


Abstract
We demonstrated previously that D3.49(164) mutations resulted in constitutive activation of the rat μ-opioid receptor and abolished receptor expression unless cells were pretreated with naloxone, an inverse agonist. In this study, we investigated the properties of the D3.49(164)Q mutant and the mechanisms underlying the effect of naloxone. Naloxone pretreatment up-regulated [3H]diprenorphine binding and protein expression of the D3.49(164)Q mutant in a time- and dose-dependent manner without affecting its mRNA level. After naloxone removal, binding and protein expression of the mutant declined with time with no effect on its mRNA level. Naloxone methiodide (a quaternary ammonium analog) caused a maximal up-regulation about 50% of the naloxone effect, indicating that naloxone acts extracellularly and intracellularly. Expression of the mutant was enhanced by inverse agonists, a neutral antagonist, and agonists, with inverse agonists being most effective. In membranes, the mutant was structurally less stable than the wild type upon incubation at 37°C, and naloxone and [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin stabilized the mutant. Coexpression of the dominant-negative mutants GRK2-K220R, arrestin-2(319–418), dynamin I-K44A, rab5A-N133I or rab7-N125I partially prevented the decline in binding of the mutant after naloxone removal. Chloroquine or proteasome inhibitor I reduced the down-regulation of the mutant. These results indicate that the D3.49(164)Q mutant is constitutively internalized via G protein coupled-receptor kinase-, arrestin-2-, dynamin-, rab5-, and rab7-dependent pathways and probably trafficked through early and late endosomes into lysosomes and degraded by lysosomes and proteasomes. Naloxone up-regulates the D3.49(164)Q mutant by stabilizing the mutant protein and blocking its constitutive internalization and down-regulation. To the best of our knowledge, this represents the first comprehensive analysis of the mechanisms involved in up-regulation of constitutively active mutants by an inverse agonist.
Opiate and opioid compounds act on opioid receptors to produce their pharmacological actions. Multiple opioid receptors (μ, δ, κ, ε) have been demonstrated from pharmacological, binding, and anatomical data (Pasternak, 1988). These opioid receptors are coupled through pertussis toxin-sensitive G proteins to affect a variety of effectors, which include adenylate cyclase, potassium channels, calcium channels, and a mitogen-activated protein kinase pathway (Law et al., 2000). μ-Opioid receptors are closely associated with analgesic and euphoric actions of opiate and opioid compounds (Pasternak, 1988). Following the cloning of the δ-opioid receptor, μ- and κ-opioid receptors were cloned (Knapp et al., 1995 and references therein). Deduced amino acid sequences of these clones display the motif of putative seven transmembrane domains (TMs) connected by alternating intracellular and extracellular hydrophilic loops, which is characteristic of G protein-coupled receptors (GPCRs). Opioid receptors belong to the rhodopsin subfamily of GPCRs.
Conformational changes are thought to underlie the activation of GPCRs. Studies have shown that movement of TMs 3, 5, and 6 and their adjoining intracellular loops are important for receptor activation (Gether and Kobilka, 1998). In recent years, constitutively active mutants (CAMs) of many GPCRs have been generated by point mutation or chimeric receptor approaches or found naturally in disease states (Lefkowitz et al., 1993; Scheer and Cotecchia, 1997; Pauwels and Wurch, 1998). Mutation in a GPCR resulting in G protein activation in the absence of an agonist was first demonstrated for α1B-adrenergic receptor (AR) (Cotecchia et al., 1990; Kjelsberg et al., 1992) and subsequently in several other receptors (Lefkowitz et al., 1993; Scheer and Cotecchia, 1997; Pauwels and Wurch, 1998). In addition to displaying agonist-independent activation, CAMs of GPCRs exhibit agonist-independent adaptive regulation, including enhanced phosphorylation, desensitization, and down-regulation of the receptors (Leurs et al., 1998).
We recently showed that mutation of Asp3.49(164) in the highly conserved DRY motif at the cytoplasmic end of the TM3 to His, Tyr, Met, or Gln resulted in constitutive activation of the μ-opioid receptor (μOR) as demonstrated by enhanced [35S]GTPγS binding, which was due to agonist-independent activation of pertussis toxin-sensitive G proteins (Li et al., 2001). These mutants seemed to assume conformations of activated states of the receptor since they exhibited higher affinity for the agonist [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO) than the wild type, which was unaffected by uncoupling of the receptor and G proteins with GTPγS (Li et al., 2001). However, unlike CAMs of other GPCRs, when transfected into human embryonic kidney 293 cells or Chinese hamster ovary (CHO) cells, these Asp3.49(164) mutants exhibited almost no binding activity and protein expression. Binding activity could be detected only after pretreatment of cells with naloxone, an inverse agonist at these mutants (Li et al., 2001). Mutations resulting in constitutive activation of GPCRs reduced, but did not abolish, their receptor binding and treatment of cells expressing the CAMs with inverse agonists increased expression levels (Pei et al., 1994; Heinflink et al., 1995; MacEwan and Milligan, 1996b;Gether et al., 1997a; Lee et al., 1997; Samama et al., 1997; Alewijnse et al., 2000) [also for reviews, see (Milligan and Bond, 1997; Leurs et al., 1998)].
The dramatic increase in the expression of the D3.49(164) mutants of the μOR by naloxone offers a unique opportunity to investigate biochemical events leading to such an effect. In this study, we examined mechanisms underlying the up-regulatory effects of naloxone pretreatment on one of the CAMs, the D3.49(164)Q mutant. By delineating the mechanisms, we gained insights into the properties of this CAM.
Experimental Procedures
Materials.
[35S]GTPγS (∼1250 Ci/mmol), [3H]diprenorphine (58 Ci/mmol), and [α-32P]dATP (∼3000 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). Naloxone was a gift from DuPont Merck Pharmaceutical Co. (Wilmington, DE). Morphine, etorphine, and diprenorphine were provided by the National Institute on Drug Abuse. Naloxone methiodide, naltrexone, chloroquine, GDP, guanosine 5′-O-(3-thiotriphosphate) (GTPγS), formamide, formaldehyde, 3-[N-morpholino]propane-sulfonic acid, NaDodSO4, and agarose were obtained from Sigma (St. Louis, MO). DAMGO was purchased from Sigma/RBI (Natick, MA). Mouse monoclonal antibodies against the hemagglutinin (HA) peptide epitope was obtained from BABCO (Berkeley, CA). Enhanced chemiluminescence Western blotting detection reagents and high-performance autoradiography film (Hyperfilm MP) were obtained from Amersham Pharmacia Biotech (Piscataway, NJ).Z-Ile-Glu(OtBu)-Ala-Leu-CHO (proteasome inhibitor I) was obtained from Calbiochem (La Jolla, CA). LipofectAMINE was purchased from Invitrogen (Gaithersburg, MD) Geneticin (G418 sulfate) was obtained from Mediatech Co. (Herndon, VA). RNAzol B reagent was purchased from Tel Test B (Friendswood, TX). RNA markers, Klenow enzyme, and dNTPs were purchased from Promega (Madison, WI). Neutral nylon membrane Biodyne B was a kind gift from Gelman Laboratory (Ann Arbor, MI). Protease inhibitor cocktail was obtained from Roche Molecular Biochemicals (Indianapolis, IN). QIAquick polymerase chain reaction purification kit was obtained from CLONTECH (Valencia, CA). Enzymes and chemicals commonly used in molecular biology experiments were purchased from Invitrogen, Promega (Madison, WI), Roche Molecular Biochemicals, QIAGEN (Valencia, CA), or Sigma (St. Louis, MO).
The rat μOR cDNA clone was a gift from Dr. Lei Yu of the University of Cincinnati. The cDNA clones of GRK2-K220R in pcDNA3.1 Zeo(+), arrestin-2(319–418) in pcDNA3 and dynamin I-K44A in pcDNA3 were donated by Dr. Jeffrey Benovic of Thomas Jefferson University. Rab5A-N133I and rab7-N125I cDNA clones were gifts from Dr. A. Wandinger-Ness of the University of New Mexico.
Numbering Schemes for Amino Acid Residues in the Rat μOR and Other GPCRs.
The numbering scheme used identifies amino acid residues in rat μOR and other GPCRs by their sequence numbers and by the generic numbering scheme proposed by Ballesteros and Weinstein (1995). This combined scheme is used to relate the results obtained for opioid receptors to equivalent positions in other GPCRs. According to the generic numbering scheme, amino acid residues in TMs are assigned two numbers (N1, N2). N1 refers to the TM number. For N2, the numbering is relative to the most conserved residue in each TM, which is assigned 50; the other residues in the TM are numbered in relation to this conserved residue, with numbers decreasing toward the N terminus and increasing toward the C terminus. The most conserved residue in the TM3 of the rat μOR is Arg165, which is referred to as R3.50(165). Asp164 is thus referred to as D3.49(164).
Oligodeoxynucleotide-Directed Mutagenesis.
The D3.49 (164)Q mutant of the HA-tagged rat μ-receptor was generated and subcloned into HindIII and XbaI sites of the mammalian expression vector pcDNA3 (Li et al., 2001).
Stable Expression of the Wild-Type Rat μOR and the D3.49(164)Q Mutant in CHO Cells.
Clonal CHO cells stably transfected with the wild-type or the D3.49(164)Q mutant (CHO-μOR cells and CHO-D3.49(164)Q cells, respectively) were established previously (Li et al., 2001). The cells were cultured in Dulbecco's modified Eagle's medium Ham's F12 supplemented with 10% fetal calf serum, 0.5 mg/ml geneticin, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere consisting of 5% CO2 and 95% air at 37°C. For most experiments, naloxone (20 μM) or other drugs were added to the medium for the indicated time period. Cells were detached by use of Versene solution (0.54 mM EDTA, 0.14 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.46 mM KH2PO4, 1 mM glucose) and collected by centrifugation at 2,000g for 15 min at 4°C. Cells were washed three times with 15 ml/100-mm dish phosphate-buffered saline by resuspension and centrifugation at 4°C. This procedure was found to be adequate for removal of naloxone and, possibly endogenous opioid peptides, since theK d values of [3H]diprenorphine binding for the wild type grown in the absence and presence of naloxone were similar (Li et al., 2001).
Membrane Preparations.
Membranes were prepared from CHO-μOR cells or CHO-D3.49(164)Q cells according to Li et al. (2001). Membranes were suspended in 50 mM Tris-HCl buffer (pH 7.4) and then aliquoted and stored at −80°C.
Opioid Receptor Binding in Membrane Preparations.
[33H]diprenorphine binding to the wild-type and D3.49(164)Q mutant on membrane was performed as described previously (Li et al., 2001). Briefly, binding was carried out in 50 mM Tris-HCl buffer (pH 7.4) containing 1 mM EGTA at room temperature for 1 h in duplicate in a final volume of 1 ml with 1 nM [33H]diprenorphine, 10 to 20 μg of membrane protein. Naloxone (10 μM) was used to define nonspecific binding.
Opioid Receptor Binding in Whole-Cell Preparations.
Binding of [33H]diprenorphine to the wild-type and D3.49(164)Q mutant μORs in whole cells was performed with 1 nM [33H]diprenorphine in Krebs' buffer (pH 7.4) at room temperature for 1 h in duplicate in a final volume of 1 ml with 50,000 to 150,000 cells (Li et al., 2001). Naloxone (10 μM) was used to define nonspecific binding. Binding results were normalized as dpm/105 cells and then calculated as percentage of the control, when necessary.
Western Blot.
Western blot was performed to examine the expression of the HA-tagged wild-type and D3.49(164)Q mutant μOR proteins as described previously (Li et al., 2001). Briefly, stably transfected CHO cells or membranes were treated as indicated and solubilized with Laemmli sample buffer and subjected to SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Nitrocellulose membranes were treated with blocking solution, incubated with monoclonal antibodies against HA and then anti-mouse IgG conjugated with horseradish peroxidase, reacted with enhanced chemiluminescence Western blotting detection reagents and exposed to X-ray films.
Northern Blot Analysis.
Total RNA was extracted from CHO-D3.49(164)Q cells using RNAzol B reagent, denatured with formamide, applied onto 1% agarose gel containing 0.66 M formaldehyde, and separated by electrophoresis in 3-[N-morpholino]propane-sulfonic acid buffer. The RNA profile was transferred by upward capillary effect to a neutral nylon membrane (Biodyne B) and fixed to the membrane by baking for 1 h at 70°C. The nylon membrane was incubated for 2 h at 68°C in prehybridization solution (0.5 M sodium phosphate, pH 7.2, 7% (w/v) SDS, 1 mM EDTA, pH 7.0). D3.49(164)Q mutant cDNA probe (1.5 kilobases) was labeled with [α-32P]dATP by random priming, purified by QIAquick polymerase chain reaction purification kit, denatured by placing in boiling water for 2 min, and chilled on ice. The labeled probe was added to the prehybridization solution, mixed thoroughly, and incubated with the membrane overnight in a hybridization oven at 68°C. After removal of the solution, the membrane was washed four times at room temperature with 2× SSC and 1% SDS, then once for 60 min at 68°C with 0.1× SSC and 0.5% SDS, and finally washed for 5 min with 2× SSC at room temperature. The membrane was then exposed to X-ray film.
[35S]GTPγS Binding Assay.
Determination of [35S]GTPγS binding to G proteins was carried out as described previously (Zhu et al., 1997) with 15 μM GDP and 0.2 nM [35S]GTPγS in reaction buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, and 0.1% bovine serum albumin) in a final volume of 0.5 ml. Nonspecific binding was determined in the presence of 10 μM GTPγS. After 60 min of incubation at 30°C, bound and free [35S]GTPγS were separated by filtration with GF/B filters under reduced pressure. Radioactivity was determined by liquid scintillation counting.
Transient Transfection of CHO-D3.49(164)Q Cells with Dominant-Negative Mutants.
CHO-D3.49(164)Q cells pretreated by 20 μM naloxone for over 96 h were transiently transfected with 8 μg/100-mm dish of bovine GRK2-K220R (Kong et al., 1994) in pcDNA3.1 Zeo(+), arrestin-2(319–418) (Krupnick et al., 1997) in pcDNA3, dynamin I-K44A (van der Bliek et al., 1993) in pcDNA3, rab5A-N133I (Bucci et al., 1992) in pcDNA3, rab7-N125I (Feng et al., 1995) in pCR3.1, or vector using LipofectAMINE (50 μl) following the manufacturer's instructions. Transfection efficiency was approximately 60%. Cells transfected with dominant-negative mutants or vectors were cultured in medium containing 20 μM naloxone for 24 h and then without naloxone for an additional 24 h. Untransfected cells grown in the presence of naloxone for the entire period served as control. Cells were washed with Krebs' buffer (pH 7.4), detached with Versene solution, pelleted, and resuspended in Krebs' buffer. Whole-cell [33H]diprenorphine binding was performed. Results were normalized to dpm/105 cells and then expressed as percentage of control.
Treatment of CHO-D3.49(164)Q Cells with Chloroquine and Proteasome Inhibitor I.
CHO-D3.49(164)Q cells pretreated by 20 μM naloxone for over 96 h were transferred into 6-well plates and cultured for another 24 h in the presence of 20 μM naloxone. Naloxone was removed by washing with cold Krebs' buffer, and cells were treated with 50 μM chloroquine, 5 μM proteasome inhibitor I, 50 μM chloroquine plus 5 μM proteasome inhibitor I, or saline for 6 h. Untreated cells cultured in the presence of naloxone for the entire period served as the control. Control and treated cells were washed three times with cold Krebs' buffer, detached with Versene solution, pelleted and resuspended in Krebs' buffer. [3H]Diprenorphine binding was performed on whole cells.
Statistical Analysis.
For comparison of multiple groups, data were analyzed with analysis of variance to determine whether there were significant differences among groups. If so, Dunnett's test was performed to determine whether there was significant difference between the control and each treatment group.
Results
Time Course of Effect of Naloxone Pretreatment on [3H]Diprenorphine Binding and mRNA and Protein Expression of the D3.49(164)Q Mutant.
CHO-D3.49(164)Q cells exhibited little [3H]diprenorphine binding and no detectable receptor protein expression (Fig. 1,A and C). Pretreatment of the cells with 20 μM naloxone increased [3H]diprenorphine binding in whole-cell preparations in a time-dependent manner, reaching a plateau at 72 h (Fig. 1A). For comparison, pretreatment with naloxone also increased [3H]diprenorphine binding of the wild type (Fig. 1B). The increase reached a plateau at 72 h, which was statistically significant and represented an increase of ∼45%. After pretreatment with naloxone for over 96 h, theK d and B maxvalues of [3H]diprenorphine binding to CHO-μOR and D3.49(164)Q cell membranes were determined to be 0.20 ± 0.01 nM and 9.9 ± 0.3 pmol/mg of protein and 0.22 ± 0.01 nM and 5.9 ± 0.2 pmol/mg of protein, respectively (mean ± S.E.M, n = 4) (Li et al., 2001).

Time course of naloxone pretreatment on [3H]diprenorphine binding of the D3.49(164)Q mutant and the wild type of the rat μOR (A and B), receptor protein expression of the D3.49(164)Q mutant (C), and mRNA of the D3.49(164)Q mutant (D). CHO-D3.49(164)Q cells and CHO-μOR cells were cultured without naloxone for at least 96 h, transferred into 6-well plates, and grown in medium with or without naloxone (20 μM) for different periods of time. The time of naloxone addition was staggered so that receptor binding, Western blot, and Northern blot were performed at the same time (at 96 h after transfer) after naloxone was removed by washing. [3H]Diprenorphine binding to whole cells, Western blot with a monoclonal antibody against the HA epitope, and Northern blot with a 32P-labeled probe were carried out as described under Experimental Procedures. A and B, results were normalized and expressed as dpm/105 cells for [3H]diprenorphine binding. Each value represents mean ± S.E.M. of six (A) and four (B) independent experiments in duplicate. ∗∗, p < 0.01 compared with that without naloxone pretreatment. C and D, each figure represents one of the two experiments performed with similar results.
Naloxone pretreatment also increased the protein level of the D3.49(164)Q mutant in a time-dependent fashion as determined by Western blot analysis using monoclonal antibodies against HA (Fig. 1C). The increase in protein expression approximated the enhancement in binding activity. In contrast, the same pretreatment did not affect the mRNA level of the D3.49(164)Q mutant (Fig. 1D).
Time Course of [3H]Diprenorphine Binding and Protein Expression of the D3.49(164)Q Mutant of the Rat μOR after Removal of Naloxone.
Cells were cultured in the presence of naloxone for ≥96 h. Following removal of naloxone from culture medium, [3H]diprenorphine binding to the D3.49(164)Q mutant declined in a time-dependent manner (Fig.2A) after a first-order kinetics witht 1/2 of ∼7 h (Fig. 2B). At 48 h after naloxone removal, only 5% binding remained (Fig. 2A), and at 96 h, virtually no binding was detected (data not shown). In contrast, [3H]diprenorphine binding to the wild type was only slightly decreased (Fig. 2A). Protein expression of the D3.49(164)Q CAM, determined by Western blot, was also reduced in a time-dependent manner after naloxone was removed (Fig. 2C). However, the mRNA level of the D3.49(164)Q mutant was unchanged after naloxone removal (Fig. 2D).
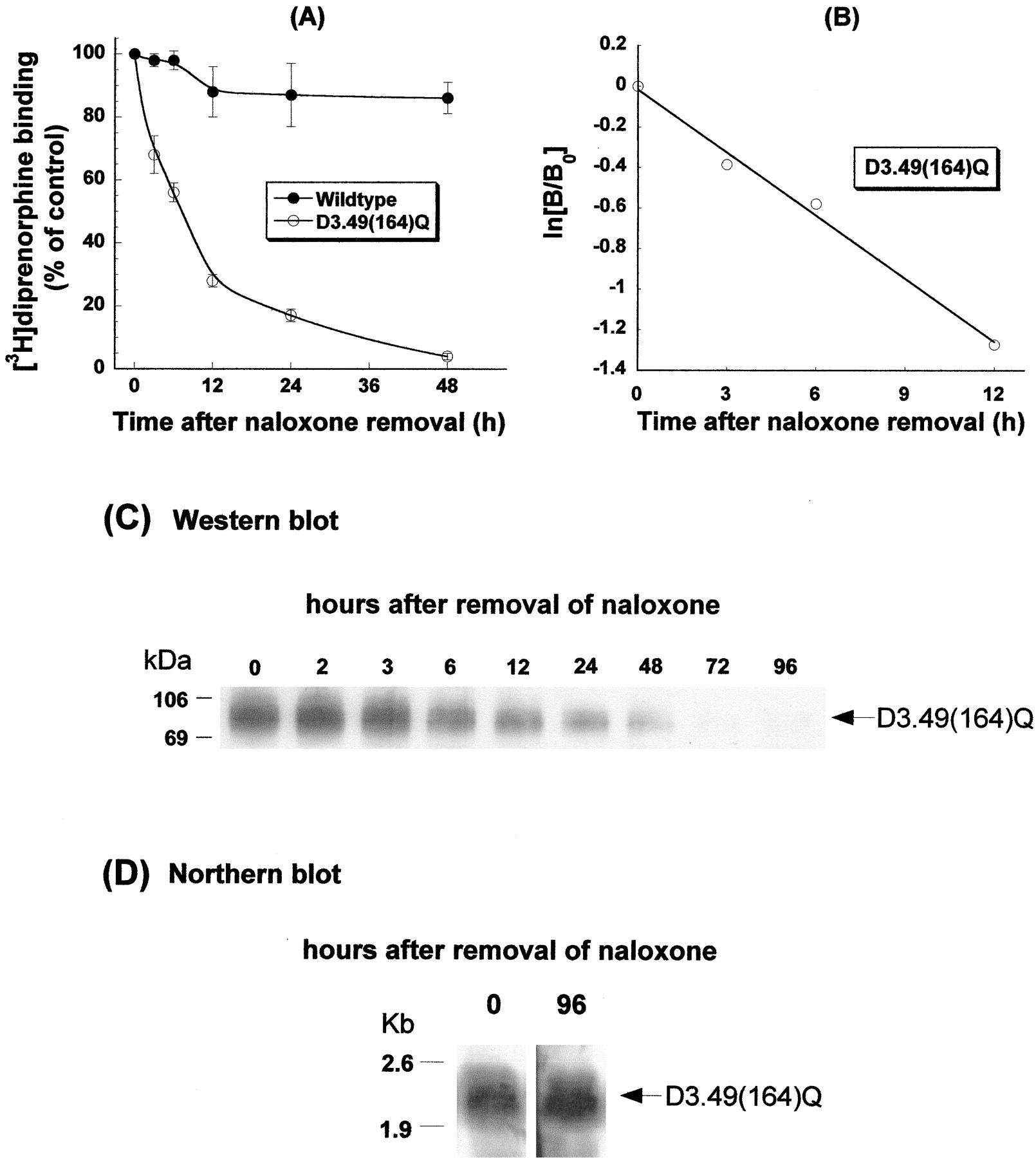
Effects of removal of naloxone on [3H]diprenorphine binding of the wild-type and D3.49(164)Q mutant of the rat μOR (A); kinetics of the decline of [3H]diprenorphine binding of the D3.49(164)Q mutant (shown in A) (B); receptor protein expression of the D3.49(164)Q mutant (C); mRNA of the D3.49(164)Q mutant (D). CHO-μOR cells and CHO-D3.49(164)Q cells were cultured in the presence of 20 μM naloxone for over 96 h. Cells were transferred into 6-well plates, and naloxone was removed from culture medium for different periods of time by washing and replenishing with medium without naloxone. The time of naloxone removal was staggered so that [3H]diprenorphine binding to the receptors in whole cells, Western blot with a monoclonal antibody against the HA epitope, and Northern blot with a32P-labeled probe were performed at the same time (48 h after the transfer). A and B, results were normalized against 105 cells for [3H]diprenorphine binding and expressed as percentage of the control (cultured in the presence of naloxone). Each value represents mean ± S.E.M. of six independent experiments in duplicate. C and D, each figure represents one of the two experiments performed with similar results.
Dose-Response Relationship of Naloxone and Naloxone Methiodide Pretreatment on [3H]Diprenorphine Binding of the D3.49(164)Q Mutant.
After cells were cultured without naloxone for ≥96 h, pretreatment with naloxone for 24 h increased expression of [3H]diprenorphine binding of the D3.49(164)Q receptor in a dose-dependent manner with an EC50of 1.8 ± 0.2 μM (n = 4) and produced the maximal effect at ∼20 μM (Fig. 3). Pretreatment with naloxone methiodide also enhanced the binding, but much higher concentrations were required (EC50 = 114.3 ± 28.2 μM, n = 4) and the maximal extent of increase was only ∼50% of that of naloxone pretreatment (Fig. 3).

Dose-response relationship of effect of pretreatment with naloxone and naloxone methiodide on expression of [3H]diprenorphine binding of the D3.49(164)Q mutant. CHO-D3.49(164)Q cells were cultured without naloxone for at least 96 h, transferred into 6-well plates, and pretreated with different concentrations of naloxone or naloxone methiodide for 24 h. [3H]Diprenorphine binding to the receptors in whole cells was performed after removal of the drugs by washing. Results were normalized to dpm/105 cells, and the control value (binding of cells cultured without any drug) was subtracted from each value. Data were expressed as percentage of the maximal response after naloxone pretreatment. Each value represents mean ± S.E.M. of four independent experiments in duplicate.
Effect of Pretreatment with Opioid Drugs on [3H]Diprenorphine Binding to the Wild-Type and D3.49(164)Q Mutant of the Rat μOR in Whole Cells.
CHO-μOR cells and CHO-D3.49(164)Q cells were cultured without naloxone for at least 96 h. Cells were then grown in the absence (control) or presence of different drugs for another 24 h at concentrations 1,000- to 10,000-fold of their K i values for the μOR.
Each drug tested increases [3H]diprenorphine binding of the D3.49(164)Q mutant, but to different degrees (Fig.4A). The order of increase was naloxone = naltrexone > naloxone methiodide, diprenorphine, morphine, and etorphine ≫ DAMGO. We did not test CTAP in this study since it may not be chemically stable during a 24-h incubation because of the presence of a disulfide bond. We then determined the efficacies of these drugs on the D3.49(164)Q mutant by examining their effects on basal [35S]GTPγS binding of the D3.49(164)Q mutant (Fig. 4A). Cells were cultured in the presence of naloxone to enhance expression of the mutant receptor so that experiments can be carried out. Cells were thoroughly washed to remove naloxone, and membranes were prepared. Naloxone and naltrexone as well as naloxone methiodide inhibited the basal [35S]GTPγS binding, indicating that the three drugs are inverse agonists of the mutant receptor, which is similar to our previous observation (Li et al., 2001). Although diprenorphine seemed to decrease the basal [35S]GTPγS binding, it was not statistically significant compared with the control, demonstrating that it may be a neutral antagonist. Morphine, etorphine, and DAMGO enhanced [35S]GTPγS binding of the D3.49(164)Q mutant, indicating that these drugs are agonists. Thus, it seems that inverse agonists are most effective in enhancing [3H]diprenorphine binding to the D3.49(164)Q mutant, followed by a neutral antagonist and agonists.
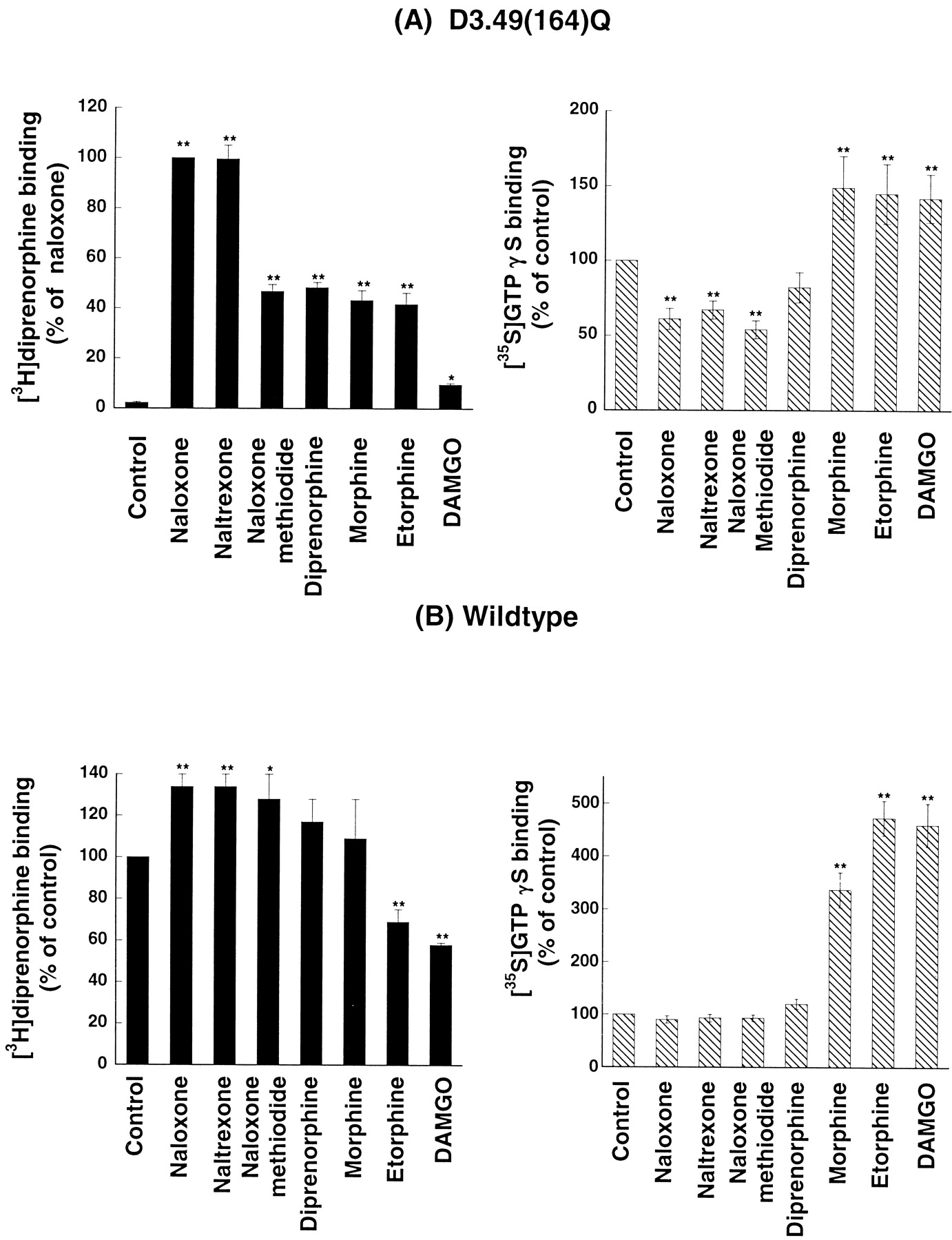
A and B, left panels: effect of pretreatment with opioid drugs on [3H]diprenorphine binding of the D3.49(164)Q mutant and the wild type of the rat μOR in whole cells. CHO-μOR cells and CHO-D3.49(164)Q cells were cultured in medium without naloxone for at least 96 h and then transferred into 6-well plates. Cells were treated for 24 h without drugs (control) or with naloxone (10 μM), naltrexone (10 μM), naloxone methiodide (200 μM), diprenorphine (10 μM), morphine (100 μM), etorphine (10 μM), or DAMGO (10 μM). After removal of the drugs by washing, [3H]diprenorphine binding (∼1 nM) to the receptor in whole cells was performed. Results were normalized to disintegrations per minute [3H]diprenorphine binding/105cells and then calculated as percentage of binding to the receptor in cells treated with 10 μM naloxone, which was ∼21,000 and ∼13,000 dpm/105 cells/nM for the wild-type and the D3.49(164)Q mutant, respectively. Each value represents mean ± S.E.M. of six independent experimentsin duplicate. ∗, p < 0.05; ∗∗, p < 0.01,compared with the control by one-way ANOVA and followed by Dunnett's multiple comparison tests. A and B, right panels: effect of opioid drugs on [35S]GTPγS binding to membranes of CHO-μOR cells and CHO-D3.49(164)Q cells. CHO-μOR cells and CHO-D3.49(164)Q cells were grown in the presence 20 μM naloxone for at least 96 h. Naloxone was removed by extensive washing, and membranes were prepared. [35S]GTPγS binding was performed in the absence (control) or presence of naloxone (10 μM), naltrexone (10 μM), naloxone methiodide (200 μM), diprenorphine (10 μM), morphine (100 μM), etorphine (10 μM), or DAMGO (10 μM). Results were expressed as percentage of the control binding. Each value represents mean ± S.E.M. of five independent experiments in duplicate. ∗∗,p < 0.01, compared with the control by one-way ANOVA followed by Dunnett's multiple comparison tests.
Effects of the same drugs on the expression and [35S]GTPγS binding of the wild type were also assessed (Fig. 4B). Naloxone, naltrexone, and naloxone methiodide pretreatment significantly increased [3H]diprenorphine binding of the wild type, whereas diprenorphine and morphine had no effect, and etorphine and DAMGO lowered the [3H]diprenorphine binding. Although naloxone, naltrexone, naloxone methiodide, and diprenorphine did not have any effects on basal [35S]GTPγS binding, morphine, etorphine and DAMGO enhanced [35S]GTPγS binding to CHO-μOR membranes. Dose-response curves of etorphine, DAMGO, and morphine were generated (data not shown). Etorphine and DAMGO stimulated [35S]GTPγS binding to higher levels than morphine, indicating that etorphine and DAMGO are full agonists, and morphine is a partial agonist at the μOR.
Structural Stability of the Wild-Type and the D3.49(164)Q Mutant of the Rat μOR in Membranes.
CHO-μOR cells or CHO-D3.49(164)Q cells were grown in the presence of 20 μM naloxone for ≥96 h, washed, and membranes were prepared. Incubation of membranes at 37°C in the presence of various protease inhibitors decreased [3H]diprenorphine binding of the wild-type and mutant receptors in a time-dependent manner, whereas incubation at 4°C did not change binding (Fig. 5A). Incubation was carried out in the presence of protease inhibitors to exclude the possibility of proteolysis of the receptor protein, particularly at 37°C. Notably, the rate and extent of the decrease were more pronounced for the D3.49(164)Q mutant than the wild type (Fig. 5A), indicating that the mutant receptor is structurally less stable. In contrast, there was no apparent change in the amount of the mutant receptor proteins in 3 h as detected by Western blot (Fig.5B). In addition, incubation of membranes for 3 h at 37°C yielded similar results for the wild-type and the D3.49(164)Q mutant with and without GTPγS (data not shown). Thus, the loss of binding activity of the D3.49(164)Q mutant represents denaturation, but not degradation of the mutant protein or dissociation of G proteins, indicating that the mutation causes instability in the receptor protein.

Stability of the wild-type and the D3.49(164)Q mutant of the rat μOR in membrane preparations. CHO-μOR or CHO-D3.49(164)Q cells were pretreated with 20 μM naloxone for at least 96 h, and membranes were prepared. A, membranes were incubated at 4°C or 37°C for 0, 1, or 3 h in the presence of protease inhibitors (5 μg/ml antipain dihydrochloride, 0.2 μg/ml aprotinin, 4 μg/ml bestatin, 0.6 μg/ml chymostatin, 0.05 μg/ml E-64, 0.02 mg/ml EDTA, 0.01 mg/ml prefabloc SC, 0.07 μg/ml pepstatin, 1 μg/ml phosphoramidon, 0.05 μg/ml leupeptin). B, Western blot was performed usingmonoclonal antibody against the HA epitope on membranes treated under the same experimental conditions as in A. The figure represents one of the two experiments performed. C, membranes were incubated at 37°C for 3 h with or without 20 μM naloxone or 10 μM DAMGO in the presence of the protease inhibitors described in A and washed thoroughly. A and C, [3H]diprenorphine binding to the wild-type and mutant receptors in membranes was performed. Results were normalized to dpm [3H]diprenorphine binding/20 μg of protein and expressed as percentage of the control (0 h). Each value represents mean ± S.E.M. of five independent experiments in duplicate. ∗∗, p < 0.01 compared with the wild-type receptor (A) and ∗∗, p < 0.01 compared with the control (without naloxone or DAMGO) (C), ##, p< 0.01 compared with the control of the wild type by one-way ANOVA followed by Dunnett's multiple comparison tests.
Incubation of CHO-D3.49(164)Q cell membranes at 37°C for 3 h in the presence of 20 μM naloxone or 10 μM DAMGO partially reversed the decrease in [3H]diprenorphine binding (Fig.5C), indicating that these ligands stabilize the structure of the D3.49(164)Q mutant receptor necessary for [3H]diprenorphine binding. Naloxone and DAMGO, however, only slightly increased [3H]diprenorphine binding of the wild type (Fig. 5C).
Effects of Coexpression of the Dominant-Negative Mutants GRK2-K220R, Arrestin-2(319–418), or Dynamin I-K44A on [3H]Diprenorphine Binding of the D3.49(164)Q Mutant Receptor after Naloxone Removal.
Coexpression of the dominant-negative mutants GRK2-K220R, arrestin-2(319–418) or dynamin I-K44A partially blocked the decrease in [3H]diprenorphine binding of the D3.49(164)Q mutant receptor after naloxone removal, compared with the vector control (Fig. 6A). These results indicate that the D3.49(164)Q mutant is constitutively internalized by GRK-, arrestin-2-, and dynamin-dependent mechanisms. Thus, naloxone acts in part by blocking constitutive internalization of the D3.49(164)Q mutant.

Effects of coexpression of the dominant-negative mutants GRK2-K220R, arrestin-2(319–418), or dynamin I-K44A (A) or the dominant-negative mutants rab5A-N133I or rab7-N125I on the decrease in [3H]diprenorphine binding of the D3.49(164)Q mutant receptor after naloxone removal (B). CHO-D3.49(164)Q cells cultured in the presence of 20 μM naloxone for over 96 h were transiently transfected with each of the cDNA constructs of the dominant-negative mutants GRK2-K220R, arrestin-2(319–418), dynamin I-K44A, or the vector (A) or each of the cDNA constructs of the dominant-negative mutants rab5A-N133I or rab7-N125I or the vector (B). Transfected cells were cultured in medium containing 20 μM naloxone for 24 h and then without naloxone for another 24 h. Untransfected cells grown in medium containing 20 μM naloxone for the entire period serve as the control. [3H]Diprenorphine binding to the mutant in whole cells was performed. Results were normalized to disintegrations per minute [3H]diprenorphine binding/105 cells and then expressed as percentage of binding in the presence of 20 μM naloxone. Each value represents mean ± S.E.M. of six independent experiments in duplicate. ∗∗, p < 0.01 compared with that of vector by ANOVA and followed by Dunnett's multiple comparison tests.
Effects of Cotransfection of the Dominant-Negative Mutants Rab5A-N133I or Rab7-N125I on [3H]Diprenorphine Binding of the D3.49(164)Q Mutant Receptor after Naloxone Removal.
Rab proteins are a family of more than 40 mammalian proteins. They are approximately 25-kDa Ras-related GTPases that are associated with distinct intracellular membranes where they control vesicle trafficking between intracellular compartments (Olkkonen and Stenmark, 1997). Rab5 is mainly involved in early endosome transport, and the fusion of endocytic vesicles with endosomes (Bucci et al., 1992). Rab7 has been implicated in membrane transport from early endosomes to late endosomes (Feng et al., 1995) or late endosomes to lysosomes (Meresse et al., 1995). Expression of the dominant-negative mutants rab5A-N133I or rab7-N125I partially blocked the decrease in [3H]diprenorphine binding to the D3.49(164)Q mutant receptor after naloxone removal, compared with the vector control (Fig. 6B). Thus, the internalized the D3.49(164)Q mutant receptors are trafficked via a rab5- and rab7-mediated pathway, probably involved early endosomes, late endosomes, and lysosomes.
Effects of Chloroquine and Proteasome Inhibitor I on the Decrease in [3H]Diprenorphine Binding to the D3.49(164)Q Mutant Receptor after Naloxone Removal.
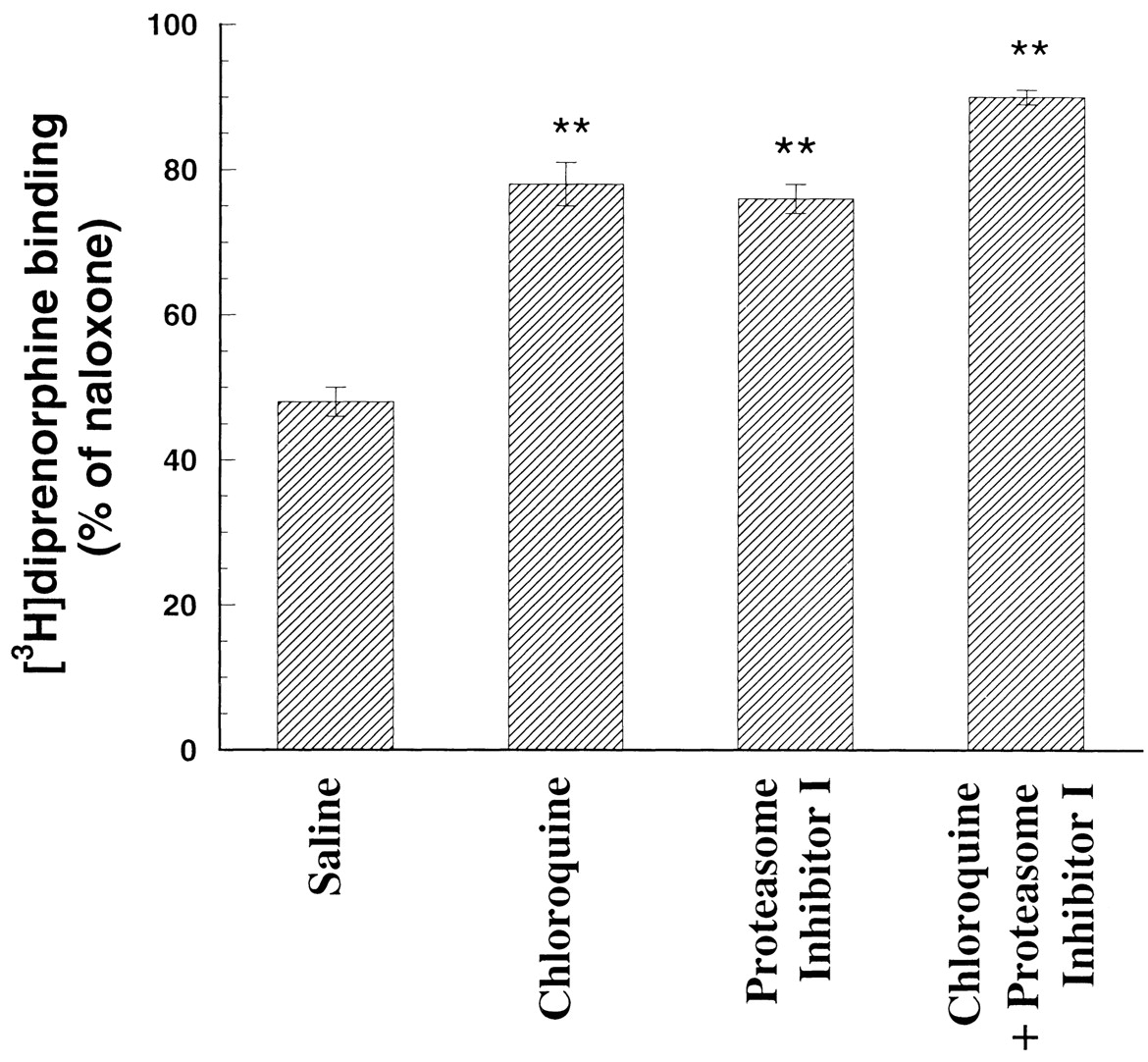
We examined effects of pretreatment of CHO-D3.49(164)Q cells with the lysosomal enzyme inhibitor chloroquine and the proteasome inhibitor I or both to determine whether lysosomes or proteasomes are involved in the degradation of the D3.49(164)Q mutant after naloxone removal. A 6-h, rather than 24-h, treatment was carried out to ensure that sufficiently high concentrations of both inhibitors are available for the entire period, even with degradation of the drugs. Chloroquine or proteasome inhibitor I partially reduced the decrease in [3H]diprenorphine binding to the D3.49(164)Q mutant receptor after naloxone removal, and the combination of the two had additive effects (Fig. 7). These results indicate that the D3.49(164)Q CAM is constitutively degraded by lysosomes and proteasomes.

Effects of chloroquine and proteasome inhibitor I on [3H]diprenorphine binding of the D3.49(164)Q mutant receptor after naloxone removal. CHO-D3.49(164)Q cells pretreated with 20 μM naloxone for at least 96 h were transferred into 6-well plates and cultured for another 24 h in the presence of 20 μM naloxone. Naloxone was removed and cells were treated with 50 μM chloroquine, 5 μM proteasome inhibitor I, 50 μM chloroquine plus 5 μM proteasome inhibitor I, or saline for 6 h. Cells grown in the presence of naloxone without either inhibitor serving as the control. Cells were washed with 3 × 3 ml/well of cold Krebs' buffer, harvested, and [3H]diprenorphine binding to the mutant opioid receptor in whole cells was performed. Results were normalized to disintegrations per minute [3H]diprenorphine binding/105 cells and then expressed as percentage of the control. Each value represents mean ± S.E.M. of four independent experiments in duplicate. ∗∗, p < 0.01 compared with that of control by ANOVA and followed by Dunnett's multiple comparison tests.
Discussion
In this study, we have demonstrated that the D3.49(164)Q CAM of the rat μOR is structurally less stable than the wild type and is constitutively internalized and down-regulated. Naloxone up-regulates the expression of the mutant primarily by two mechanisms: stabilization of the structure and blockade of its constitutive internalization and down-regulation.
Naloxone Acts Extra- and Intracellularly.
Incubation of cells with either naloxone or naloxone methiodide increased the expression of [3H]diprenorphine binding of the D3.49(164)Q mutant in a dose-dependent manner (Fig. 3). However, the maximal effect of naloxone methiodide was only 50% of that of naloxone. Since naloxone methiodide is permanently positively charged, which limits its permeability through membranes, these results indicate that naloxone acts on extracellular and intracellular sites to up-regulate the mutant. This is consistent with the observation that treatment of cells harboring CAMs of α1B- and β2-ARs with inverse agonists increased plasma membrane and diffuse intracellular staining of the receptors (McLean et al., 1999; Stevens et al., 2000).
In intact cells, after removal of naloxone, binding and protein level of the D3.49(164)Q mutant decreased with time (Fig. 2, A and C). In contrast, incubation of membranes at 37°C in the presence of protease inhibitors decreased binding, without changing the receptor protein level (Fig. 5, A and B) and incubation with or without GTPγS yielded similar results. Thus, the decrease in binding activity of the CAM in membranes is due to denaturation of the protein, but not degradation of the receptor or uncoupling of the receptor from G proteins. The difference in the actions of naloxone on intact cells and on membranes suggests that there are cellular mechanisms involved in the decline of the D3.49(164)Q mutant in cells. Naloxone pretreatment of cells caused a much greater enhancement in the binding of the D3.49(164)Q mutant than DAMGO (Fig. 4A), although both stabilized the D3.49(164)Q mutant protein to similar extents (Fig. 5C). Hence, naloxone and DAMGO have different effects on the cellular mechanisms.
Structural Instability of the D3.49(164)Q Mutant.
Upon incubation of membranes at 37°C, the D3.49(164)Q mutant lost binding activities at a greater rate and to a greater extent than the wild type, indicating that the mutant is structurally less stable than the wild type. These results are in accord with previous reports on CAMs of β2-adrenergic and H2histamine receptors (Gether et al., 1997a; Samama et al., 1997;Rasmussen et al., 1999; Alewijnse et al., 2000). The observation that naloxone and DAMGO stabilized the structure of the D3.49(164)Q mutant in membranes (Fig. 5C) is consistent with the reports that the structures of CAMs of β2-adrenergic and H2 histamine receptors, either purified or, in membranes, were stabilized by agonists, antagonists, and inverse agonists (Gether et al., 1997a; Samama et al., 1997; Rasmussen et al., 1999; Alewijnse et al., 2000).
Up-Regulation of the D3.49(164)Q Mutant by Ligands.
We found that ligands with different efficacies, including inverse agonists, a neutral antagonist, and agonists, up-regulated the D3.49(164)Q mutant. These findings are similar to those of Gether et al. (1997a) andAlewijnse et al. (2000), but slightly different from that of Samama et al. (1997). Gether et al. (1997a) showed that treatment with an inverse agonist, a neutral antagonist, or an agonist increased the expression of a CAM β2-AR in insect Sf9 cells. Histamine enhanced the expression of a CAM of the H2histamine receptor but to a lesser extent than ranitidine, an inverse agonist (Alewijnse et al., 2000). Samama et al. (1997) reported that in transgenic mice expressing a CAM β2-AR, treatment with inverse agonists, neutral antagonists, or a partial agonist, but not full agonists, profoundly up-regulated the CAM receptor.
The observation that pretreatment with inverse agonists enhanced the expression of the D3.49(164)Q CAM is similar to previous findings showing that inverse agonists increased expression levels of CAMs of β2-adrenergic, H2histamine, and thyrotropin-releasing hormone receptors (Pei et al., 1994; Heinflink et al., 1995; MacEwan and Milligan, 1996b; Gether et al., 1997a; Lee et al., 1997; Milligan and Bond, 1997; Samama et al., 1997; Alewijnse et al., 2000) [also see reviews, (Milligan and Bond, 1997; Leurs et al., 1998)]. Only a few studies examined effects of agonists and neutral antagonists. Agonist treatment was found to double the density of the CAM T6.34(373)K α2A-AR (Betuing et al., 1997). In contrast, neutral antagonists did not enhance the expression of a CAM β2-AR and the wild-type H2 histamine receptor, which has constitutive activity (MacEwan and Milligan, 1996a,b; Smit et al., 1996). Stevens et al. (2000) reported that different CAMs of the α1B-AR responded differently to ligands, adding complexity to regulation of CAMs. Although the R6.29(288)K/K6.31(290)H/A6.34(293)L CAM was up-regulated by ligands with antagonist/inverse agonist properties, both the D3.49(142)A and A6.34(293)E CAMs were not (Lee et al., 1997; Stevens et al., 2000). In contrast, phenylephrine, an agonist, increased the expression of all three CAMs (Stevens et al., 2000).
Naloxone up-regulated the D3.49(164)Q CAM in a dose-dependent manner with an EC50 of ∼1.8 μM and reaching a maximal response at 20 μM. These concentrations are about 100 and 1,000 times its K i value for the mutant, respectively. Even higher concentrations of naloxone methiodide were required. Similarly, up-regulation of a CAM of the α1B-AR by inverse agonists/antagonists occurred with EC50 values in μM, despite having low nanomolar affinity (Stevens et al., 2000). The reasons for the requirement for such high concentrations are not clear. It is likely that since a 24-h incubation at 37°C was carried out, the drugs might not be stable throughout the incubation period. In addition, the drugs might bind to bovine serum albumin in fetal bovine serum, thus reducing their effective concentrations.
Constitutive Internalization of the D3.49(164)Q Mutant.
Coexpression of each of the dominant-negative mutants GRK2-K220R, arrestin-2(319–418), or dynamin I-K44A in CHO-D3.49(164)Q cells partially blocked the decrease in receptor level after naloxone removal. These results indicate that in the absence of naloxone, the D3.49(164)Q mutant undergoes constitutive internalization via a GRK-, arrestin-2-, and dynamin-dependent pathway. This is the same pathway by which the wild-type μOR is internalized upon activation by some agonists. After stimulation by agonists such as etorphine and peptides, the μOR undergoes phosphorylation by GRKs followed by plasma membrane translocation of β-arrestin- and dynamin-dependent receptor internalization (Whistler and von Zastrow, 1998; Zhang et al., 1998). Activation of the μOR by morphine or levorphanol, however, did not promote internalization (Keith et al., 1996; Sternini et al., 1996). Thus, the D3.49(164)Q mutant seems to adopt conformations that are similar to those induced by DAMGO or etorphine, but not those caused by morphine.
The observation that GRK and arrestin-2 were involved in constitutive internalization of the D3.49(164)Q mutant is in accord with the report of Pei et al. (1994) and Mhaouty-Kodja et al. (1999).Pei et al. (1994) found that a CAM of the β2-AR was phosphorylated by GRK2 in the absence of agonist. Mhaouty-Kodja et al. (1999) reported that the A6.34(293)E CAM of the α1B-AR seemed to cause a higher degree of constitutive arrestin-2-green fluorescent protein translocation to membranes than the wild-type receptor. In contrast, the D3.49(142)A CAM of the α1B-AR was found not to have enhanced constitutive phosphorylation or internalization (Mhaouty-Kodja et al., 1999). Whether the D3.49(164)Q mutant of the μOR is constitutively phosphorylated is being investigated.
Trafficking of the D3.49(164)Q Mutant to Endosomes and Lysosomes.
Expression of the dominant-negative mutants rab5A-N133I (Bucci et al., 1992) or rab7-N125I (Feng et al., 1995) in CHO-D3.49(164)Q cells partially blocked the decrease in [3H]diprenorphine binding after naloxone removal. These results indicate that the D3.49(164)Q mutant is constitutively internalized and trafficked in a rab5- and rab7-dependent pathway, probably via endocytic vesicles to early endosomes to late endosomes probably to lysosomes. Etorphine and various peptide agonists promote internalization of the μOR to transferrin-containing endosomes (Arden et al., 1995; Keith et al., 1996). We have reported previously that U50,488H-induced down-regulation of the human κOR involves trafficking of the receptor by rab5- and rab7-dependent mechanisms (Li et al., 2000). However, whether rab5 and rab7 are involved in agonist-induced down-regulation of the μ-opioid receptor has not been reported.
Constitutive Degradation of the D3.49(164)Q Mutant by Lysosomes and Proteasomes.
Pretreatment of CHO-D3.49(164)Q cells with chloroquine or proteasome inhibitor I reduced the down-regulation after naloxone removal, and the two drugs have an additive effect. These results indicate that both lysosomes and proteasomes are involved in the degradation of the D3.49(164)Q mutant after naloxone removal. A likely scenario is that a fraction of the receptors was degraded in lysosomes, whereas others were degraded in proteasomes. Another possibility is that proteasome degradation of a protein or proteins other than the receptor is required for targeting and transport of the receptor to lysosomes and degradation of the receptor by lysosomes, as suggested by Hicke (1999). A third possibility is that because of the conformational instability, some D3.49(164)Q receptor proteins become denatured and are degraded by proteasomes before being trafficked to membranes. We have previously shown that both lysosomes and proteasomes participate in the degradation of the human κOR during agonist-induced down-regulation (Li et al., 2000). Proteasomes have been demonstrated to be involved in agonist-induced down-regulation of the μ- and δ-opioid receptors (Chaturvedi et al., 2001).
Proposed Actions of Naloxone.
Only after naloxone pretreatment could the D3.49(164)Q mutant protein be detected, indicating that new receptor protein synthesis is involved. The continued presence of naloxone is required to maintain high levels of expression, since removal of naloxone led to decreased and eventually undetectable receptor levels. Naloxone seems to act on multiple sides. We propose that as receptors are synthesized and processed, naloxone binds to the receptor proteins and stabilizes the structures. Without naloxone, some newly synthesized mutant receptors become denatured and degraded. Naloxone binding enhanced the amount of the mutant receptors being trafficked to plasma membranes. After naloxone removal, some of the D3.49(164)Q mutant receptors in plasma membranes are denatured and some are constitutively internalized. Degradation of internalized receptors and denatured receptors seem to occur in lysosomes and proteasomes. Naloxone binding to receptors in membranes stabilizes the structure and inhibits constitutive internalization and down-regulation of the mutant.
Differential Effects of Drugs on Up-Regulation of the D3.49(164)Q Mutant.
At saturation concentrations, the inverse agonists naloxone and naltrexone are more effective in enhancing the expression of the mutant than the neutral antagonist diprenorphine and the agonists morphine, etorphine, and DAMGO (Fig. 4A). It seems that ligands, regardless of their efficacies, can stabilize the CAM receptor protein, but only inverse agonists and not agonists, can block internalization and down-regulation. Whether neutral antagonists can inhibit internalization and down-regulation of the mutant is not clear. It is likely that the distinct conformations induced by inverse agonists are more resistant to the actions of cellular machinery leading to internalization and down-regulation than those induced by neutral antagonists or agonists. The finding of Gether et al. (1997b)supports this argument. These researchers showed that in the wild-type and a CAM of the β2-AR labeled with a fluorophore, inverse agonists caused changes in fluorescence that are opposite to agonist-induced alterations.
Although pretreatment with etorphine and DAMGO caused down-regulation of the wild type, morphine had no significant effects. Based on these results, it is inferred that the effects of etorphine and DAMGO on the expression of the D3.49(164)Q mutant are the combined result of two opposing actions: stabilizing the CAM structure and promoting down-regulation, the proportion of which may vary with the drug. In contrast, since morphine did not down-regulate the wild type, its effect on the expression of the D3.49(164)Q mutant is probably due to stabilization of the receptor protein only.
DAMGO treatment of cells seems to be less effective in enhancing the expression the D3.49(164)Q CAM than etorphine and morphine although they all act as agonists. The reasons for this difference may be 2-fold. DAMGO is much more hydrophilic than etorphine and morphine and thus has much lower permeability through plasma membranes, which limits its action to extracellular receptors. In addition, since incubation was performed at 37°C for 24 h, DAMGO, a peptide, may be less stable than etorphine and morphine under such conditions.
Different Efficacies of Drugs at the Wild-Type and the D3.49(164)Q Mutant.
Naloxone, naltrexone, and naloxone methiodide displayed inverse agonist activities at the D3.49(164)Q mutant but not at the wild-type receptor. Morphine stimulated the D3.49(164)Q mutant to a similar level as etorphine and DAMGO, demonstrating that it is a full agonist at the mutant, whereas it is a partial agonist at the wild type. Thus, drugs have different efficacies at the wild-type and the D3.49(164)Q mutant. These results are consistent with the finding that both agonist and antagonist elicit more profound structural changes in CAM of the β2-AR than in the wild type (Gether et al., 1997a).
Use of [3H]Diprenorphine for Opioid Receptor Binding.
We used [3H]diprenorphine for opioid receptor binding in whole cells to assess the total receptor, both intracellular and extracellular, since [3H]diprenorphine is a hydrophobic ligand. Approximately 15% of the total receptors were intracellular for both the wild-type and the D3.49(164)D mutant when total and extracellular receptors were assessed by use of naloxone and DAMGO to define nonspecific binding, respectively (J. L. and L.-Y. L.-C., unpublished observation).
Conclusion.
The D3.49(164)Q CAM of the μOR is inherently unstable and undergoes tonic agonist-independent internalization and down-regulation via GRK-, arrestin-2-, dynamin-, rab5-, and rab7-dependent pathway. Because of these properties, the constitutive activities that we detected for this CAM may represent an underestimate of the actual activities. Naloxone up-regulates the D3.49(164)Q mutant by stabilizing the mutant protein and inhibiting its constitutive internalization and down-regulation. These two mechanisms are probably to be involved in up-regulation of other GPCR CAMs by inverse agonists (Leurs et al., 1998). The proportion of the contribution from either mechanism depends on the inherent property of the CAM. For example, CAMs of the β2-adrenergic and H2 histamine receptors had reasonable, albeit lower, expression levels compared with the wild type, suggesting that they are down-regulated but to a lesser degree. The up-regulatory effect of inverse agonists is attributed more to their stabilization effects (Gether et al., 1997a; Samama et al., 1997; Rasmussen et al., 1999; Alewijnse et al., 2000). In contrast, the expression of the D3.49(164)Q mutant of the μOR was undetectable, indicating that this CAM is profoundly down-regulated. Naloxone, an inverse agonist, caused a dramatic increase in the expression of this CAM. In addition to stabilizing the mutant protein structure, the inhibitory effect of naloxone on constitutive down-regulation of this CAM becomes evident.
Acknowledgments
We thank the following researchers who have generously given us the cDNA constructs used in this study: Dr. Lei Yu of the University of Cincinnati, OH, Dr. Jeffrey Benovic of Thomas Jefferson University (Philadelphia, PA), and Dr. A. Wandinger-Ness of the University of New Mexico.
Footnotes
- Received March 13, 2001.
- Accepted July 12, 2001.
-
This work was supported by National Institutes of Health Grants DA04745, DA10702 and DA11263.
Abbreviations
- TM
- transmembrane domain
- GPCR
- G protein coupled-receptor
- AR
- adrenergic receptor
- CAM
- constitutively active mutant
- CHO cells
- Chinese hamster ovary cells
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly-ol]-enkephalin
- GTPγS
- guanosine 5′-O-(3-thiotriphosphate)
- HA
- hemagglutinin
- μOR
- μ-opioid receptor
- SSC
- standard saline citrate
- ANOVA
- analysis of variance
- GRK
- G protein-coupled receptor kinase
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}