


Abstract
Agonist-selective actions of opioids on the desensitization of μ-opioid receptors (MORs) have been well characterized, but few if any studies have examined agonist-dependent recovery from desensitization. The outward potassium current induced by several opioids was studied using whole-cell voltage-clamp recordings in locus ceruleus neurons. A brief application of the irreversible opioid antagonist β-chlornaltrexamine (β-CNA) was applied immediately after treatment of slices with saturating concentrations of opioid agonists. This approach permitted the measurement of desensitization and recovery from desensitization using multiple opioid agonists, including [Met]5enkephalin (ME), [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO), etorphine, fentanyl, methadone, morphine, morphine-6-glucuronide, oxycodone, and oxymorphone. The results indicate that desensitization protects receptors from irreversible antagonism with β-CNA. The amount of desensitization was measured as the decrease in current during a 10-min application of a saturating agonist concentration and was a good predictor of the extent of receptor protection from irreversible inactivation with β-CNA. After desensitization with ME or DAMGO and treatment with β-CNA, there was an initial profound inhibition of MOR-induced current that recovered significantly after 45 min. There was, however, no recovery of MOR-mediated current with time after treatment with agonists that did not cause desensitization, such as oxycodone. These results demonstrate that desensitization prevents irreversible inactivation of receptors by β-CNA.
Opiates are the most effective analgesics known. Activation of the μ-opioid receptor (MOR) belies their therapeutic efficacy as well as the euphoria and rewarding properties that lead to their abuse. Agonist-bound MORs activate G proteins and signal through downstream effectors. They also become substrates for the regulatory machinery responsible for agonist-induced MOR desensitization, endocytosis, and recovery from desensitization (Williams et al., 2001; Connor et al., 2004; von Zastrow, 2004; Bailey and Connor, 2005). Different opioid agonists have widely varying signaling efficacies in each of these processes. For example, the highly efficacious endogenous peptide agonist [Met]5enkephalin (ME) causes both profound desensitization and internalization. However, morphine and some other alkaloid opiates are regarded as partial agonists that induce desensitization and endocytosis to a lesser degree (Yu et al., 1997; Keith et al., 1998; Whistler and von Zastrow, 1998; Alvarez et al., 2002; Borgland et al., 2003; Celver et al., 2004; Schulz et al., 2004; Dang and Williams, 2005; Koch et al., 2005; Johnson et al., 2006).
It is widely speculated that differences in short-term MOR regulatory events underlie the profound agonist-selective differences observed in the development of tolerance in vivo (Whistler et al., 1999; Stafford et al., 2001; Walker and Young, 2001; Patel et al., 2002; Grecksch et al., 2006; Pawar et al., 2007). There is no consensus, however, on which elements of MOR regulation—signaling efficacy, desensitization, internalization, or resensitization—are most directly correlated with tolerance (Whistler et al., 1999; Bohn et al., 2000; Williams et al., 2001; Connor et al., 2004; von Zastrow, 2004; Bailey and Connor, 2005; Koch et al., 2005). Understanding how MOR agonists, particularly those used in pain management, differ with respect to these fundamental aspects of MOR regulation, particularly in neurons, will contribute to the development of effective analgesic therapy.
Short-term MOR regulation is characterized by the receptor-mediated components of desensitization and recovery from desensitization that occur within minutes of agonist exposure. Receptor-specific desensitization is believed to be dependent on agonist binding, phosphorylation, and binding to β-arrestin followed by sequestration to clathrin-coated pits and dynamin-dependent endocytosis (Connor et al., 2004; von Zastrow, 2004). These rapid receptor-specific events are separate from the opioid-induced increase in activity of adenylyl cyclase after 1 to 2 h of agonist treatment (Avidor-Reiss et al., 1997). One possible mechanism that may account for differences between DAMGO- and morphine-induced desensitization includes MOR phosphorylation by distinct kinases, G protein receptor kinase-2 and protein kinase C, respectively (Johnson et al., 2006). It remains unclear whether differences in agonist-specific desensitization affect the rate and extent of recovery of MOR signaling.

Treatment with β-CNA after ME-induced desensitization decreased the absolute MOR-mediated current but did not inhibit recovery from desensitization. A, current trace from control experiment where a saturating concentration of ME (30 μM, 10 min) resulted in an outward current that peaked and declined during the application. After the wash, ME (30 μM) was applied at 5 and 45 min. The resulting current partially recovered after 5 min and recovered completely after 45 min. At the end of the experiment the α2-adrenoceptor agonist UK14304 (3 μM) was applied to control for changes in the recording after the prolonged washout. B, β-CNA (500 nM, 2 min) was applied immediately after ME (30 μM, 10 min) induced desensitization, and recovery was again measured with ME test pulses at 5 and 45 min. In this experiment, the current induced by ME (30 μM) was almost eliminated after 5 min and recovered significantly after 45 min.
In the present study, several different opioid agonists were used to measure potassium current (GIRK) amplitude, short-term desensitization, and recovery from desensitization using whole-cell recording from locus ceruleus neurons in brain slices. An experimental protocol that used the treatment of brain slices with the irreversible opioid antagonist β-CNA was used to measure recovery of functional receptors after desensitization. Application of β-CNA resulted in a dramatic inhibition of MOR-mediated current after the pronounced desensitization induced by some agonists (ME, DAMGO, fentanyl, etorphine, and methadone). This inhibition was transient and recovered substantially after 45 min. There was less recovery after treatment with agonists that caused an intermediate amount of desensitization (morphine and morphine-6-glucuronide). Oxycodone or a low concentration of ME (300 nM, EC50) did not cause desensitization, and there was no recovery of signaling after treatment with β-CNA. This suggests that whether or not agonist-specific mechanistic differences govern desensitization, the degree of recovery is directly proportional to the amount of desensitization.
Materials and Methods
Tissue Preparation and Recording. Adult (150-250 g) male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were used for all experiments. Details of the method of slice preparation and recording have been published previously (Williams et al., 1984). In brief, rats were anesthetized with halothane and killed. The brain was dissected, blocked, and mounted in a vibrating microtome chamber to cut horizontal slices (260 μm thick) containing locus ceruleus (LC). Slices were cut in cold (4°C) artificial cerebrospinal fluid containing 126 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 21.4 mM NaHCO3, and 11 mM d-glucose while being continuously equilibrated with 95% O2/5% CO2. Slices were subsequently incubated in a 25-ml glass tube at 35°C for a minimum of 30 min before experiments and constantly equilibrated with 95% O2/5% CO2. Slices were then hemisected and transferred to the recording chamber (0.5 ml), where they were superfused with 35°C artificial cerebrospinal fluid at a rate of 1.5 ml/min. Whole-cell recordings were made from LC neurons with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) in the voltage-clamp mode (cells held at -55 mV). Pipettes (1.7-2.1 MΩ) were filled with an internal solution containing the following: 115 mM methyl potassium sulfate, 20 mM NaCl, 1.5 mM MgCl2, 10 mM HEPES, 10 mM BAPTA, 2 mM Mg-ATP, 0.5 mM Na-GTP, and 10 mM phosphocreatine, pH 7.3. Data were collected with PowerLab (chart version 4.2.3) and sampled at 100 Hz. Analysis was performed with Prism software (GraphPad Software Inc., San Diego, CA) and Kaleidagraph software (Abelbeck/Synergy, Reading, PA). Values are presented as arithmetic mean ± S.E.M. One-way analysis of variance was performed, and differences for which p < 0.05 were considered significant.
Drugs. Drugs were applied by bath superfusion. The following drugs were superfused: [Met5]enkephalin, DAMGO, oxycodone, UK14304, yohimbine, bestatin, thiorphan (Sigma, St. Louis, MO), etorphine, methadone, fentanyl, oxymorphone, morphine, morphine-6-glucuronide (National Institute on Drug Abuse, Neuroscience Center, Bethesda, MD), β-CNA (Tocris Cookson, Ellisville, MO). Some compounds (UK14304, thiorphan, and β-CNA) were dissolved in dimethyl sulfoxide, ethanol, or methanol. The final concentrations of these solvents did not exceed 0.01% DMSO, 0.00001% ethanol, 0.05% methanol, respectively. All other drugs were dissolved in water.
Results
Protection from Antagonist Binding and Recovery from Desensitization. A saturating concentration of ME (30 μM) evoked an outward current that desensitized rapidly over the course of a 10-min application, as has been shown previously (Fig. 1) (Harris and Williams, 1991; Osborne and Williams, 1995; Fiorillo and Williams, 1996). Peak currents measured 461 ± 28 pA (n = 17) and decreased to 65 ± 1% of the peak (295 ± 18 pA) after 10 min. After washout of the desensitizing stimulus (ME, 30 μM, 10 min), short pulses of ME (30 μM) were applied after 5 and 45 min to assay recovery from desensitization (Fig. 1A). The current activated by ME recovered from 65 ± 1% at the end of the desensitization period to 75 ± 2% at 5 min and 96 ± 6% at 45 min (n = 7). Thus, as reported previously, recovery from desensitization was complete by 45 min (Dang and Williams, 2004). A saturating concentration of the α2-adrenergic agonist UK14304 (3 μM) was superfused at the conclusion of the each experiment to activate the same potassium conductance by another G-protein-coupled receptor. This allowed for the comparison between multiple opioid agonists that evoked different maximum currents and to detect heterologous desensitization.
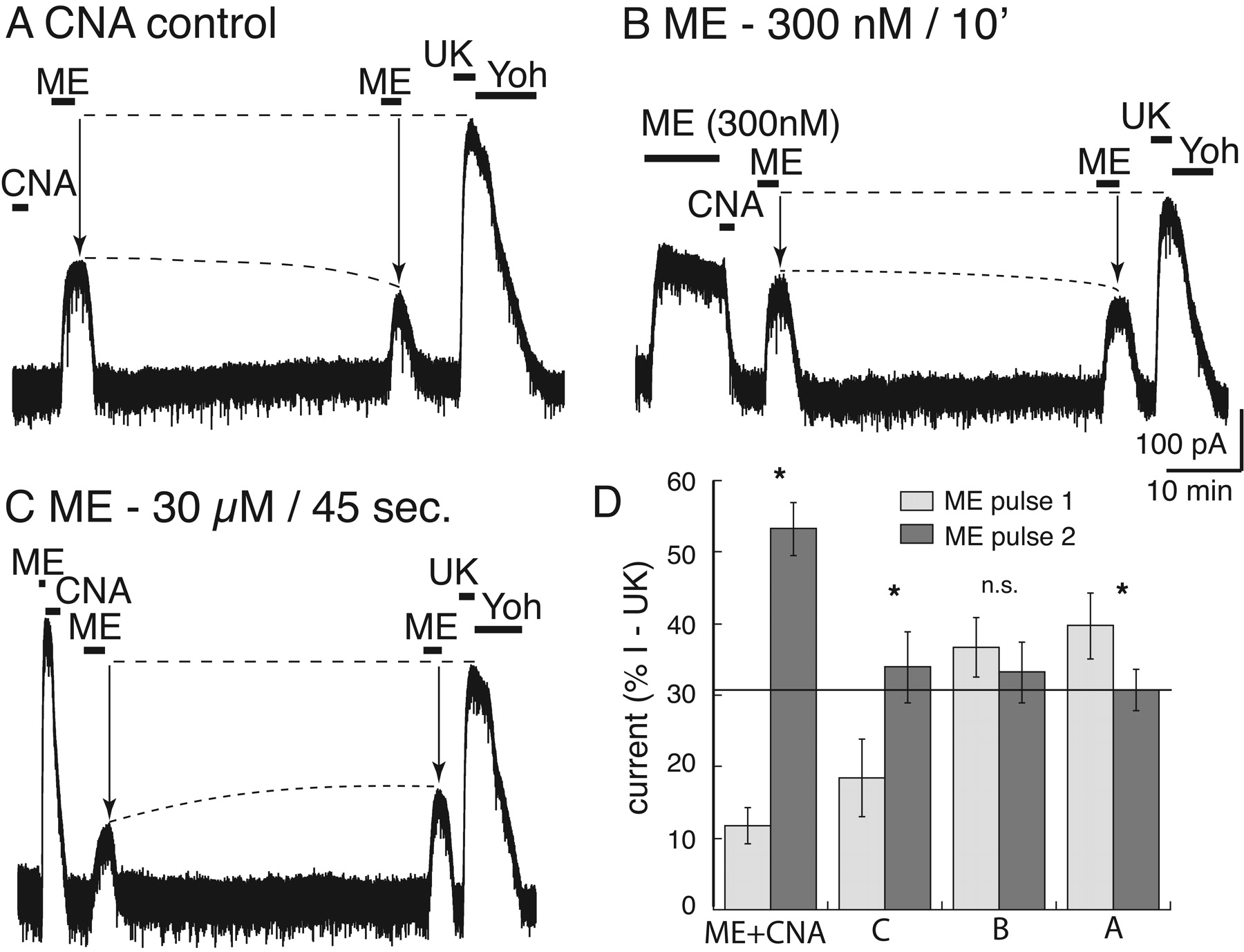
The amount of desensitization determines the degree of MOR protection from β-CNA blockade. A, current trace of a control experiment in which β-CNA (500 nM, 2 min) was applied without prior treatment of the preparation with ME. Application of ME (30 μM) at 5 and 45 min after the washout resulted in current that decreased after 45 min. At the end of the experiment, UK14304 (3 μM) was applied such that the currents induced by ME could be normalized. B, an experiment using an EC50 concentration of ME (300 nM, 10 min) applied before treatment with β-CNA. In this case the amplitude of the current induced by ME (30 μM) after 5 and 45 min did not change. C, an experiment in which a saturating concentration of ME (30 μM) was applied for only 45 s followed by treatment with β-CNA (500 nM, 2 min). In this case, the amplitude of the ME (30 μM) current increased between the 5- and 45-min test points but not to the same extent as was observed with a longer (10 min) ME-desensitizing application. D, summarized results after treatment of slices with β-CNA (500 nM, 2 min) showing the change in the size of the current induced by the first test pulse (5 min, gray bar) and the second test pulse (45 min, black bar) of ME (30 μM) in different experiments. The current amplitudes were normalized to the current induced by UK14304 that was measured at the end of each experiment. The bars labeled ME + CNA are taken from the experiments illustrated in Fig. 1B; CNA control is illustrated in trace A; ME 300 nM/10 min is illustrated in trace B; ME 30 μM/45 s is illustrated in trace C. * over the bars indicates a significant difference between the amplitude of the current induced by ME (30 μM) on first (5 min) and second (45 min) test pulse (p < 0.05). The only experiment in which there was no difference between the two pulses was low-concentration ME (EC50, 300 nM/10 min) (B). The only experiment in which there was a decrease between the two pulses was without prior ME treatment (A).
To determine whether recovery from desensitization involved reactivation of desensitized receptors, the irreversible opioid antagonist β-CNA was applied immediately after the desensitizing agonist application. In this experiment, ME (30 μM) was applied for 10 min followed by treatment with β-CNA (500 nM, 2 min; Fig. 1B). As in the control experiment, two pulses of ME (30 μM) were applied 5 and 45 min after the end of the desensitizing agonist application. At 5 min, the current measured 10 ± 2% (n = 10) of the initial ME-induced current and recovered to 43 ± 2% by 45 min. The two test pulses measured activation of receptors that were no longer desensitized and that were also protected from β-CNA binding.
Three additional experiments were performed to characterize the interaction between desensitization and antagonism by β-CNA (Fig. 2). In all experiments, test pulses of ME (30 μM) were applied at 5 and 45 min to assay the state of MOR signaling. The current induced by the test pulses was then expressed as a percentage of the peak α2-adrenergic-mediated current. In the first experiment (Fig. 2A), β-CNA (500 nM) was tested in the absence of a prior agonist application. In this experiment, the current amplitude induced by ME (30 μM) after 5 min was 40 ± 5% of the current induced by UK14304, indicating that the short treatment with β-CNA blocked a significant number of receptors. When the second ME (30 μM) test pulse was applied after 45 min there was a further decrease in the current amplitude to 31 ± 3% (total change, -9 ± 2%; n = 7). The decrease in current suggests that β-CNA remaining in the slice continued to react with MORs after the first test pulse was delivered, such that more receptors were removed by the time of the second test pulse. Therefore, the increase in test pulse current amplitude that was observed after ME-induced desensitization (30 μM/10 min) and treatment with β-CNA was an underestimate of the total extent of desensitization-induced protection and, ultimately, of MOR recovery (Fig. 1B).
In the second experiment, ME (300 nM EC50 concentration) was superfused for 10 min before the application of β-CNA (500 nM, 2 min; Fig. 2B). The peak current amplitude was 214 ± 19 pA (n = 8) and was 204 ± 20 pA after 10 min, indicating that no significant desensitization occurred. Test pulses at 5 and 45 min after treatment with β-CNA were unchanged at 37 ± 4 and 33 ± 4% (total change, -3 ± 3%). The results of this experiment illustrate two important measurements: first, the amplitude of the current induced by ME test pulses (5 and 45 min) were the same as those observed in the experiment when β-CNA was applied without prior exposure to ME (Fig. 2A); and second, there was no increase between the current induced by the first and second test pulse. Thus, without prior desensitization, there was no protection from β-CNA.
In the third experiment, a short application of high-concentration ME (30 μM) was applied for 45 s before β-CNA (500 nM, 2 min; Fig. 2C). This short treatment has been shown previously to induce a moderate amount of desensitization (Dang and Williams, 2004). The peak current amplitude induced by this brief application period was 400 ± 62 pA. After β-CNA, the amplitude of the current induced by ME test pulses was 18 ± 5% of the UK14304 current at 5 min and increased to 34 ± 5% after 45 min (total recovery, 15 ± 1%; n = 6). The total recovery of 15 ± 1% was considerably less than the 42 ± 4% (12 ± 3 to 53 ± 4%) after the 10-min application of ME (30 μM).
The summarized results are presented in Fig. 2D, where the amplitude of the current induced by the two tests with ME (30 μM) are plotted as a percentage of the current induced by UK14304 (3 μM). The results show that without desensitization there was a decrease or no change in the relative amplitude of the current induced by ME measured at 5 and 45 min (Fig. 2D: A, -9 ± 2%; B, -3 ± 3%). Test pulses applied at these two time points served as a measure of MOR resensitization. With increasing amount of desensitization, the current induced by the ME test pulse at 5 min was depressed, and there was more recovery of the ME-induced current after 45 min (Fig. 2D: ME + CNA, 42 ± 4%; C, 15 ± 1%). Taken together, these results suggest that ME-induced desensitization protected MORs from irreversible antagonism by β-CNA and that as MOR desensitization increased, the amount of resensitization measured also increased.
Recovery from Desensitization Using Other Opioid Agonists. Several opioid agonists were tested to compare agonist-induced desensitization and recovery specific to each compound (Fig. 3). For each agonist, desensitization and recovery were measured as described in the previous section: a saturating concentration was applied for 10 min (“desensitizing stimulus”) followed by β-CNA (500 nM, 2 min), and the recovery from desensitization was assayed with test pulses of ME (30 μM) at 5- and 45-min after the end of the desensitizing stimulus. Test pulse amplitudes were expressed as a percentage of the peak UK14304 current (Fig. 3F). DAMGO (10 μM) evoked a peak current of 549 ± 68 pA (n = 5) that desensitized to 62 ± 5% of peak after 10 min. Test pulse amplitudes increased by 37 ± 6% during the 45-min recovery period (from 9 ± 2% at 5 min to 46 ± 4% at 45 min; Fig. 3A). Although an accurate measurement of desensitization induced by methadone (15 μM, 10 min) was not possible because of slow rise to peak, test pulse amplitudes after the desensitizing stimulus increased by 28 ± 1% (from 12 ± 2 to 40 ± 3%, n = 5; Fig. 3B). This change was smaller than that observed with ME or DAMGO but was larger than any other alkaloid agonists tested.
Morphine (15 μM/10 min) and M-6-G (15 μM/10 min) evoked peak currents of 263 ± 24 pA (n = 7) and 254 ± 45 pA (n = 6) that desensitized by 25 ± 5% and 23 ± 2%, respectively. Moreover, the two alkaloid agonists were similar with respect to the recovery from desensitization, because test pulses after morphine desensitization increased by 17 ± 2% (from 47 ± 4% at 5 min to 64 ± 4% at 45 min), whereas those after M-6-G increased by 13 ± 5% (5 min, 39 ± 2%; 45 min, 53 ± 6%). The peak current induced by oxycodone (15 μM, 278 ± 29 pA; n = 8) was the same as that induced by morphine and M-6-G; however, it failed to desensitize after 10 min (1 ± 2%). Likewise, there was no significant change between the test pulse delivered at 5 min (45 ± 4%) and 45 min (47 ± 3%). Thus, despite evoking a large GIRK current, there was no evidence that oxycodone induced any MOR desensitization.
Desensitization Using Fentanyl, Etorphine, and Oxymorphone. Fentanyl (10 μM/10 min), etorphine (1 μM/10 min), and oxymorphone (15 μM/10 min) were also used as desensitizing agonists. All three agonists evoked large outward currents with rapid onset and subsequent desensitization. Fentanyl (10 μM/10 min) evoked a peak current of 338 ± 24 pA (n = 16) that desensitized to 70 ± 3% of the peak; etorphine (1 μM) evoked a peak current of 359 ± 25 pA (n = 9) that desensitized to 74 ± 1% of the peak; and oxymorphone (15 μM/10 min) evoked a 348 ± 54 pA (n = 6) current that desensitized to 69 ± 2% of the peak. Measuring recovery with these drugs in the brain slice preparation presented challenges that excluded them from the same analysis as the other ligands. To reverse the current evoked by these agonists, the concentration of β-CNA (1 μM, 2 min) was increased, whereas the duration was kept the same. This treatment was sufficient to reverse the fentanyl-induced current (Fig. 4, A and C); however, reversal was incomplete for etorphine (Fig. 4D). This concentration of β-CNA (1 μM, 2 min) had almost no effect on the current induced by oxymorphone (data not shown).
In experiments with fentanyl and etorphine, after wash of β-CNA, an outward current developed in the absence of any applied agonist (Fig. 4, A, C, and D). In control experiments in which slices were not treated with β-CNA, a stable outward current was maintained for 45 min, indicating that the high affinity, lipophilicity, and efficacy of these agonists sustained signaling (Fig. 4B and Supplemental Fig. S1). Thus, the increase in outward current that followed treatment with β-CNA resulted from reactivated receptors by agonist that remained present in the slice, suggesting that the desensitization induced by these agonists resulted in protection from β-CNA. As receptors recovered from desensitization, a sufficient concentration of each drug remained in the slice to activate those receptors. The recovery of the outward current is therefore similar to the recovery observed after desensitization induced by other agonists that was measured using the ME test-pulse protocol. Although the amount of recovery is complicated by the fact that the agonist was present as the receptors recover from desensitization, it was possible to obtain a rough estimate of the rate of recovery. This rate was determined by fitting the increase in outward current to a single exponential to estimate a time constant. The time constant for recovery after desensitization with fentanyl was 7.1 ± 0.6 min (n = 5) and for etorphine was 4.6 ± 1.1 min (n = 4). This suggests that there is a fast phase of MOR recovery, analogous to the fast receptor desensitization that results from saturating concentrations of agonist applied for 10 min.

Agonist-selective protection from blockade by β-CNA. The protocol used for these experiments was the same of each agonist and is identical with that illustrated in Fig. 1B. The indicated agonist (A-E) was applied at a saturating concentration for 10 min followed by treatment with β-CNA (500 nM, 2 min), and then test pulses of ME (30 μM, 2 min) were applied after 5 and 45 min. At the end of each experiment, UK14304 (3 μM) was applied and used to normalize the opioid currents. F, a summary of the results obtained for each agonist. * over the bars indicates a significant difference between the amplitude of the current induced by ME (30 μM) on the first (5 min) and second (45 min) test pulse (p < 0.05). The only agonist in which there was no difference between the two pulses was oxycodone.
Summary. The results show that each agonist caused a different maximal activation of outward GIRK current and capacity to induce desensitization (Fig. 5A). ME and DAMGO induced the largest outward currents and caused the greatest amount of desensitization, whereas morphine and M-6-G are less effective at both. Oxycodone stands alone in that it evoked a current as large as that induced by morphine and M-6-G but induced no detectable desensitization (Fig. 5A). In fact, the results with oxycodone were comparable with the experiments using the EC50 concentration of ME (300 nM/10 min) in that there was no desensitization (Fig. 5A). The concentration of oxycodone (15 μM) was saturating because the outward current induced by concentrations ranging from 10 to 30 μM were the same (percentage of the maximum UK14304 current; 10 μM, 68 ± 3.4%, n = 6; 15 μM, 72 ± 6.4%, n = 6; 30 μM, 69 ± 3.2, n = 6). When agonists were compared based on the degree of desensitization and recovery measured by the two ME test pulses delivered at 5 min (ME pulse 1) and 45 min (ME pulse 2, Fig. 5B), the two measurements correlated well.
Discussion
The development of a protocol using the irreversible MOR antagonist β-CNA made the acquisition of the present results possible. Saturating concentrations of alkaloid agonists normally require an extended duration to wash from brain slices such that measuring the recovery from desensitization was not possible previously. The present results suggest that recovery from desensitization may be nearly complete within 15 min after the end of the desensitizing treatment. The use of β-CNA reduced the number of active receptors. After a 10-min application of ME or DAMGO, the reduction in active receptors by β-CNA resulted in a dramatic inhibition of ME current induced at the 5-min test point. The only way that the test pulse amplitude could increase after 45 min after β-CNA treatment is if unoccupied receptors became available throughout this period. Desensitization therefore resulted in a state in which β-CNA was not able to bind to MORs, perhaps as a result of receptor endocytosis or because desensitized receptors have an increased affinity for receptor bound agonist. Finally, β-CNA probably remained in the slice and continued to inactivate MORs beyond the initial 2-min application because in β-CNA control experiments (no prior desensitizing stimulus), the current induced by ME at the 45-min test was smaller than that measured after 5 min (Fig. 2A). Thus, the measurement of MOR recovery from desensitization was probably an underestimate.

Desensitization with fentanyl and etorphine protects receptors from blockade by β-CNA. A, fentanyl (FEN, 10 μM) was applied for 10 min followed by treatment with β-CNA (1 μM, 2 min). The current induced by fentanyl declined during the 10-min application and was almost completely reversed after treatment with β-CNA. After the washout of β-CNA, an outward current was observed that was blocked by naloxone (NLX, 1 μM). B, a control experiment showing that the outward current induced by fentanyl did not decline upon washout. C, summarized results of experiments using fentanyl with and without β-CNA (1 μM/2 min). D, summarized results from experiments using etorphine (1 μM/10 min) in the same experimental protocol as with fentanyl.

The amplitude of the current induced by agonists does not always correlate with the ability to induce desensitization; however, the extent of desensitization and the recovery from desensitization do correlate. A, summary of results plotting the amount of desensitization as a function of the mean current induced by a series of agonist; 1, M-6-G; 2, morphine; 3, fentanyl; 4, oxymorphone; 5, etorphine; 6, ME; 7, DAMGO; Oxyc, oxycodone; ME-L, ME (EC50, 300 nM). B, the pulse 2/pulse 1 ratio of the current induced by test pulses of ME (30 μM) plotted as a function of the amount of desensitization induced by a series of agonists.
The results suggest that treatment with β-CNA resulted in a distribution of receptors into three possible configurations: free/unbound, irreversibly inactivated by β-CNA, and desensitized/internalized (Fig. 6). The treatment with β-CNA was short enough that not all receptors were inactivated. This was demonstrated in experiments in which β-CNA was applied without a prior desensitizing stimulus and subsequent ME test pulses resulted in a reduced but measurable current (Fig. 2A). The depression of the maximum current indicated that a substantial pool of receptors was inactivated by β-CNA, whereas others remained unbound (Christie et al., 1986). The third pool of receptors were desensitized and protected from binding to β-CNA and were thus capable of recovery and subsequent activation. This pool of receptors may have been agonist-bound and desensitized on the plasma membrane and therefore neither capable of signaling nor available for binding to β-CNA. It is also possible that desensitized receptors were internalized and thus physically inaccessible to β-CNA. Without high-resolution imaging of the receptors, it is not possible to distinguish the two possibilities. In cultures of mouse LC neurons, however, desensitization or the recovery from desensitization was not changed after blockade of internalization of a fluorescent opioid agonist with concanavalin A (Arttamangkul et al., 2006). This experiment indicated that internalization was not required for desensitization and suggests that the results using β-CNA could result from a process in which receptors remain on the plasma membrane.
The degree of MOR recovery was directly related to the amount of initial desensitization before treatment of the tissue with β-CNA. Furthermore, the rank order of this series of agonists in the recovery process correlates with their ability to induce receptor internalization (Keith et al., 1998; Koch et al., 2005). In either case, within 45 min, the pool of receptors recovered to a state that permitted agonist activation.

A schematic illustrating the three pools of MORs resulting from desensitization, treatment with β-CNA, and recovery from desensitization. A significant number of receptors are irreversibly bound to β-CNA. The pools of active/free receptors and those that are desensitized or internalized vary based on the agonist used for desensitization and the time (5 or 45 min) after the induction of desensitization.
The results of this study indicate that the peak amplitude of the current evoked by a saturating concentration of several agonists generally correlated with the amount of acute desensitization (Fig. 5). DAMGO and ME evoked the largest current and induced the greatest amount of desensitization, whereas the smaller maximum current induced by morphine and M-6-G caused significantly less desensitization. These results are in agreement with those reported previously in heterologous systems (Yu et al., 1997). There were, however, two notable exceptions illustrated in experiments using either a saturating concentration of oxycodone (15 μM) or a low concentration of ME (300 nM). Oxycodone and ME (300 nM) evoked currents similar in amplitude to morphine and M-6-G but induced no desensitization. Thus, the amount of desensitization induced by many, but not all, agonists can be predicted by the efficacy in activation of the GIRK conductance.
These results demonstrate that the amount of short-term desensitization induced by a saturating concentration of any given agonist can be used to predict the amount of recovery from desensitization. When there is more desensitization, more recovery was obtained. Although the degree of agonist-specific desensitization may be governed by different mechanisms, the present results indicate that MOR resensitization is directly related to the degree of desensitization. Moreover, the temporal component of these experiments suggests that there is a rapid phase of receptor resensitization that is analogous to rapid MOR desensitization. Desensitization was induced by a short agonist exposure (10 min), and the rate of recovery in the fentanyl and etorphine experiments was greatest in the first 15 min. These observations probably reflect short-term MOR regulatory processes.
This is in contrast to the results from other studies showing that morphine and DAMGO induced the same degree of MOR desensitization but that morphine-exposed receptors failed to recover after 60 min, whereas the DAMGO-treated receptors recovered completely after 40 min (Koch et al., 2004; Schulz et al., 2004). Although our results agree qualitatively in that MORs recover to a greater extent after DAMGO exposure than after morphine exposure, important differences prohibit direct comparison of our results. One significant difference between the present results and those obtained in HEK293 cells expressing MORs was the duration of agonist exposure. The desensitization induced by a 4-h exposure used in experiments with the HEK293 cells may have resulted in downstream adaptations that decreased signaling rather than direct receptor-dependent desensitization observed in the present study. It is possible that these experiments address separate phenomena.
Oxycodone Is Different. Oxycodone is a frequently prescribed opiate analgesic used to control moderate to severe pain. It has approximately the same lipophilicity as morphine (partition coefficients of 0.91 and 1.07, respectively), but lower MOR affinity (Ki, 1.7 ± 0.5 and 43.9 ± 7 nM) (Peckham and Traynor, 2006). Oxycodone has approximately the same efficacy as morphine as determined by a guanosine 5′-3-O-(thio)triphosphate stimulation assay in rat thalamic brain slices (36.6 ± 4.9 and 42.8 ± 5.3% of the DAMGO-induced activation, respectively) (Peckham and Traynor, 2006). Its analgesic efficacy is probably the consequence of high bioavailability and the potency, affinity, and efficacy of its primary active metabolite oxymorphone (Lemberg et al., 2006). Results presented here show that oxymorphone evoked a large outward current that desensitized extensively, whereas oxycodone-evoked currents are similar to morphine but, in contrast to all other agonists tested, did not desensitize at all. Thus, the properties of both oxycodone and oxymorphone must be taken into account for experiments involving long-term treatment via systemic administration because the properties of each are so different.
With respect to efficacy, desensitization, and recovery, oxycodone is most similar to low-concentration ME (300 nM), not morphine or M-6-G. The significance of this deviation is that oxycodone-bound MORs may elicit robust GIRK signaling but somehow manage to elude desensitization machinery. It is the only agonist tested here that is capable of discriminating between these elements of short-term agonist-specific MOR signaling and receptor regulation. Furthermore, it has been reported that oxycodone and morphine trigger the same amount of internalization in HEK293 cells (Koch et al., 2005), although it remains unknown how the two drugs compare with respect to internalization in neurons. If oxycodone triggered less endocytosis than morphine in neurons, in the same way that it induced significantly less desensitization, it may prove to be an important tool for experiments testing the relative activity versus endocytosis hypothesis (Whistler et al., 1999).
Receptor Number and ME Concentration Affect Desensitization. The three experiments done with ME indicate that a saturating concentration is required to induce desensitization (Figs. 2 and 6). A saturating concentration of ME (30 μM) induced desensitization to approximately 65% of peak current amplitude after 10 min. When an EC50 concentration of ME (300 nM) was used, the amplitude of the peak current was approximately 50% of that induced by a saturating ME (30 μM) concentration (215 and 460 pA, respectively), and this caused no desensitization (95% current remaining after 10 min). These results demonstrate that desensitization is dependent on both receptor occupancy and receptor number, as has been shown for MOR and other G-protein-coupled receptors (Zhang et al., 1997; Law et al., 2000).
Conclusions
Several strategies are currently used in the clinic to effectively manage pain and to treat opiate addiction. Regardless of the indication or treatment desired, tolerance is the central complication in opiate therapy, because compensation by dose escalation often results in toxic consequences. It is now appreciated that agonist-specific differences in the development of tolerance in vivo are profound and that there are cellular correlates of agonist-specific MOR regulation. Effective therapy can be maximized only by creating a thorough pharmacological profile of each opiate agonist, knowing how each differentially regulates MOR and understanding how differential MOR regulation influences the development of tolerance.
Footnotes
-
This work was supported by National Institutes of Health grants 1F30-DA021466 (to M.S.V.) and DA08163 (to J.T.W.), and by the NARSAD-Ritter Foundation (to J.T.W.).
-
ABBREVIATIONS: MOR, μ-opioid receptor; LC, locus ceruleus; β-CNA, β-chlornaltrexamine; ME, [Met]5enkephalin; DAMGO, [d-Ala2,N-Me-Phe4-Gly5-ol]-enkephalin; M-6-G, morphine-6-glucuronide; GIRK, G protein-gated inwardly rectifying potassium; UK14304, 5-bromo-6-[2-imidazolin-2-ylamino]quinoxaline; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; HEK, human embryonic kidney.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received October 25, 2007.
- Accepted January 14, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}