


Abstract
Fusion proteins were generated by attachment of green fluorescent protein (GFP) to the C-terminal tail of either the wild-type human β2-adrenoceptor or a form with enhanced constitutive activity. Sustained treatment of HEK293 cells stably expressing the constitutively active mutant (CAM) β2-adrenoceptor-GFP with the inverse agonist betaxolol resulted in a marked up-regulation of the fusion protein that could be monitored by both fluorescence and immunoblotting of membrane fractions. This was not observed for the wild-type β2-adrenoceptor-GFP. Addition of the agonist isoprenaline to CAM β2-adrenoceptor-GFP expressing cells previously treated with betaxolol resulted in rapid internalization of the receptor into punctate intracellular vesicles in a manner similar to wild-type β2-adrenoceptor-GFP. A range of “β-blockers” replicated the up-regulation of the CAM β2-adrenoceptor-GFP, although pharmacological specificity was maintained, as it was not produced by α1- and α2-adrenoceptor-selective antagonists/inverse agonists. Parallel intact cell binding studies with [3H]dihydroalprenolol confirmed up-regulation of the CAM β2-adrenoceptor-GFP by betaxolol but failed to predict the optically monitored up-regulation produced by high concentrations of alprenolol. The cellular distribution of the up-regulated CAM β2-adrenoceptor-GFP was not identical after sustained treatment of the cells with different β-blockers. Inverse agonists, able to reduce basal intracellular cAMP levels, such as betaxolol and ICI118551, resulted in both increased plasma membrane receptor and increased diffuse intracellular staining. In contrast, treatment with labetolol and alprenolol resulted in a significant fraction of the intracellular receptor displaying a punctate distribution pattern. These ligands displayed substantial agonism to stimulate intracellular cAMP levels via the CAM β2-adrenoceptor-GFP.
Considerable interest has recently been afforded to studies in which certain mutations have been introduced into G protein-coupled receptors (GPCRs) to result in agonist-independent signaling (also described as constitutive activity) of the expressed mutant GPCR (Lefkowitz et al., 1993; Scheer and Cotecchia, 1997; Leurs et al., 1998). Such experiments have been considered to shed light on possible structural alterations in the GPCR that occur upon agonist binding to result in activation of a cognate G protein and thus regulate the activity of downstream effector enzymes. Such strategies appear to possess validity because, in the case of the human β2-adrenoceptor, for example, one of the structural modifications associated with agonist binding to the wild-type GPCR is a movement of transmembrane helix VI that can be measured by the positioning of residue Cys285 (Gether et al., 1997b). In a constitutively active mutant (CAM) form of this GPCR, this same Cys residue is closer to the ligand-binding pocket than in the ligand-unoccupied wild-type receptor (Javitch et al., 1997).
Perhaps the most studied of the CAM GPCRs is a form of the human β2-adrenoceptor in which a short segment of the third intracellular loop was replaced with the corresponding region from the α1B-adrenoceptor (Lefkowitz et al., 1993; Samama et al., 1993, 1994). We have previously examined a number of features of this modified GPCR following stable expression in neuroblastoma × glioma hybrid, NG108-15, cells (MacEwan and Milligan, 1996 a,b; Stevens and Milligan, 1998). We have been most interested by observations that sustained treatment of cells expressing the CAM β2-adrenoceptor with certain “β-blockers”, including betaxolol, results in the subsequent detection of higher levels of3H-β2-adrenoceptor antagonist binding sites in membrane preparations derived from these cells (Pei et al., 1994; MacEwan and Milligan, 1996a,b). Betaxolol has the characteristics of an inverse agonist (Pei et al., 1994; Samama et al., 1994; MacEwan and Milligan, 1996a,b), i.e., a ligand that suppresses the basal signaling capacity of a GPCR. This effect appeared to be selective, because certain other β-blockers, including alprenolol, did not mimic these effects. Furthermore, the effects of the inverse agonists on CAM β2-adrenoceptor levels were not related to their capacity to suppress the GPCR-mediated activation of basal adenylyl cyclase activity (MacEwan and Milligan, 1996b).
In contrast to the effects on cellular levels of the CAM β2-adrenoceptor, we have noted that equivalent treatment with betaxolol had a much less dramatic effect on cellular levels of the wild-type human β2-adrenoceptor when it too was expressed stably in NG108-15 cells (MacEwan and Milligan, 1996a). Because betaxolol treatment of CAM β2-adrenoceptor–expressing cells had little effect on the levels of mRNA encoding this receptor (MacEwan and Milligan, 1996a), we have suggested that inverse agonists of the CAM β2-adrenoceptor function to stabilize an inherently unstable protein and thus decrease its rate of degradation. In the face of an apparently unchanged rate of synthesis, then, the inverse agonist causes an increase in steady-state levels of the GPCR. Kobilka and colleagues have gone further, indicating that any appropriate receptor ligand, whether agonist, neutral antagonist, or inverse agonist, may act to stabilize the structure of the purified CAM β2-adrenoceptor and slow its denaturation (Gether et al., 1997a,b).
In the current study, we have constructed and stably expressed forms of the wild-type and CAM β2-adrenoceptor that have had a modified form of the 27-kDa green fluorescent protein (GFP) derived from Aequorea victoria added in-frame to their C-terminal tail. We use these constructs in an intact cell setting to directly visualize ligand regulation of the levels and cellular distribution of the CAM β2-adrenoceptor and to explore whether ligand-induced up-regulation of this receptor is restricted to compounds that display inverse agonism.
Materials and Methods
[3H]dihydroalprenolol (DHA; 64 Ci/mmol), [3H]CGP-12177 (44 Ci/mmol), [3H]adenine, and [3H]cAMP were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). All reagents for cell culture were obtained from Life Technologies, Inc. (Paisley, Strathclyde, UK). Receptor ligands were obtained from RBI (Gillingham, Dorset, UK). All other reagents were obtained from Sigma or Fisons (Loughborough, UK) and were of the highest purity available.
Construction of GFP-Tagged Forms of the β2-Adrenoceptor.
Human wild-type β2-adrenoceptor in pcDNA3 was amplified by polymerase chain reaction (PCR) using a HindIII-FLAG forward primer, 5′-AAAAAAAAGCTTGCCACCATGGACTACAAGGACGACGATGATAAGGGGCAACCCGGGAACGGC-3′, and a BamHI reverse primer, 5′-AAAAAGGATCCTCCCGCCAGCAGTGAGTCATTTGTA-3′. This removed the stop codon and the initiating methionine (start codon) of the β2-adrenoceptor, with an initiator ATG being present in the N-terminally added FLAG epitope tag (ATG GAC TAC AAG GAC GAC GAT GAT AAG). The PCR product was digested with HindIII and BamHI and the resulting fragment ligated into pcDNA3. The sequence encoding amino acids 172 to 291 was restricted from this construct using KpnI/HpaI and replaced by the equivalent region of the CAM β2-adrenoceptor (Samama et al., 1993, 1994). A modified form of GFP (Zernicka-Goetz et al., 1997) was also amplified by PCR using a BamHI forward primer, 5′-AAAAAGGATCCAGTAAAGGA GAAGAACTTTTC-3′, and an XbaI reverse primer, 5′-TGCTCTAGATTATTTGTATAGTTCATCCATGCCATG-3′. This removed the initiating methionine of GFP, and the resulting PCR product was digested and linked in frame to generate the wild-type or CAM β2-adrenoceptor-GFP constructs.
Transient and Stable Transfection of HEK293 Cells.
HEK293 cells were maintained in minimum essential medium (MEM; Sigma) supplemented with 0.292 g/l l-glutamine and 10% newborn calf serum at 37°C. Cells were grown to 60 to 80% confluency before transient transfection. Transfection was performed using LipofectAMINE reagent (Life Technologies, Inc.) according to the manufacturer's instructions. To generate cell lines stably expressing the various constructs, cells were seeded/diluted and maintained in MEM supplemented with 1 mg/ml Geneticin (Life Technologies, Inc.) 2 days after transfection. The medium was replaced every 3 days with MEM containing 1 mg/ml Geneticin. Clonal expression was initially examined by fluorescence microscopy for the GFP-containing constructs. Clones for further study, and those expressing the non-GFP-tagged forms of the receptors, were selected and expanded, and3H-ligand-binding studies were performed to examine the receptor expression levels.
Confocal Laser Scanning Microscopy.
Cells were observed using a laser scanning confocal microscope (Zeiss Axiovert 100; Zeiss, Oberkochen, Germany) with a Zeiss Plan-Apo 63 × 1.40 NA oil immersion objective, pinhole of 35, and electronic zoom 1 or 3. The GFP was exited using a 488-nm argon/krypton laser and detected with a 515- to 540-nm band pass filter. The images were manipulated with Zeiss LSM or MetaMorph (Universal Imaging Corporation, West Chester, PA) software. Two different protocols for preparation of cells were used. When examining the time course of internalization and recycling, live cells were used. The cells were grown on glass coverslips and mounted on the imaging chamber. The cells were maintained in Krebs-Ringer-HEPES buffer (KRH) and the temperature was maintained at 37°C. In other studies, fixed cells were used. The cells on glass coverslips were washed with PBS and fixed for 20 min at room temperature using 4% paraformaldehyde in PBS/5% sucrose, pH 7.2. After one wash with PBS, the coverslips were mounted on microscope slides with 40% glycerol in PBS.
3H-Ligand-Binding Studies.
CAM β2-adrenoceptor-GFP cells were grown in 6-cm dishes and treated with or without 10 μM betaxolol or various concentrations of alprenolol for 24 h. In some cases, betaxolol-treated cells were subsequently exposed to 10 μM isoprenaline for 30 min. After treatment, the cells were washed 3 times with ice-cold PBS (2.7 mM KCl, 137 mM NaCl, 1.5 mM KH2PO4, 8 mM Na2HPO4, pH 7.4). Cells were then detached from plates with PBS/0.5 mM EDTA, pelleted, and resuspended in ice-cold KRH (130 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM CaCl2, 20 mM HEPES, 1.2 mM Na2PO4, 10 mM glucose, 0.1% BSA; pH 7.4). After the cells were counted in a hemocytometer, approximately 100,000 cells were added to each assay tube.
For binding studies, a single concentration of [3H]DHA (2 nM) or [3H]CGP-12177 (10 nM) was used to measure total cell receptor and cell surface receptor, respectively. Parallel studies with 10 μM propranolol allowed assessment of nonspecific binding. [3H]DHA binding assays were performed at 30°C for 45 min and [3H]CGP-12177 binding was performed at 14°C for 2.5 h in KRH. All experiments were terminated by rapid filtration through Whatman GF/C filters (Brandel Inc., Gaithersburg, MD) followed by three washes with ice-cold TE (75 mM Tris, 1 mM EDTA; pH 7.4) buffer.
Intact Cell Adenylyl Cyclase Activity Measurements.
Intact cell adenylyl cyclase activity measurements were performed essentially as described previously (Wong, 1994; Merkouris et al., 1997). Cells were split into wells of a 12-well plate, and the cells were allowed to reattach. Cells were then incubated in medium containing [3H]adenine (1.5 μCi/well) for 16 to 24 h. The generation of [3H]cAMP in response to the treatment of the cells with various ligands and other reagents was then assessed.
Electrophoresis and Immunoblot Analysis.
A borate-based electrophoretic buffer system (Poduslo, 1981) was used, with some modifications. The resolving polyacrylamide gel was made of 10% acrylamide, 0.0625% bisacrylamide, 0.1 M Tris (pH 8.5), 0.1 M boric acid, 0.0025 M EDTA, 0.1% SDS, 0.005%N,N,N,N′tetramethylethylenediamine, and 0.1% ammonium persulfate. The stacking gel was of the same composition, except that it contained 4% acrylamide. The borate electrophoresis running buffer was composed of 0.1 M Tris, 0.1 M boric acid, 0.0025 M EDTA, and 0.1% SDS (pH 8.5). Standard and borate electrophoresis were run for 1 h at 200 and 150 V, respectively, using a Mini Protean II gel kit (Bio-Rad, Richmond, CA). After SDS-polyacrylamide gel electrophoresis, proteins were electrophoretically transferred to nitrocellulose. The membrane was blocked for 1 h in 3% fat-free milk in PBS-T buffer (PBS containing 0.1% Tween 20). After a brief wash in PBS-T buffer, the membrane was incubated overnight at 4°C with an appropriate primary antibody diluted in PBS-T buffer containing 1% fat-free milk. A GFP polyclonal antibody (Clontech Laboratories, Palo Alto, CA) was used for the detection of the constructs. The primary antibody was then removed, and the blot was washed extensively in PBS-T buffer. Subsequent incubation with secondary antibody (donkey anti-rabbit IgG conjugated with horseradish peroxidase, Scottish Antibody Production Unit, Carluke, Scotland) proceeded for 2 h at room temperature, and after extensive washing in PBS-T buffer, the blot was visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech). Quantitative analysis of specific bands was performed by scanning with an imaging densitometer, GS-670 (Bio-Rad).
Studies in Microtiter Plates.
Cells were seeded into black Costar view plates on the day before the experiment. On the day of the experiment, the media were removed from the cells and drug was added to the well in a final volume of 100 μl. The experiments were performed in phenol red-free F12 media containing 10% fetal calf serum. A Spectrafluor Plus fluorimeter was used to read the plates, reading from the bottom at a gain of 100. A blank plate was initially read on the flourimeter, and then the plates of cells were read at time 0 and after a 22-h incubation at 37°C with drug. Results were calculated by subtracting the blank plate from the fluorescence values obtained to control for plate autofluorescence.
Miscellaneous.
All experiments were performed at least three times using different batches of cells.
Results
A PCR-based strategy was used to link a cDNA encoding a modified form of the GFP from A. victoria with enhanced autofluorescence properties (Zernicka-Goetz et al., 1997) with cDNAs encoding both the wild-type β2-adrenoceptor and a CAM form of this GPCR, produced by replacement of a small segment of the distal end of the third intracellular loop with the equivalent segment of the hamster α1B-adrenoceptor (Samama et al., 1993). These fusion proteins were anticipated to encode single open reading frames in which the C terminus of the GPCR was linked directly to the N terminus of GFP. Following transient transfection of these constructs and visualization on a fluorescence microscope to confirm successful expression and autofluorescence (data not shown), both of these constructs were expressed stably in HEK293 cells. Individual clones were identified based on a combination of appropriate autofluorescence and specific binding of the β-adrenoceptor antagonist [3H]DHA and subsequently expanded. In clones expressing the wild-type β2-adrenoceptor-GFP construct, confocal microscopy performed on intact cells grown on a glass coverslip demonstrated substantial amounts of the GFP-derived autofluorescence to be plasma membrane delineated (Fig. 1). Addition of the β-adrenoceptor agonist isoprenaline (10−5 M) resulted in a time-dependent internalization of the construct into discrete, punctate intracellular vesicles (Fig. 1) as has previously been reported for similar constructs (Barak et al., 1997; Kallal et al., 1998). The wild-type β2-adrenoceptor-GFP construct that had been internalized during a 30-min treatment with isoprenaline could be recycled to the plasma membrane following removal of isoprenaline and its replacement by the β-adrenoceptor blocker alprenolol (10−5 M) (data not shown). Saturation binding experiments with [3H]DHA on membranes of cells expressing either unmodified wild-type β2-adrenoceptor or the wild-type β2-adrenoceptor-GFP construct indicated that both forms of the receptor bound this ligand with similar affinity (Table 1).

Plasma membrane location of wild-type-β2-adrenoceptor-GFP and internalization in response to isoprenaline. The wild-type β2-adrenoceptor-GFP construct was expressed stably in HEK293 cells, and individual clones were isolated. A patch of cells was imaged in the confocal microscope in the absence of agonist (A) and following addition of 10 μM isoprenaline for 10 (B) and 30 (C) min.
Ligand-binding characteristics of wild-type and CAM β2-adrenoceptor-GFP constructs
Clones expressing the CAM β2-adrenoceptor-GFP construct were also isolated. Saturation binding experiments with [3H]DHA indicated that the CAM β2-adrenoceptor-GFP construct bound this ligand with an affinity similar to the wild-type β2-adrenoceptor-GFP (Table 1). However, these clones did not, in general, display the same level of GFP autofluorescence as the clones expressing the wild-type β2-adrenoceptor-GFP construct. Such observations were consistent with routinely lower levels of steady-state expression of the CAM β2-adrenoceptor-GFP construct. This was confirmed by the lower levels of [3H]DHA-specific binding to membrane fractions isolated from these cells compared with clones expressing the wild-type β2-adrenoceptor-GFP construct (Table 1). Competition for the specific binding of [3H]DHA to membranes expressing wild-type β2-adrenoceptor-GFP or CAM β2-adrenoceptor-GFP by isoprenaline indicated that this agonist had substantially higher affinity for CAM β2-adrenoceptor-GFP than for wild-type β2-adrenoceptor-GFP (Fig.2). As such, the previously noted high affinity of this agonist for the CAM β2-adrenoceptor compared with wild-type β2-adrenoceptor (Samama et al., 1993) was preserved following addition of GFP to the C-terminal tail of both of these GPCR variants.

High affinity of isoprenaline for the CAM β2-adrenoceptor is retained after addition of GFP. Competition between [3H]DHA (0.34 nM) and varying concentrations of isoprenaline for specific binding to membranes expressing either wild-type (WT)-β2-adrenoceptor-GFP (▾) or CAM β2-adrenoceptor-GFP (■) was assessed. Data are presented as means ± S.E. n = 3.
Although clear plasma membrane-localized CAM β2-adrenoceptor-GFP could be observed in stably expressing clones, there was a greater fraction of the GFP autofluorescence located intracellularly than for wild-type β2-adrenoceptor-GFP (Fig.3A). When we treated cells expressing the CAM β2-adrenoceptor-GFP construct with the β-blocker betaxolol (24 h, 10−5 M) and then visualized the cells, a marked increase in both plasma membrane-delineated and diffuse intracellular fluorescence was observed (Fig. 3A). Washing of the cells followed by an intact cell ligand binding experiment with [3H]DHA indicated a 3-fold up-regulation of CAM β2-adrenoceptor-GFP in response to betaxolol (Fig. 3B). That the increased GFP autofluorescence in response to treatment with betaxolol in cells expressing CAM β2-adrenoceptor-GFP represented up-regulation of the GPCR-GFP fusion protein was further confirmed by immunoblotting studies on membranes of untreated and betaxolol-treated cells (Fig. 4). Antibodies against both the β2-adrenoceptor and against GFP indicated that betaxolol treatment substantially increased levels of a family of poorly resolved polypeptides that are likely to represent differentially glycosylated forms of the CAM β2-adrenoceptor-GFP (Fig. 4), although the current studies cannot exclude that a degree of proteolytic degradation has occurred to produce this pattern.
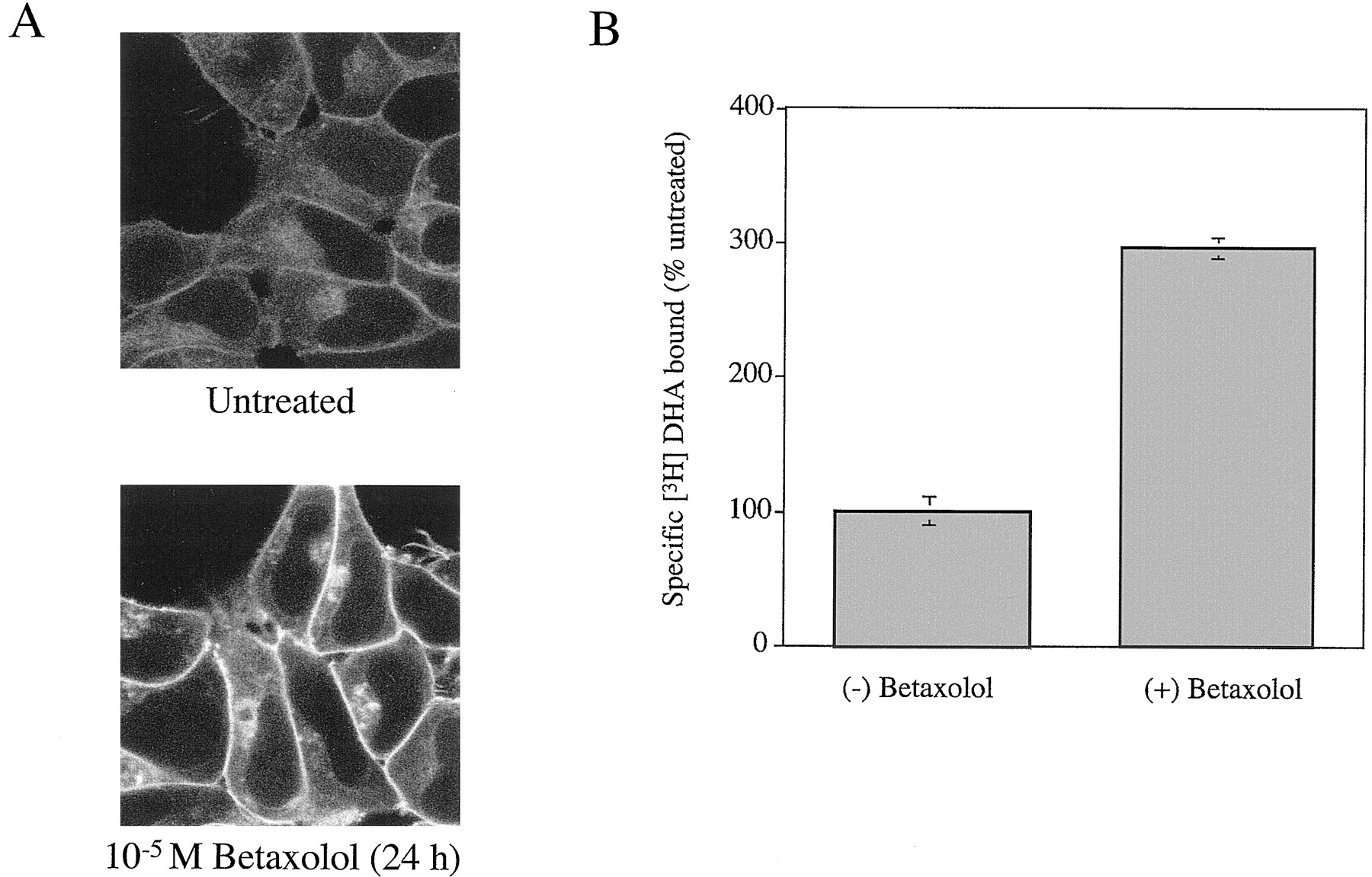
Expression of CAM β2-adrenoceptor-GFP and up-regulation by betaxolol: A, fluorescence studies; B, binding studies. The CAM β2-adrenoceptor-GFP construct was expressed stably in HEK293 cells, and individual clones were isolated. A, cells of a single clone were grown on glass coverslips in the absence (upper panel) or presence (lower panel) of betaxolol (10−5 M) for 24 h. These cells were then visualized. B, cells of this clone, which were untreated or treated with betaxolol (10−5 M, 24 h) and then washed, were used to measure the specific binding of [3H]DHA in intact cells ([3H]DHA is a lipophilic antagonist that crosses the plasma membrane and thus provides a measure of total cell levels of β2-adrenoceptor-binding sites). Data are presented as means ± S.E. n = 5.

Up-regulation of CAM β2 -adrenoceptor-GFP by betaxolol: immunoblot studies. Membrane fractions were prepared from cells expressing the CAM β2-adrenoceptor-GFP, which had been maintained for 24 h in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of betaxolol (10−5 M). These were resolved by SDS-polyacrylamide gel electrophoresis. After transfer to nitrocellulose, the samples were immunoblotted using a polyclonal anti-GFP antibody (lanes 1 and 2) or an antibody to the β2-adrenoceptor (lanes 3 and 4). Two additional experiments produced similar results.
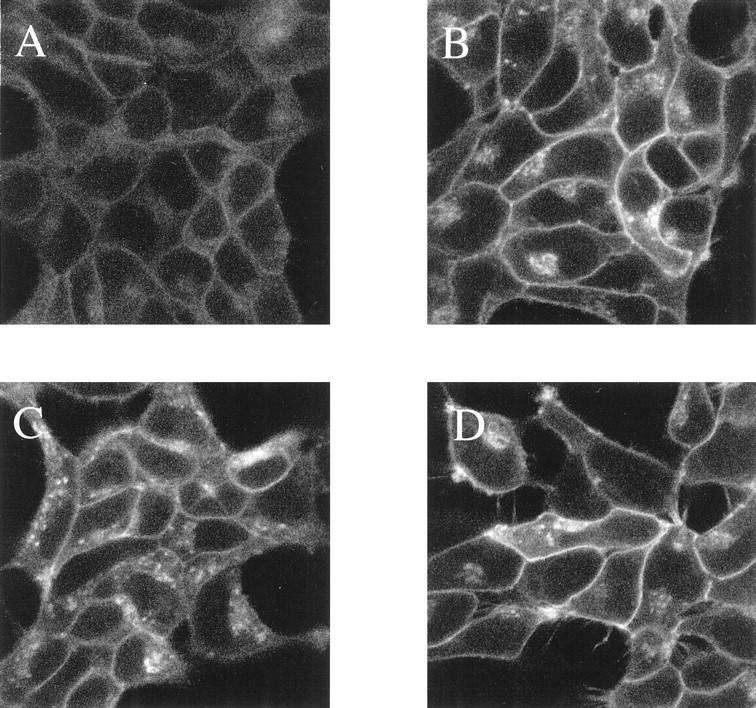
Up-regulation of GFP-autofluorescence was also observed by treatment of the CAM β2-adrenoceptor-GFP–expressing cells with a range of β-blockers including DHA, labetolol, and ICI118551 (24 h, each at 10−5 M) (Fig.5). Pharmacological selectivity of this effect was apparent because it was not produced by treatment with the α1-adrenoceptor antagonist prazosin or the α2-adrenoceptor antagonist yohimbine (24 h, each at 10−5 M; data not shown). Although each of the β-blockers described above resulted in greater levels of autofluorescent signal, the pattern of distribution of cellular CAM β2-adrenoceptor-GFP was not identical. Both betaxolol and ICI118551 resulted in a large increase in homogenous plasma membrane-delineated fluorescence (Fig. 5). However, as with the untreated cells, a significant amount of predominantly diffuse, intracellular staining was observed, the level of which was greater than in the untreated cells (Fig. 5). By contrast, after treatment with labetolol, although a substantial increase in plasma membrane CAM β2-adrenoceptor-GFP was observed, there was also an increase in intracellular signal. A significant fraction of this autofluorescent signal displayed a subplasma membrane, distinctly punctate localization (Fig. 5C) that appeared similar to the pattern produced by short-term treatment with the agonist isoprenaline (see Fig. 1 and later). To explore a possible basis for these differences, basal intact cell adenylyl cyclase activity and its regulation by a variety of ligands was assessed. Although basal cAMP levels in these cells were low, both ICI118551 and betaxolol were able to reduce them further, indicating that these ligands function as inverse agonists at the CAM β2-adrenoceptor-GFP. By contrast, alprenolol and DHA displayed partial agonism, and, in this system, a maximally effective concentration of labetolol was able to elevate cAMP levels to the same extent as isoprenaline (Fig.6). Sustained treatment of cells expressing wild-type β2-adrenoceptor-GFP with betaxolol or the other ligands described above failed to result in a significant up-regulation of the construct, as fluorescence intensity and distribution pattern was little modified by the drug treatments (data not shown).

Up-regulation of CAM β2-adrenoceptor-GFP by other β-adrenoceptor ligands: fluorescence studies. The CAM β2-adrenoceptor-GFP–expressing cells used in Fig. 3 were exposed to no ligand (A), DHA (B), labetolol (C), or ICI118551 (D) (each at 10−5 M) for 24 h. The cells were then imaged. Results from a typical experiment are displayed.
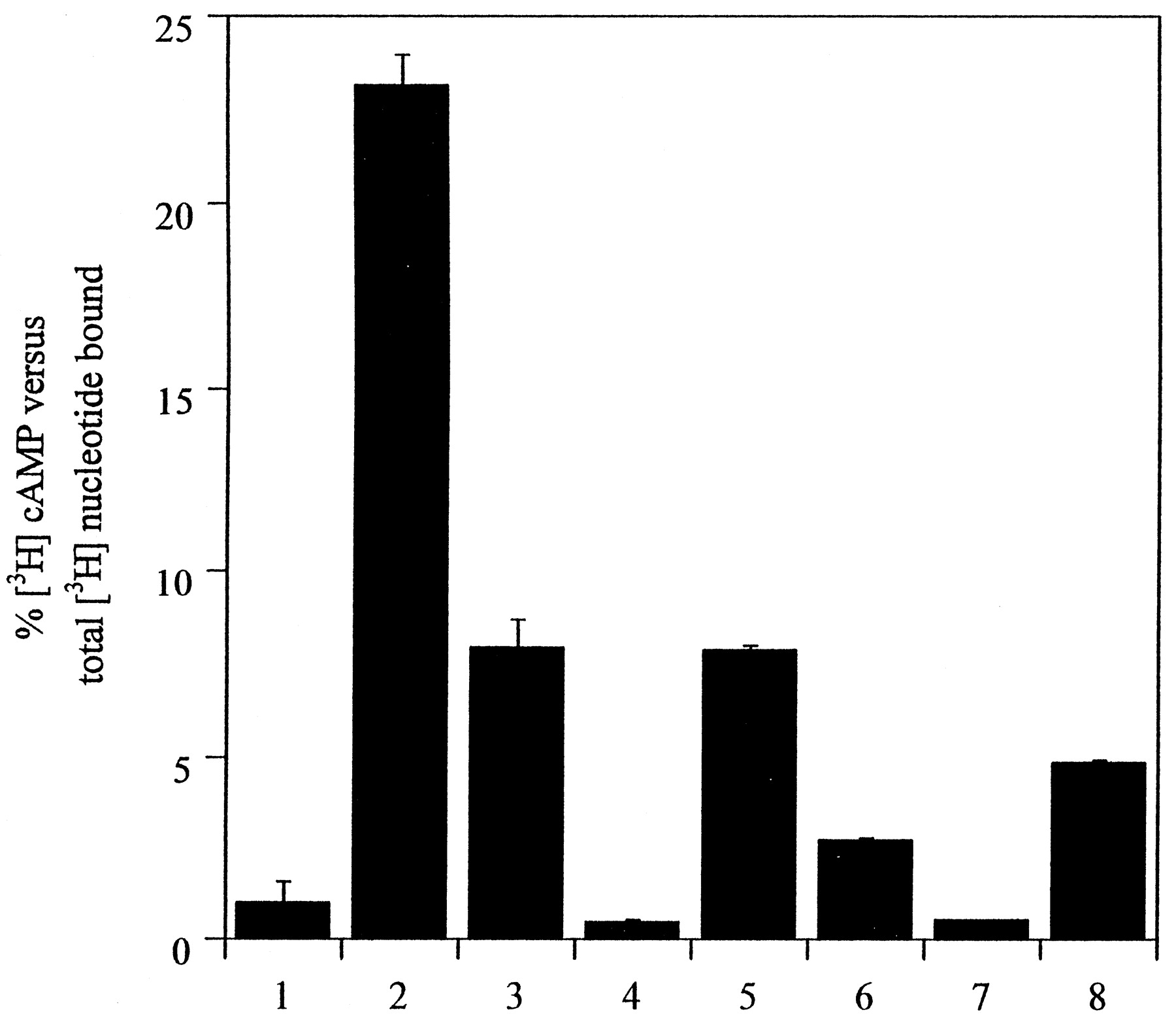
Ligand regulation of adenylyl cyclase activity in intact cells expressing CAM β2 -adrenoceptor-GFP. Basal (1) adenylyl cyclase activity and its regulation by forskolin (5 × 10−5 M; 2), isoprenaline (10−5 M; 3), betaxolol (4), and the range of β-blockers used in Fig. 5 [labetolol (5), DHA (6), ICI118551 (7)] and alprenolol (8; all at 10−5 M) were assessed as described in Materials and Methods. Data represent means ± S.E. of triplicate assays from a single representative experiment. Three additional assays produced similar results, although the extent of [3H] nucleotide conversion varied over a 2-fold range between the experiments.
The capacity of betaxolol to alter the fluorescence intensity of CAM β2-adrenoceptor-GFP–expressing cells could be detected and directly quantitated in a spectrofluorimeter after seeding of cells into wells of a 96-well microtiter plate (Fig.7). This allowed concentration-response curves to betaxolol to be calculated conveniently, something which was impractical by confocal visualization of sets of coverslips. Twenty-two hours after addition of the ligand, fluorescence intensity had increased in a concentration-dependent manner, with EC50 = 0.17 μM. This value was in good accordance with the measured Ki of betaxolol to bind to this GPCR-GFP construct as determined from competition binding experiments between [3H]DHA and betaxolol (0.23 μM). This enhanced fluorescent signal was not simply due to the addition of the ligand, because no alteration in fluorescence intensity was recorded when betaxolol was added and fluorescence was measured immediately (Fig. 7).

Concentration dependence of the up-regulation of CAM β2 -adrenoceptor-GFP by betaxolol. Cells expressing CAM β2-adrenoceptor-GFP were grown in wells of a 96-well microtiter plate. The cells were then exposed to varying concentrations of betaxolol, and fluorescence was measured on a Spectrofluor Plus fluorimeter either at 0 h (●) or after 22 h (▴). Values are the mean percentages ± S.E of basal fluorescence from six experiments performed in duplicate.
The betaxolol-up-regulated CAM β2-adrenoceptor-GFP was sensitive to agonist treatment. After the removal of betaxolol and its replacement by isoprenaline (10−5 M), rapid internalization of the construct into intracellular, punctate vesicles was observed. This process could be visualized by confocal microscopy (Fig.8, A–D) and was indistinguishable in phenotype from that recorded above for wild-type β2-adrenoceptor-GFP (Fig. 1). [3H]CGP-12177 is a hydrophillic β-adrenoceptor antagonist that is unable to cross the plasma membrane. Therefore, in intact cell-specific binding experiments, it identifies only the cell surface population of forms of β-adrenoceptors. Such intact cell-binding studies were performed on naive cells expressing CAM β2-adrenoceptor-GFP, those that had been pretreated with betaxolol (24 h, 10−5 M), and such cells after replacement of betaxolol with isoprenaline (10−5 M) for 30 min. Cell surface up-regulated CAM β2-adrenoceptor-GFP was essentially all internalized by short-term agonist treatment (Fig. 8E).

Internalization of up-regulated CAM β2 -adrenoceptor-GFP by isoprenaline. Confocal studies: CAM β2-adrenoceptor-GFP–expressing cells were untreated (A) or exposed to betaxolol (10−5 M, 24 h; B–D). After betaxolol treatment, the cells were washed, and isoprenaline (10−5 M) was added for 0 (B), 10 (C), or 30 (D) min. [3H]CGP12177 binding studies: E, cells, as above, were untreated, exposed to betaxolol (10−5 M, 24 h), or exposed to betaxolol followed by further exposure to isoprenaline (10−5 M, 30 min). Intact cells were then washed and used to measure the specific binding of [3H]CGP12177. Data are presented as means ± S.E.n = 3.
In previous studies using 3H-ligand binding studies, sustained treatment of cells expressing the CAM β2-adrenoceptor with alprenolol did not apparently produce an increase in cellular levels of the mutant protein (MacEwan and Milligan, 1996a,b). However, exposure of CAM β2-adrenoceptor-GFP–expressing cells to a high concentration of alprenolol (24 h, 10−5 M) caused a clear increase in cellular fluorescence (Fig.9A). It was noted that alprenolol treatment also resulted in a distinctly punctate appearance of a fraction of the intracellularly located GPCR, as observed earlier following treatment with labetolol. To explore the basis for the apparent discrepancy of up-regulation of CAM β2-adrenoceptor-GFP by alprenolol in the current studies but not CAM β2-adrenoceptor in previous work, CAM β2-adrenoceptor-GFP–expressing cells were exposed to a range of concentrations of alprenolol. The cellular autofluorescence pattern (data not shown) and, after extensive washing, the measured specific binding of [3H]DHA to intact cells were monitored. The 3H-ligand binding studies demonstrated a clear increase in levels of the construct after treatment with concentrations of alprenolol between 10−10 M and 10−8 M. This reached a plateau with treatment with 10−7 M alprenolol and was greatly reduced by pretreatment with 10−5 M alprenolol (Fig. 9B). Equivalent results were produced when using [3H]CGP-12177 as radioligand (Fig. 9B). Using concentrations of alprenolol up to 10−7 M, the ratios of specific binding of [3H]CGP-12177 to [3H]DHA were no different from those of the untreated cells (Fig. 9B). Such observations suggest that the overall cell-surface to total-cell expression levels of CAM β2-adrenoceptor-GFP are not modified substantially by alprenolol treatment and that there are equivalent increases in amounts of the GPCR in both intracellular and plasma membrane compartments.

Apparent concentration dependence of the up-regulation of CAM β2 -adrenoceptor-GFP by alprenolol. A, confocal studies: CAM β2-adrenoceptor-GFP–expressing cells were untreated or exposed to alprenolol (10−5 M, 24 h). B, binding studies: CAM β2-adrenoceptor-GFP–expressing cells were untreated or exposed to varying concentrations of alprenolol for 24 h. They were subsequently washed, and intact cell-specific binding of single concentrations of either [3H]DHA (■) or [3H]CGP12177 (▾) were measured as described inMaterials and Methods to ascertain apparent levels of total cell receptor and cell surface receptor, respectively. Data represent means ± S.E. of triplicate assays from a single experiment that was representative of three performed.
Discussion
The generation and expression of fusion proteins containing modified forms of the GFP from A. victoria have recently revolutionized protein imaging studies in single cells and provided a means to interlink biochemical and cell biological studies on the kinetics and regulation of protein distribution and redistribution in intact living cells. GPCRs represent a family of proteins, many of which internalize in response to binding of agonist ligands. These processes have been actively imaged in real time after expression of forms of GPCRs with GFP attached to their C-terminal tail (Barak et al., 1997; Tarasova et al., 1997; Awaji et al., 1998; Kallal et al., 1998; Drmota et al., 1998, 1999). Although it might be anticipated that attachment of a 27-kDa polypeptide to the end of a GPCR could significantly interfere with function, a series of reports have indicated that the modified GPCRs display essentially unaltered pharmacology and interact with G proteins to initiate second messenger regulation (Barak et al., 1997; Tarasova et al., 1997; Awaji et al., 1998; Drmota et al., 1998, 1999; Kallal et al., 1998). Furthermore, agonist-induced internalization and recycling to the plasma membrane have been recorded for a range of such constructs (Barak et al., 1997; Tarasova et al., 1997; Awaji et al., 1998; Drmota et al., 1998; Kallal et al., 1998).
An area of considerable interest in GPCR biology has been the observations that many GPCRs are not silent in the absence of agonist ligands but display constitutive activity (Lefkowitz et al., 1993;Scheer and Cotecchia, 1997; Leurs et al., 1998). A range of mutations of GPCRs have been reported to enhance the degree of constitutive activity. Such modified forms of the GPCR are thus believed to offer insights into conformational changes that may occur upon agonist binding to a wild-type GPCR (Gether et al., 1997a; Javitch et al., 1997). One of the most studied CAM GPCRs is a form of the human β2-adrenoceptor in which a short segment of the distal region of the third intracellular loop was replaced by the equivalent section of the α1B-adrenoceptor (Samama et al., 1993, 1994; Gether et al., 1997b; Javitch et al., 1997). As well as producing considerably greater agonist-independent stimulation of adenylyl cyclase activity than the wild-type GPCR, this CAM β2-adrenoceptor has been shown to denature more readily than the wild-type β2-adrenoceptor when purified and potentially to have a markedly lower functional half-life (Gether et al., 1997a,b). In the present study, we have constructed and stably expressed a C terminally GFP-tagged form of this CAM β2-adrenoceptor to directly address such issues and to re-examine a series of reports that indicated that inverse agonists, but not neutral antagonists, cause up-regulation of the CAM β2-adrenoceptor (MacEwan and Milligan, 1996a,b). Such GFP-tagged constructs also could be used to explore the cellular distribution of the CAM β2-adrenoceptor compared with the wild-type β2-adrenoceptor.
We have previously noted that prolonged treatment with either betaxolol or sotolol results in a substantial up-regulation of a CAM β2-adrenoceptor expressed stably in NG108-15 cells (MacEwan and Milligan, 1996a,b). Such conclusions were based on detection of increased levels of specific binding of [3H]DHA after washing of the cells and membrane preparation. However, an equivalent up-regulation was not observed after pretreatment with alprenolol (MacEwan and Milligan, 1996a). Because betaxolol and sotolol both display characteristics of inverse agonists at the modified GPCR, whereas alprenolol displays weak partial agonist function, an obvious conclusion was that the up-regulation reflected stabilization of the CAM-GPCR in a manner dependent on the inverse agonist characteristics of the ligands. However, in the current studies, fluorescence analysis clearly indicated the CAM β2-adrenoceptor-GFP construct to be up-regulated by alprenolol (Fig. 9A) as well as by betaxolol (Fig. 3A) and a range of other β-blockers. The most likely explanation for this discrepancy is that the current fluorescence studies provide a direct monitor of the effect of the added ligands. By contrast, the previous work required removal of the ligand, membrane preparation, and subsequent 3H-ligand-binding studies. Betaxolol, as a β1-adrenoceptor-selective ligand, has relatively low affinity for the CAM β2-adrenoceptor, whereas alprenolol has high affinity. It could thus be anticipated that betaxolol would be effectively removed in washing regimens, whereas this would be more difficult to achieve with a high-affinity ligand. As such, it was possible that residual alprenolol would compete with [3H]DHA in the subsequent binding experiments, thus reducing the measured binding of a single concentration of [3H]DHA. To approach this directly, we treated CAM β2-adrenoceptor-GFP–expressing cells for 24 h with differing concentrations of alprenolol, subsequently washed the cells, and measured the specific binding of [3H]DHA. Clear concentration-dependent up-regulation of [3H]DHA binding was observed by prior treatment of the cells with concentrations of alprenolol up to 10−8 M. This plateaued at 10−7 M but at 10−5 M was essentially nonexistent (Fig. 9B). Such results would indeed be consistent with the competition model outlined above. Furthermore, equivalent results were obtained when the specific binding of a single concentration of the membrane impermeant antagonist [3H]CGP-12177 was measured after cellular pretreatment with varying concentrations of alprenolol (Fig. 9B).
The concentrations of many of the ligands used in these studies are very high when compared with their affinity to bind the β2-adrenoceptor. However, this represented a deliberate policy, because it is possible to envisage use of this approach to identify novel ligands at either this or similarly modified GPCRs. In such initial screens, it is normal to use ligands at concentrations between 1 and 10 μM. We also wished to explore whether the up-regulation was dependent on the ligand's being membrane permeable. However, equivalent treatments with CGP-12177 also produce marked up-regulation of the construct when monitored optically (data not shown). Importantly, ligand-induced up-regulation of fluorescence associated with the CAM β2-adrenoceptor-GFP retained pharmacological specificity. Levels of the GPCR construct were unaltered by sustained treatment of the cells with either the α1-adrenoceptor antagonist/inverse agonist prazosin or the α2-adrenoceptor antagonist/inverse agonist yohimbine.
Levels of the wild-type β2-adrenoceptor-GFP construct were little affected by sustained treatment with β-blockers, but as previously reported by others (Barak et al., 1997;Kallal et al., 1998), the agonist isoprenaline caused internalization of the construct into punctate vesicles, and recycling of this construct to the plasma membrane could be achieved in rapid order by removal of the agonist and replacement with alprenolol. It is well established that the CAM β2-adrenoceptor does not function in an entirely agonist-independent manner (Samama et al., 1993, 1994; Stevens and Milligan, 1998), thus after betaxolol-mediated up-regulation of the CAM β2-adrenoceptor-GFP, isoprenaline was also able to cause rapid internalization of the construct into punctate vesicles in a manner similar to the wild-type β2-adrenoceptor-GFP (Fig. 8, A–D).
Treatment of CAM β2-adrenoceptor-GFP–expressing cells with a range of β-blockers resulted in increased brightness of the cells as monitored in the confocal microscope, with substantial increases in plasma membrane-delineated signal produced by all the ligands. However, careful examination of the cells demonstrated differences in the distribution pattern of the intracellular up-regulated GPCR (Figs. 5and 9A). Treatment with both betaxolol and ICI118551 resulted in a diffuse, largely uniform pattern of intracellular-delineated GPCR fluorescence. In contrast, alprenolol, to some degree, and more markedly labetolol, produced a pattern in which a fraction of the GPCR signal was present with a punctate, intracellular location, somewhat akin to the pattern observed after short-term treatment with the agonist isoprenaline. It is known that compared with full agonists such as adrenaline or isoprenaline, the relative intrinsic activity of partial agonists is more pronounced at the β2-adrenoceptor as expression levels are increased (MacEwan et al., 1995), and for the CAM β2-adrenoceptor compared with the wild-type β2-adrenoceptor at equal levels of expression (Samama et al., 1993). Relatively few ligands traditionally described as “antagonists” appear to be purely neutral in effect after binding within the crevice formed from the topological architecture of the seven transmembrane domains of GPCRs for catecholamines (Milligan et al., 1995; Milligan and Bond, 1997). Indeed, ligand stabilization of particular conformations of a GPCR may be considered in a similar manner to the induced-fit models of enzyme-substrate interactions. Therefore, the bulk of antagonists will favor production of conformations less or more similar to agonist-induced conformations than the mean spectrum of populations present in the absence of ligand. They will therefore behave as either inverse agonists or partial agonists. If partial agonists, however, it might be expected that they would display poor intrinsic activity relative to classical agonists for that GPCR, or they would previously have been characterized as agonists rather than antagonists. We therefore measured the capacity of the β-blockers used in this study to regulate cAMP levels in intact cells expressing CAM β2-adrenoceptor-GFP (Fig. 6). Although the basal level of cAMP was relatively low in these cells, both betaxolol and ICI118551 reduced this level further, a property consistent with their classification as inverse agonists. However, alprenolol displayed a clear ability to increase cAMP levels in intact CAM β2-adrenoceptor-GFP–expressing cells, acting as a partial agonist when compared with isoprenaline. Perhaps surprisingly, at maximally effective concentrations, labetolol produced as large an increase in intracellular cAMP levels as isoprenaline (Fig.6). The apparent high efficacy of labetolol in this assay was unexpected but may reflect a combination of the levels of CAM β2-adrenoceptor-GFP expressed in these cells and that only a small degree of G protein activation is required to regulate the full cellular population of adenylyl cyclase that can be accessed by receptor. It thus appears that visual examination of the distribution of up-regulated CAM β2-adrenoceptor after sustained exposure to a selection of β-blockers can provide a useful indication of the ligand's functional pharmacological properties.
Visual examination of the fluorescence of cells grown on individual coverslips with and without sustained treatment with the β-adrenoceptor ligands was appropriate when examining a single concentration of ligand but not very suitable to attempt to generate quantitative concentration-response curves. However, the increase in cellular fluorescence of CAM β2-adrenoceptor-GFP–expressing cells in response to treatment with betaxolol could also be monitored in a spectrofluorimeter. As such, cells grown in a 96-well microtiter plate could be used to generate EC50 values for the effect of betaxolol (Fig. 7). The values obtained were in good accord with previous estimates for the up-regulation of non-GFP-tagged CAM β2-adrenoceptor, inhibition of basal adenylyl cyclase activity in membranes expressing the CAM β2-adrenoceptor (MacEwan and Milligan, 1996a), and the Ki of betaxolol estimated from ligand binding experiments in these cells. Importantly, the increase in cellular fluorescence was not observed by simply adding betaxolol to the cells and immediately monitoring fluorescence intensity. As such, it requires the time-dependent up-regulation of CAM β2-adrenoceptor-GFP levels.
These studies markedly extend the use of GFP tagging of GPCRs in that they have allowed analysis of the regulation of cellular distribution of a CAM form of a GPCR and the regulation in cellular levels of the protein in response to sustained challenge with receptor ligands. The capacity of a series of pharmacologically selective ligands to specifically regulate cellular levels of the CAM GPCR in a manner that can be easily detected or visualized without further manipulation of the cells also hints at approaches to the identification of ligands at novel GPCRs.
Footnotes
- Received July 19, 1999.
- Accepted September 21, 1999.
-
Send reprint requests to: Dr. Graeme Milligan, Davidson Bldg., University of Glasgow, Glasgow G12 8QQ, Scotland, UK. E-mail:g.milligan{at}bio.gla.ac.uk
-
Financial support for this work was provided by the Medical Research Council and the European Union Biomed II program, Inverse Agonism: Implications for Drug Design. A.J.M. received a studentship from the Biotechnology and Biosciences Research Council.
Abbreviations
- GPCR
- G protein-coupled receptor
- CAM
- constitutively active mutant
- DHA
- dihydroalprenolol
- GFP
- green fluorescent protein
- MEM
- minimum essential medium
- KRH
- Krebs-Ringer-HEPES buffer
- PBS-T buffer
- PBS containing 0.1% Tween 20
- PCR
- polymerase chain reaction
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}