activity affects β-adrenergic mediated ion channel activity, we examined the effects of PVN on the Iso-stimulated Ca2+ current. Because preparation of PVN involved the use of H2O2 and catalase (see under Materials and Methods), we first determined whether this mixture alone had any effect on the Iso-stimulated current. Exposure of myocytes to a catalase and H2O2 containing solution had no effect on the response to Iso. The current measured in the presence of Iso plus catalase and H2O2 was 97 ± 7.3% (n = 3) of that measured in the presence of Iso before exposure to catalase and H2O2. This is consistent with the reported inability of this catalase and H2O2 mixture to inhibit Iso stimulated Cl− channel activity (Hool et al., 1998). However, exposure to 100 μM PVN consistently resulted in a significant inhibition of the Iso-stimulated Ca2+current (Fig. 3). Superfusion of myocytes with 100 μM PVN in the presence of 30 nM Iso inhibited the Iso stimulated current by 73 ± 6.4% (n = 5, P < .01), and this inhibitory effect was at least partially reversible. After washout of PVN, the Iso response was restored to 51 ± 7.3% of that observed before PVN exposure. This ability of PVN to inhibit the Iso-stimulated Ca2+ current is consistent with the previous report that PVN also inhibits the Iso-stimulated Cl− current in these same cells (Hool et al., 1998).

The tyrosine phosphatase inhibitor PVN selectively antagonizes the Iso-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 30 nM Iso alone, Iso plus 100 μM PVN, and PVN alone. B, current traces recorded at time points indicated in A. C, cumulative responses (n = 5) normalized to the magnitude of the Ca2+ current recorded under control conditions. **, the magnitude of the current measured in the presence of Iso plus PVN was significantly smaller than the magnitude of the current measured in the presence of Iso alone (P < .01). ns, the magnitude of the current measured in the presence of PVN alone was not significantly different from the magnitude of the current measured in the absence of PVN.
To determine whether PTP inhibition really does exert effects at the level of the β-AR, we monitored the effects of PVN on forskolin and histamine stimulated currents. As with vanadate, PVN had no effect on the Ca2+ current stimulated by forskolin. The Ca2+ current measured in the presence of 3 μM forskolin plus 100 μM PVN was 91 ± 5.5% (n = 5) of that measured in the presence of forskolin before PVN exposure. Similar results were obtained when we evaluated the ability of PVN to inhibit the forskolin stimulated Cl− current. The Cl− current measured in the presence of 3 μM forskolin plus 100 μM PVN was 98 ± 4.7% (n = 5) of that measured in the presence of forskolin before exposure to PVN. The consistent absence of an effect of PVN on forskolin-stimulated currents further supports the idea that inhibition of PTP activity exerts its effect at a point upstream of adenylate cyclase.
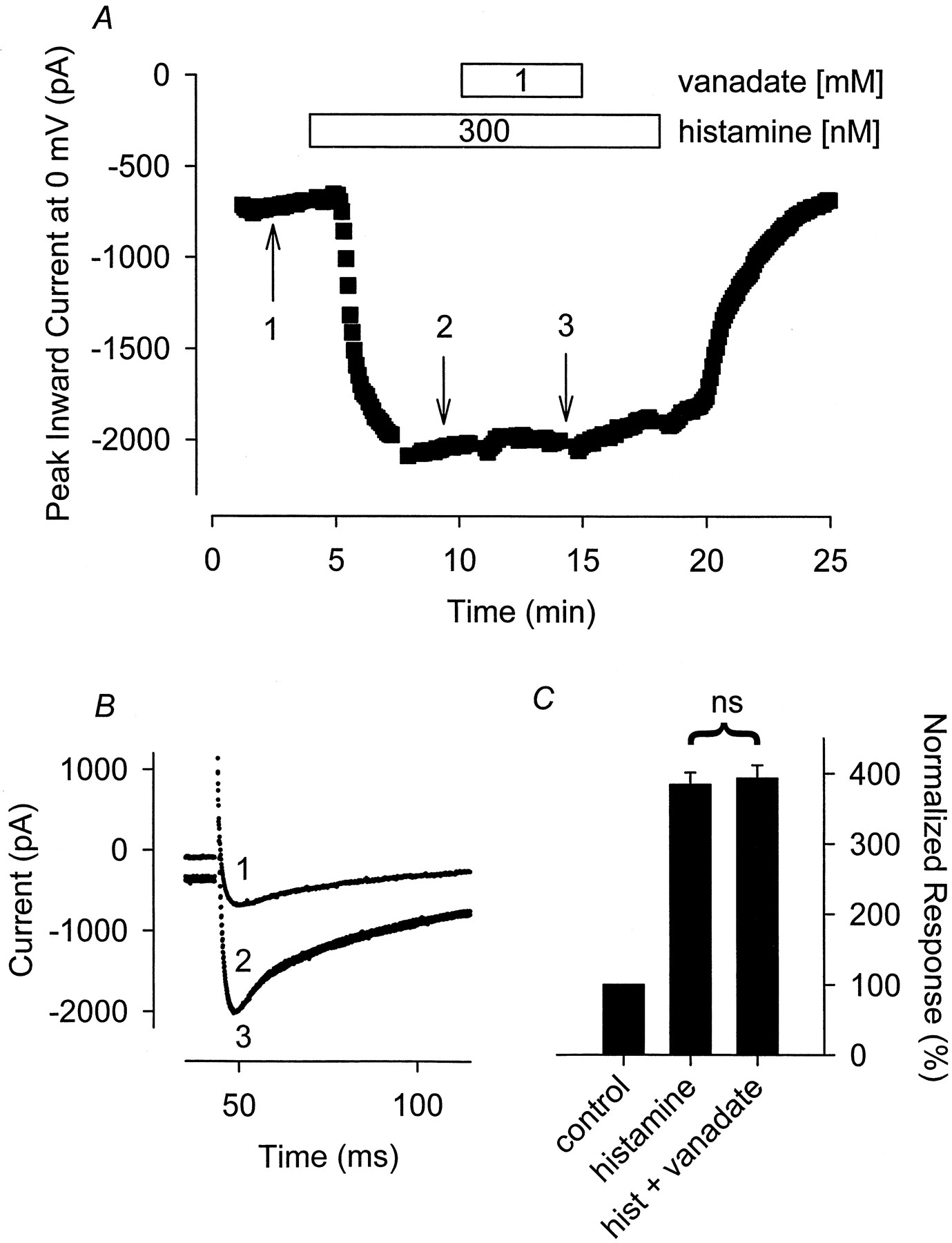
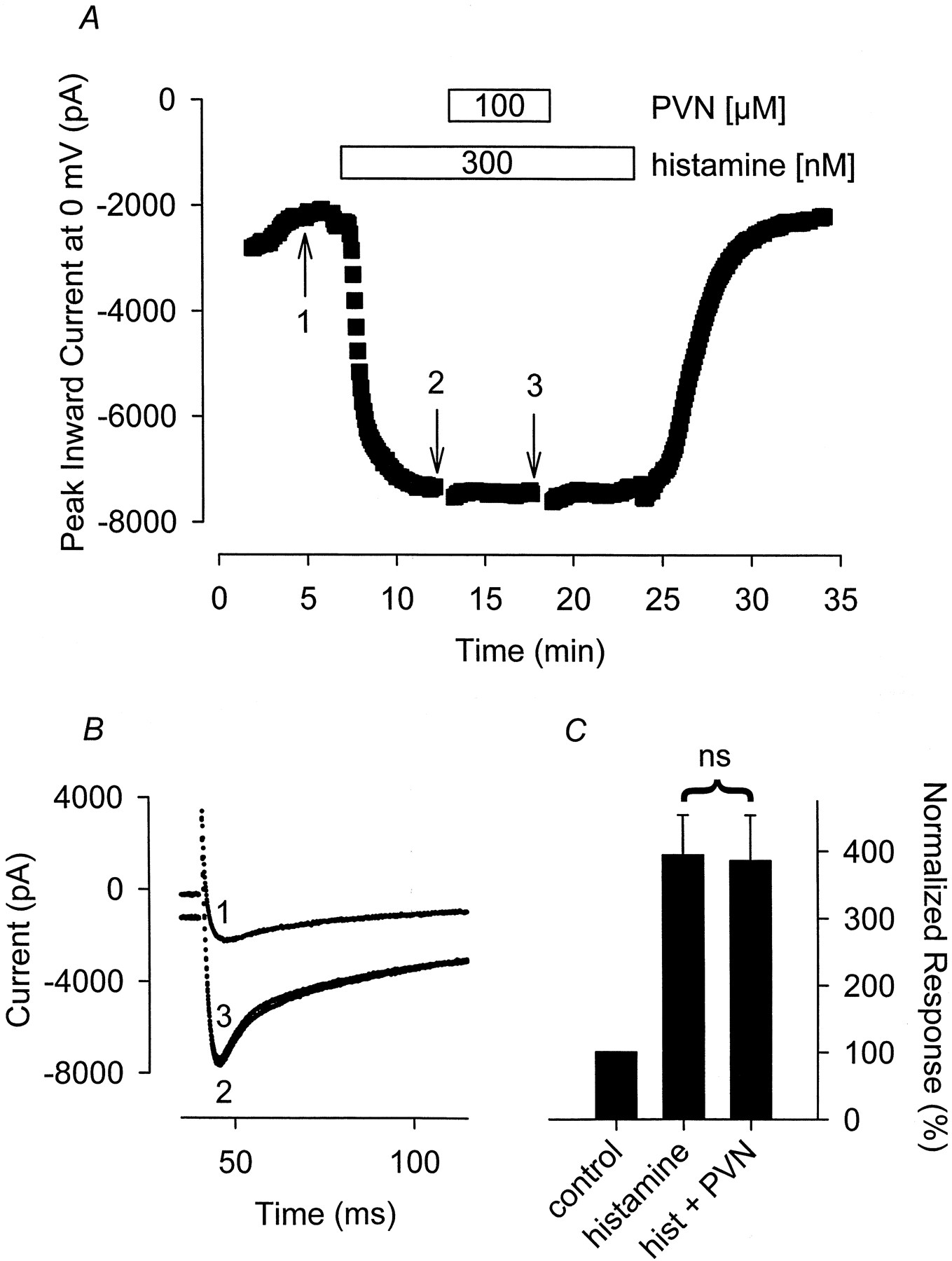
PVN also failed to inhibit the Ca2+ and Cl− currents stimulated by histamine. The Ca2+ current measured in the presence of 300 nM histamine plus 100 μM PVN was 103 ± 6.5% (n = 4) of that measured in the presence of histamine before adding PVN (Fig. 4). The Cl−current measured in the presence of 300 nM histamine plus 100 μM PVN was 97 ± 20% (n = 5) of that recorded in the presence of histamine before exposure to PVN.

The tyrosine phosphatase inhibitor PVN has no effect on the hist-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 300 nM histamine alone and histamine plus 100 μM PVN. B, representative current traces recorded at the time points indicated in A. Note, changes in prepulse current at −30 mV reflect parallel regulation of the time-independent CFTR Cl− current present in this cell. C, cumulative responses (n = 4) normalized to the magnitude of the peak Ca 2+ current recorded under control conditions. ns, the magnitude of the current measured in the presence of PVN plus histamine was not significantly different from the magnitude of the current measured in the presence of histamine alone.
Effect of bpV(phen) on L-type Ca2+ Channel Regulation.
Although PVN is considered a more potent PTP inhibitor than vanadate, it actually consists of a mixture of peroxovanadate species, and under the conditions used to prepare PVN in our experiments, the final mixture would be expected to consist of > 80% vanadate (Huyer et al., 1997). Therefore, we next examined the effects of bpV(phen), one of a novel series of highly purified synthetic peroxovanadium derivatives containing a central vanadium atom, an oxo- group, two peroxo- ligands, and a bidentate ancillary ligand (1,10-phenanthroline) in the inner coordination sphere that confers stability, specificity, and potency to its role as a PTP inhibitor (Posner et al., 1994).
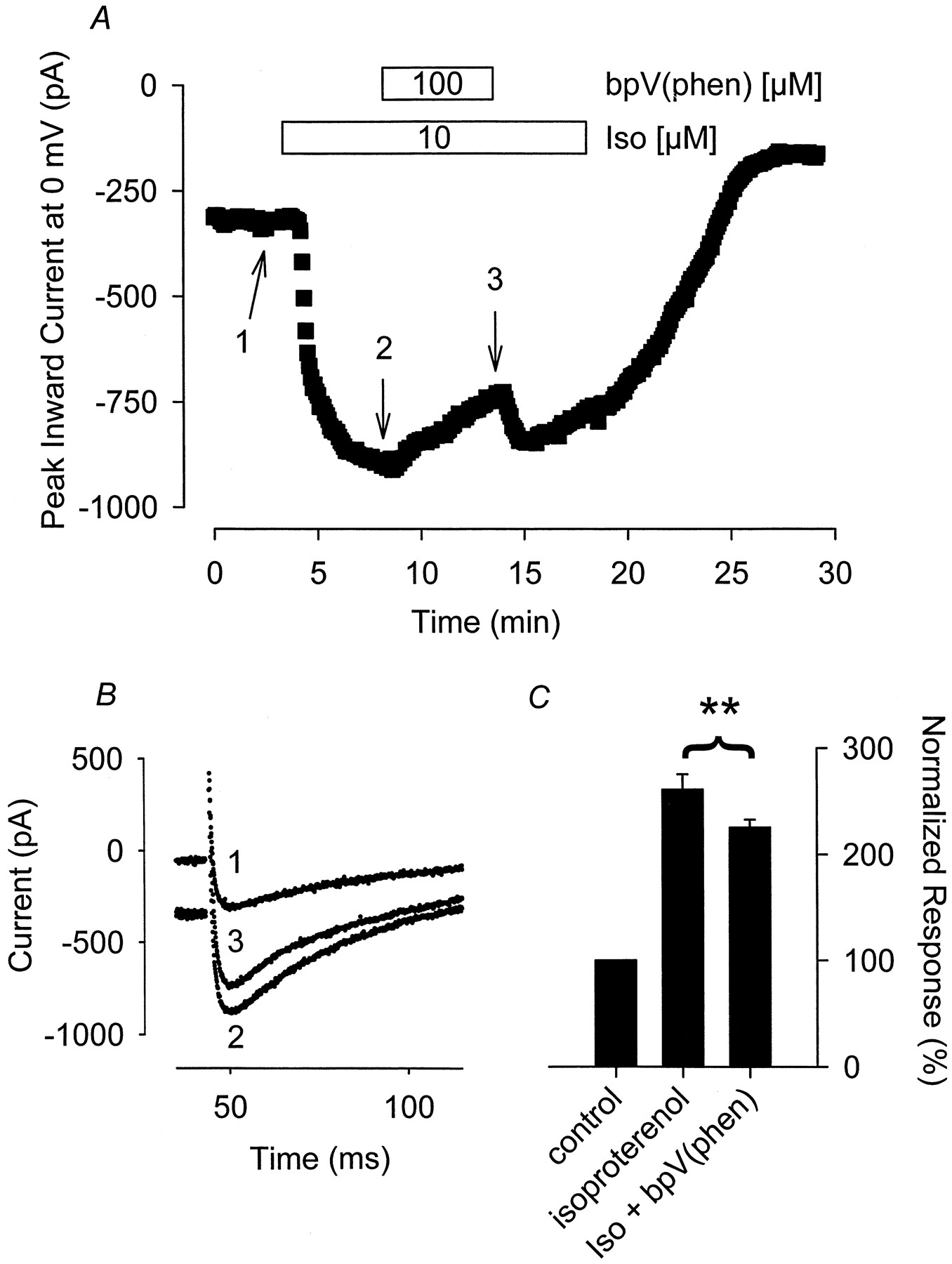
As demonstrated in Fig. 5, application of 100 μM bpV(phen) alone had no inhibitory effect on the Ca2+ current in the absence of Iso. The Ca2+ current measured in the presence of bpV(phen) alone was 99 ± 12% (n = 4) of that observed before exposure to this PTP inhibitor. However, bpV(phen) consistently and completely inhibited the effect that 30 nM Iso had on the Ca2+ current (Fig. 5). Exposure of myocytes to 100 μM bpV(phen) inhibited the Iso-stimulated current by 96 ± 8.6% (n = 5, P < .01). The reversibility of the bpV(phen) response was similar to that observed with PVN. After washout of bpV(phen), the Iso response was restored to only 52 ± 11% of that observed before exposure to bpV(phen). Unlike its ability to inhibit the response to Iso, bpV(phen) had no effect on the Ca2+ current stimulated by histamine (Fig. 6). The Ca2+ current measured in the presence of 300 nM histamine plus 100 μM bpV(phen) was 101 ± 4.4% (n = 5) of that measured in the presence of histamine before exposure to bpV(phen).

The tyrosine phosphatase inhibitor bpV(phen) selectively antagonizes the Iso-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 30 nM Iso alone, Iso plus 100 μM bpV(phen), and bpV(phen) alone. B, current traces recorded at time points indicated in A. C, cumulative responses (n = 4–5) normalized to the magnitude of the Ca2+ current recorded under control conditions. **, the magnitude of the current measured in the presence of Iso plus bpV(phen) was significantly smaller than the magnitude of the current measured in the presence of Iso alone (P < .01). ns, the magnitude of the current measured in the presence of bpV(phen) alone was not significantly different from the magnitude of the current measured in the absence of bpV(phen).

The tyrosine phosphatase inhibitor bpV(phen) has no effect on the hist-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 300 nM histamine alone and histamine plus 100 μM PVN. B, representative current traces recorded at the time points indicated in A. Note, changes in prepulse current at −30 mV reflect parallel regulation of the time-independent CFTR Cl−current present in this cell. C, cumulative responses (n = 5) normalized to the magnitude of the peak Ca2+ current recorded under control conditions. ns, the magnitude of the current measured in the presence of PVN plus histamine was not significantly different from the magnitude of the current measured in the presence of histamine alone.
Effect of PTP Inhibitors on the Sensitivity to β- Adrenergic Stimulation.
Based on the results presented so far, it is unclear whether PTP inhibitors cause a shift in the sensitivity to β-adrenergic stimulation or whether they inhibit β-adrenergic responses in a noncompetitive manner. To address this question, we looked at the effect of increasing the concentration of Iso on the response to bpV(phen) (Fig. 7). We found that exposure of myocytes to 100 μM bpV(phen) inhibited the Ca2+ current response to 10 μM Iso by only 20 ± 3.9% (n = 7, P < .01). This is significantly smaller than the nearly 100% inhibition that the same concentration of bpV(phen) had on the response to 30 nM Iso (see Fig. 5). This suggests that PTP inhibitors decrease the sensitivity of the L-type Ca2+ current to β-AR stimulation in a competitive manner, which is consistent with our previous observation that the PTK inhibitor genistein increases the sensitivity of cardiac myocytes to β-AR stimulation (Hool et al., 1998).
The magnitude of inhibition of the β-adrenergic stimulated Ca2+ current by the tyrosine phosphatase inhibitor bpV(phen) is attenuated in the presence of a supermaximal stimulating concentration of Iso. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 10 μM Iso alone and Iso plus 100 μM bpV(phen). B, current traces recorded at time points indicated in A. C, cumulative responses (n = 7) normalized to the magnitude of the Ca2+ current recorded under control conditions. **, the magnitude of the current measured in the presence of Iso plus bpV(phen) was significantly smaller than the magnitude of the current measured in the presence of Iso alone (P < .05).
Effect of Receptor Tyrosine Kinase Agonists on L-type Ca2+ Channel Regulation.
The PTP inhibitors employed in this study are known insulinomimetic compounds (Posner et al., 1994). Insulin and IGF-1 cause tyrosine phosphorylation of a number of cellular proteins via activation of their respective receptors, which have intrinsic tyrosine kinase activity. PTP inhibitors enhance the tyrosine phosphorylation of similar pools of cellular proteins by blocking tyrosine phophatase activity. In fact, insulin and IGF-1 have specifically been reported to cause tyrosine phosphorylation of β2-ARs that is associated with inhibition of cAMP production in noncardiac preparations (Hadcock et al., 1992;Baltensperger et al., 1996; Karoor and Malbon, 1996). To determine whether PTP inhibitors are mimicking the action that activation of these growth factor receptors has on modulation of β-adrenergic responses in cardiac myocytes, we looked for effects of insulin and IGF-1 on L-type Ca2+ channel activity. However, exposure of myocytes to insulin and IGF-1 did nothing to either basal (data not shown) or Iso stimulated Ca2+ channel activity in guinea pig ventricular myocytes.
Hadcock et al. (1992) found that insulin induced maximal tyrosine phosphorylation of β2-ARs in smooth muscle cells at concentrations as low as 1 nM, and phosphorylation occurred in as little as 5 min at higher concentrations. Furthermore, Bahouth and Lopez (1992) observed that insulin caused maximal attenuation of β1-AR stimulated adenylate cyclase activity SK-M-MC cells within 10 min. We found that the Ca2+ current response to 30 nM Iso after 5 min of exposure to 100 nM insulin was 96 ± 2.9% (n = 6) of that measured before exposure to insulin (Fig.8). Karoor and Malbon (1996) found that IGF-1 induced tyrosine phosphorylation of β2-ARs at concentrations as low as 0.1 nM, and maximal phosphorylation occurred in as little as 2 min at higher concentrations. We found that the Ca2+ current response to 30 nM Iso after 5 min of exposure to 4 nM IGF-1 was 98 ± 3.7% (n = 9) of that measured before exposure to IGF-1 (Fig. 9). Comparable results were obtained after exposure to either agonist for up to 10 min (data not shown). These results suggest that the effect that PTP inhibitors have on β-adrenergic regulation of L-type Ca2+channel activity in cardiac myocytes is not associated with insulin and IGF-1 receptor tyrosine kinase activity.

Insulin has no effect on the Iso-stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 30 nM Iso alone and Iso plus 100 nM insulin. B, current traces recorded at the time points indicated in A. Note, changes in prepulse current at −30 mV reflect parallel regulation of the time-independent CFTR Cl− current present in this cell. C, cumulative responses (n = 6) normalized to the magnitude of the peak Ca2+ current recorded under control conditions. ns, the magnitude of the current measured in the presence of insulin plus Iso was not significantly different from the magnitude of the current measured in the presence of Iso alone.

IGF-1 has no effect on the isoproterenol (Iso) stimulated Ca2+ current. A, time course of changes in peak Ca2+ current recorded during depolarizing voltage-clamp steps to 0 mV, applied once every 5 s, after exposure to 30 nM Iso alone and Iso plus 4 nM IGF-1. B, current traces recorded at the time points indicated in A. Note, changes in prepulse current at −30 mV reflect parallel regulation of the time-independent CFTR Cl− current present in this cell. C, cumulative responses (n = 9) normalized to the magnitude of the peak Ca2+ current recorded under control conditions. ns, the magnitude of the current measured in the presence of IGF-1 plus Iso was not significantly different from the magnitude of the current measured in the presence of Iso alone.
Discussion
In the present study, we found that neither vanadate, PVN, nor bpV(phen) had any effect on basal L-type Ca2+channel activity in guinea pig ventricular myocytes. However, all three were capable of inhibiting β-adrenergic regulation of this current. These results are consistent with the idea that basal tyrosine kinase and phosphatase activities exert inhibitory and stimulatory effects, respectively, on β-adrenergic regulation of cardiac ion channels. This conclusion is also consistent with data from our previous study demonstrating that the PTK inhibitor genistein is capable of increasing the sensitivity of cardiac ion channels to β-AR stimulation (Hool et al., 1998). In that study, we found that the facilitating effect of genistein was not mimicked by the inactive structural analog daidzein and that this effect could be blocked by PVN. Taken together with the results of the present study, these observations suggest that under basal conditions, PTK and PTP activities exist in an equilibrium that is capable of both up-regulating and down-regulating β-adrenergic responses in cardiac myocytes.
Vanadate, PVN, and bpV(phen) are all capable of inhibiting PTP activity (Posner et al., 1994; Crans et al., 1995; Huyer et al., 1997). All three compounds can inhibit PTP activity because of their ability to adopt a coordinate trigonal bipyramidal structure that binds as a transition state analog of phosphate to phosphoryl transfer enzymes (Shaver et al., 1995). Furthermore, they can readily cross the plasma membrane of intact cells. This is apparently attributable to their structural similarity to phosphate, which allows them to enter via phosphate or anion carrier systems (Kustin and Robinson, 1995). As a phosphate analog, vanadate can also exert effects on a number of other enzymes, including certain serine-threonine kinases and phosphatases. In addition to acting as a phosphate analog, vanadate and peroxovanadate compounds can also exert effects by acting as oxidants (Stankiewicz et al., 1995). Therefore, an important question to consider is whether or not their ability to inhibit Iso responses can be explained by some mechanism other than inhibition of PTP activity. For example, oxidation of Iso by vanadate could explain the apparent selective inhibition of β-adrenergic responses. However, this is unlikely, because vanadate should not be involved in oxidative reactions at physiologic pH (Crans et al., 1995). This is consistent with our own verification of the stability of Iso in PVN containing solutions by directly measuring its presence via electrochemical detection after separation on a reverse-phase HPLC column (C. Sims and R.D. Harvey, unpublished observation). It is also unlikely that these compounds are acting as β-AR antagonists, because Krawietz et al. (1982) demonstrated that vanadate, at concentrations higher than those used in the present study, did not affect the binding of Iso to β-ARs.
Vanadate has also been reported to affect adenylate cyclase activity (Krawietz et al., 1982; Aiton and Cramb, 1985). However, an effect on adenylate cyclase is not likely to explain the results we have observed, because the concentration of vanadate we used was reported to stimulate, not inhibit, its activity. The effect of vanadate on adenylate cyclase activity is also reported to be minimal in intact cardiac myocytes (Aiton and Cramb, 1985). Consistent with this, none of the compounds we used exhibited any significant stimulatory effect on the basal current. Furthermore, if any of these compounds were inhibiting β-adrenergic responses by acting directly on adenylate cyclase activity, they should have also affected histamine and possibly even forskolin responses, which they did not. The fact that β-adrenergic responses were selectively inhibited also rules out unlikely effects on PKA or serine-threonine phosphatase activity.
The ability of vanadate, PVN, and bpV(phen) to inhibit Iso responses can be clearly distinguished from artifacts such as rundown based on the fact that inhibition was only observed when currents were activated by Iso. Furthermore, the inhibitory effects of these compounds were at least partially reversible upon washout. Although all of the compounds we used can inhibit PTP activity by acting as phosphate transition analogs, the peroxovanadate derivates in PVN and bpV(phen) are also reportedly able to oxidize the cysteine residue found in the catalytic site of PTPs in vitro (Huyer et al., 1997). This might explain the slightly greater degree of irreversibility observed in the presence of PVN and bpV(phen) experienced in our hands.
To verify that the effects of vanadate, PVN, and bpV(phen) were caused by inhibition of PTP activity, and not by direct effects on components of the cAMP signaling pathway, we demonstrated that these compounds did not inhibit forskolin and histamine responses. These results provided important insight into the mechanism by which inhibition of PTP activity inhibits β-adrenergic responses. Because H2 histaminergic and β-ARs regulate cardiac ion channels through the same Gs-mediated cAMP/PKA-dependent signaling pathway, the ability of PTP inhibitors to affect Iso but not histamine responses suggests that basal tyrosine kinase activity exerts an inhibitory effect at the level of the β-AR. However, the lack of an effect on histamine responses might also be explained if: 1) the sensitivity of the Ca2+current to PTP inhibitors could be overcome by increasing the level of cAMP-dependent stimulation; and 2) the level of cAMP-dependent stimulation produced by histamine were significantly greater than that produced by Iso. Although our results do demonstrate that the response to PTP inhibitors can be overcome by increasing the level of β-AR stimulation, it seems unlikely that the concentration of histamine used in our study (300 nM) produced responses that were significantly different from those produced by the concentration of Iso that was used (30 nM). In fact, the concentrations of the agonists used in the present study were specifically chosen because they have been reported to produce approximately equivalent, submaximal Ca2+ current responses in guinea pig ventricular myocytes (Kameyama et al., 1985; Hescheler et al., 1987). Consistent with this idea, the magnitude of the response to histamine that we observed was not significantly different (P > .05) than the magnitude of the response to Iso. Therefore, the more likely explanation for the results we have obtained is that the PTP inhibitors were selectively affecting β-adrenergic responses.
One possible explanation for the selective inhibition of β-adrenergic responses could be that there is direct tyrosine phosphorylation of the β-AR. Consistent with this hypothesis, it has been demonstrated that insulin and IGF-1 can antagonize β2-AR mediated responses in noncardiac preparations by directly phosphorylating the receptor protein (Hadcock et al., 1992; Karoor et al., 1995;Baltensperger et al., 1996; Karoor and Malbon, 1996). Whether such a mechanism can explain the results of the present study is yet to be determined. Although Iso has been reported to mediate cAMP-dependent responses by acting at both β1- and β2-ARs in some cardiac preparations (Xiao et al., 1999), we have previously demonstrated that in guinea pig ventricular myocytes Iso regulates ion channel function solely through β1-ARs (Hool and Harvey, 1997). Therefore, an important question to address is whether or not direct tyrosine phosphorylation can regulate β1-ARs in cardiac myocytes the same way that it regulates β2-ARs in noncardiac preparations.
The feasibility of such a mechanism is supported by the report that insulin desensitizes β1-AR mediated activation of adenylate cyclase in SK-N-MC neuroepithelioma cells (Bahouth and Lopez, 1992). Also, IGF-1 stimulated tyrosine phosphorylation of the β2-AR was mapped to tyrosyl residues in the second intracellular loop of the receptor protein (Karoor and Malbon, 1996). The potential exists then, for tyrosine phosphorylation of analogous sites in the second intracellular loop of the β1-AR, because this region is highly conserved among β-AR subtypes (Frielle et al., 1987). However, the results of our present study suggest that acute exposure to insulin and IGF-1 does not activate the tyrosine kinase dependent mechanism that attenuates the β-adrenergic responsiveness of cardiac Ca2+channels.
An alternative explanation for the results we have obtained is that tyrosine kinase activity could be indirectly affecting β1-AR function by regulating the activity of a G protein-coupled receptor kinase. Consistent with such a hypothesis is the recent report that the nonreceptor tyrosine kinase src can phosphorylate and stimulate G protein-coupled receptor kinase 2 activity (Sarnago et al., 1999), which is important in regulating β-AR function in cardiac myocytes (Lohse et al., 1996). Whatever molecular mechanism is responsible for the results we have obtained, our present study suggests that tyrosine phosphorylation may play an important role in regulating β-adrenergic responses in cardiac myocytes. It has yet to be determined whether the PTK activity responsible is associated with a receptor and/or a nonreceptor tyrosine kinase.
Acknowledgments
We thank M. Sanders for expert technical assistance, A. Belevych for helpful discussions, and L. Szweda for assistance with HPLC measurements.
Footnotes
- Received April 24, 2000.
- Accepted August 30, 2000.
-
Send reprint requests to: Dr. Robert Harvey, Department of Physiology and Biophysics, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106–4970. E-mail: rdh3{at}po.cwru.edu
-
This work was supported by grants from the National Institutes of Health (AG16658) and the American Heart Association.
Abbreviations
- AR
- adrenergic receptor
- Gs
- stimulatory G protein
- PKA
- protein kinase A
- CFTR
- cystic fibrosis transmembrane conductance regulator
- PTK
- protein tyrosine kinase
- PTP
- protein tyrosine phosphatase
- PSS
- physiological salt solution
- Iso
- isoproterenol
- IGF-1
- insulin-like growth factor-1
- PVN
- peroxovanadate
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}