


Abstract
Morphine has been widely accepted as the opioid agonist that sustains signaling because it does not cause receptor desensitization or internalization. This notion has led to the hypothesis that long-term morphine treatment initiates downstream adaptations that underlie tolerance and dependence. This study uses whole-cell recordings from neurons in the locus ceruleus to measure the potassium current induced by morphine. The results show that morphine does cause short-term desensitization. The desensitization induced by morphine was slower and smaller then that induced by [MET]5-enkephalin (ME). After a brief application of a saturating concentration of ME, the current induced by morphine was smaller, and desensitization was not observed. In tissue taken from morphine-treated animals, the peak current induced by morphine was the same as in untreated animals, but morphine-induced desensitization was facilitated. The results suggest that morphine, like other agonists, can initiate receptor desensitization to decrease signaling.
Morphine is one of a very few opioids that cause little or no desensitization, internalization, and recycling of μ-opioid receptors (MOR) (Sternini et al., 1996; Yu et al., 1997; Kovoor et al., 1998; Whistler and von Zastrow, 1998; von Zastrow, 2001; Alvarez et al., 2002; Bailey et al., 2003; Minnis et al., 2003). This property of morphine has led to the hypothesis that continued signaling by morphine results in downstream adaptations that mediate tolerance to morphine. Upon removal of morphine, these adaptive changes result in a rebound or withdrawal (Whistler et al., 1999; Finn and Whistler, 2001; He et al., 2002). It is therefore important to determine whether morphine is truly unique among opioid agonists. This study re-examines short-term desensitization induced by morphine in neurons of the locus ceruleus (LC).
Studies examining morphine-induced desensitization and receptor trafficking have yielded inconsistent and often contradictory results. Many studies have reported that morphine causes little or no desensitization or internalization, making it unique among opioid agonists (Sternini et al., 1996; Kovoor et al., 1998; Whistler and von Zastrow, 1998; Whistler et al., 1999; Finn and Whistler, 2001; Alvarez et al., 2002; Blanchet and Luscher, 2002; He et al., 2002; Bailey et al., 2003). However, several reports suggest that under certain experimental conditions, morphine can cause desensitization and receptor internalization. For example, when G-protein receptor kinase 2 (GRK2) was overexpressed in human embryonic kidney 293 cells, morphine was able to cause desensitization and internalization (Whistler and von Zastrow, 1998; Zhang et al., 1998). Morphine has also been shown to cause MOR internalization in proximal dendrites but not at the soma of cultured nucleus accumbens neurons expressing both endogenous and epitope-tagged MORs (Haberstock-Debic et al., 2003). In addition, a recent study in LC neurons reported that after activation of PKC morphine caused significant desensitization (Bailey et al., 2004). Finally, with the use of a sensitive assay, morphine-induced desensitization but not internalization was detected in AtT20 cells (Borgland et al., 2003, but see Celver et al., 2004). Together, these results all suggest that morphine, under some experimental circumstances, can cause desensitization and internalization.
The failure to observe morphine-induced desensitization under control conditions in LC neurons may be the result of the protocols used to examine the effect of morphine. Morphine-induced desensitization was often examined after the maximal opioid current was determined using a brief test with a saturating concentration of ME. Recent work indicates that even a brief treatment with ME resulted in significant desensitization (Dang and Williams, 2004). It is possible that the failure to detect morphine-induced desensitization in previous studies resulted from the desensitization induced by the prior test with ME. This study examines the current induced by morphine before exposure to ME, and the results indicate that morphine does cause desensitization.
Materials and Methods
Whole-cell recordings were done in either coronal or horizontal brainstem slices (250–270 μm) containing the LC prepared from adult male Sprague-Dawley rats (140–200g; Charles River Laboratories, Inc., Wilmington, MA) as described previously (Ishimatsu and Williams, 1996). Extracellular solution contained 126 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 21.4 mM NaHCO3, 11.1 mM glucose, equilibrated with 95% O2/5% CO2 at 35°C. Whole-cell recordings were made using Nomarski optics and infrared illumination. Recordings were made with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA) voltage-clamp mode. Pipettes (2–3 MΩ) were filled with internal solution containing 115 mM MES potassium salt, 20 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 5 mM HEPES, 4 mM Mg-ATP, and 0.4 mM Na-GTP, pH 7.3.
Data collection was done with a PowerLab (Chart ver. 4.1) sampled at 100 Hz. Data analysis was done with Prism software (GraphPad Software, San Diego, CA). Values are given as mean ± S.E.M. For all experiments, p < 0.05 was considered significant. Multiple group comparisons were made with two-way analysis of variance or unpaired t test. Paired t test was used to determine significance within groups.
Morphine Treatment. Rats were anesthetized with halothane or isoflurane and given one placebo/morphine pellet (75 mg/pellet) on day 1, two pellets on day 3, and two pellets on day 5. Experiments were done on days 6 or 7. Control animals in this study consist of naive and placebo-treated animals.
Materials. [Met]5-Enkephalin, bestatin, and yohimbine were obtained from Sigma (St. Louis, MO). Naloxone and UK14304 were obtained from RBI/Sigma (Natick, MA). Thiorphan was obtained from Bachem California (Torrance, CA). Morphine wash was obtained from the National Institute on Drug Abuse.
Results
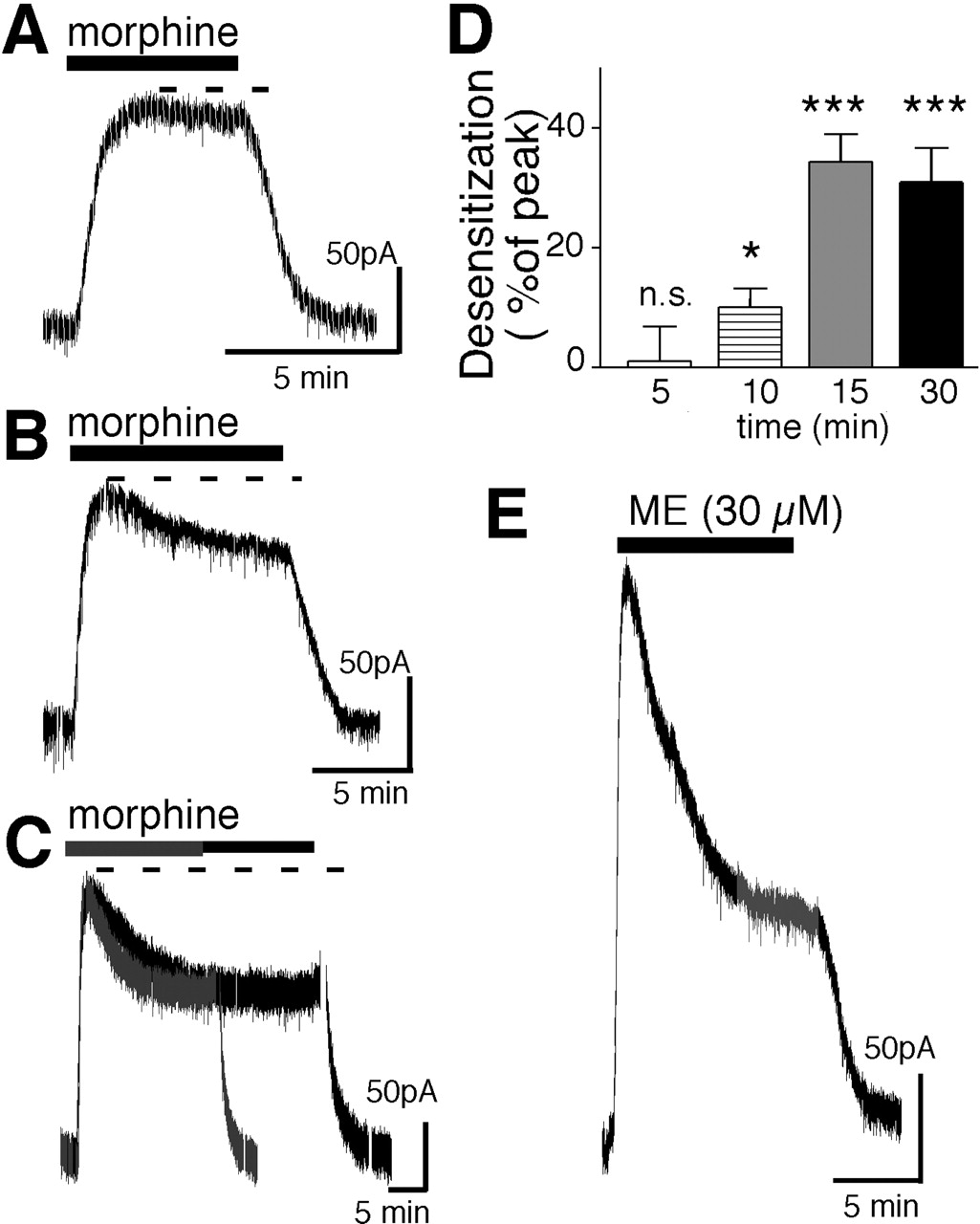
Morphine (15 μM) caused an outward potassium current with a peak amplitude of 113 ± 9 pA (n = 22). The current did not decline for the first 5 min but decreased by 10 ± 3% after 10 min (n = 5) and 34 ± 5% after 15 min (n = 12). Longer treatment with morphine (15 μM) caused no further reduction in current. After 30 min, the morphine-induced current decreased by 31 ± 6%, which was not different from that after 15 min (Fig. 1). The morphine-induced desensitization was slower than that caused by ME. The rate of morphine-induced desensitization was estimated by fitting the curve in experiments where the decline in current had reached equilibrium (Fig. 1C). The estimated t1/2 of desensitization induced by morphine was 5.3 ± 0.3 min compared with 3.3 ± 0.8 min for ME. The decline in peak current induced by ME (30 μM) was 55 ± 2% (n = 17) after 5 min (Fig. 1E) and did not change with longer applications of ME. This is consistent with previous reports demonstrating that ME-induced desensitization reached a steady state after 5 min (Dang and Williams, 2004). Thus, the decline in current induced by morphine was slower and smaller than that induced by ME.
Several studies have reported that morphine caused little or no desensitization (Alvarez et al., 2002; Blanchet and Luscher, 2002; Bailey et al., 2003). The experiments in those studies were often done after the maximal opioid current was determined with a saturating concentration of ME. Given the observation that even a brief application of ME (10 μM) causes transient receptor desensitization (Dang and Williams, 2004), the inability to detect morphine-induced desensitization may have resulted from occlusion induced by the test pulse of ME. When ME (10 μM, 2 min) was applied before morphine (15 μM, 15 min), the current induced by morphine was smaller and did not decline during the application (Figs. 2 and 3A). The morphine-induced current was normalized to the current induced by a saturating concentration of the α-2-adrenoceptor agonist UK14304 (3 μM). The morphine-evoked current was 72 ± 7% of the current induced by UK14304 (3 μM) when tested without prior application of ME. After a test pulse of ME (10 μM), the current evoked by morphine was reduced to 47 ± 4% of the UK14304 current (unpaired t test; P < 0.004). After pre-exposure to ME (10 μM, 1–2 min), the morphine-induced current did not decline over a 15-min application (a change of 4 ± 4%, n = 10; Fig. 2, A and B). This result indicates that even a short exposure to ME applied at a saturating concentration caused enough desensitization to decrease the current induced by morphine and occlude morphine-induced desensitization. To further investigate the decrease in the morphine current induced by ME, experiments were done with a protocol that allowed the current caused by morphine to be determined before and after application of ME. In these experiments, morphine (15 μM) was applied for 2 min, the superfusion solution was changed to ME (30 μM) for 5 min and then returned to morphine (15 μM). The morphine-induced current that remained after treatment with ME was 38 ± 3% of control (n = 5, Fig. 5C). Although the desensitization induced by ME with this protocol would be expected to be different (because of the presence of morphine), the results of this experiment indicate that the decline in the current induced by morphine was dependent on the extent of desensitization induced by ME.

Morphine-induced desensitization is slower and smaller than that induced by ME. A, morphine (15 μM, 5 min) caused an outward current that was sustained throughout the application period. B, application of morphine for 10 min resulted in an outward current that declined. C, experiments from two cells in which morphine was applied for 15 and 30 min to illustrate that the decline of the current had reached steady state after 15 min. D, summary experiments measuring the decline of the morphine-induced current during different application periods. E, the outward current induced by ME (30 μM, 10 min) is larger and declines more rapidly than the current induced by morphine.
Previous work showed that the desensitization induced by a brief treatment with ME (10 μM, 2 min) recovered completely after 20 min (Dang and Williams, 2004). Morphine-induced desensitization was tested 5, 10, 20, and 30 min after a brief exposure to ME (10 μM, Fig. 3). Morphine-induced desensitization was not observed 10 min after ME treatment. After 20 min, the current induced by morphine declined by 15 ± 4% of the peak over 15 min (n = 6, Fig. 3B). After a recovery period of 30 min, the morphine-induced current declined by 22 ± 3% over 15 min (n = 9) compared with 35% in control. Thus, the recovery of the ability for morphine to cause desensitization was slow compared with the previously reported recovery from desensitization (Dang and Williams, 2004).
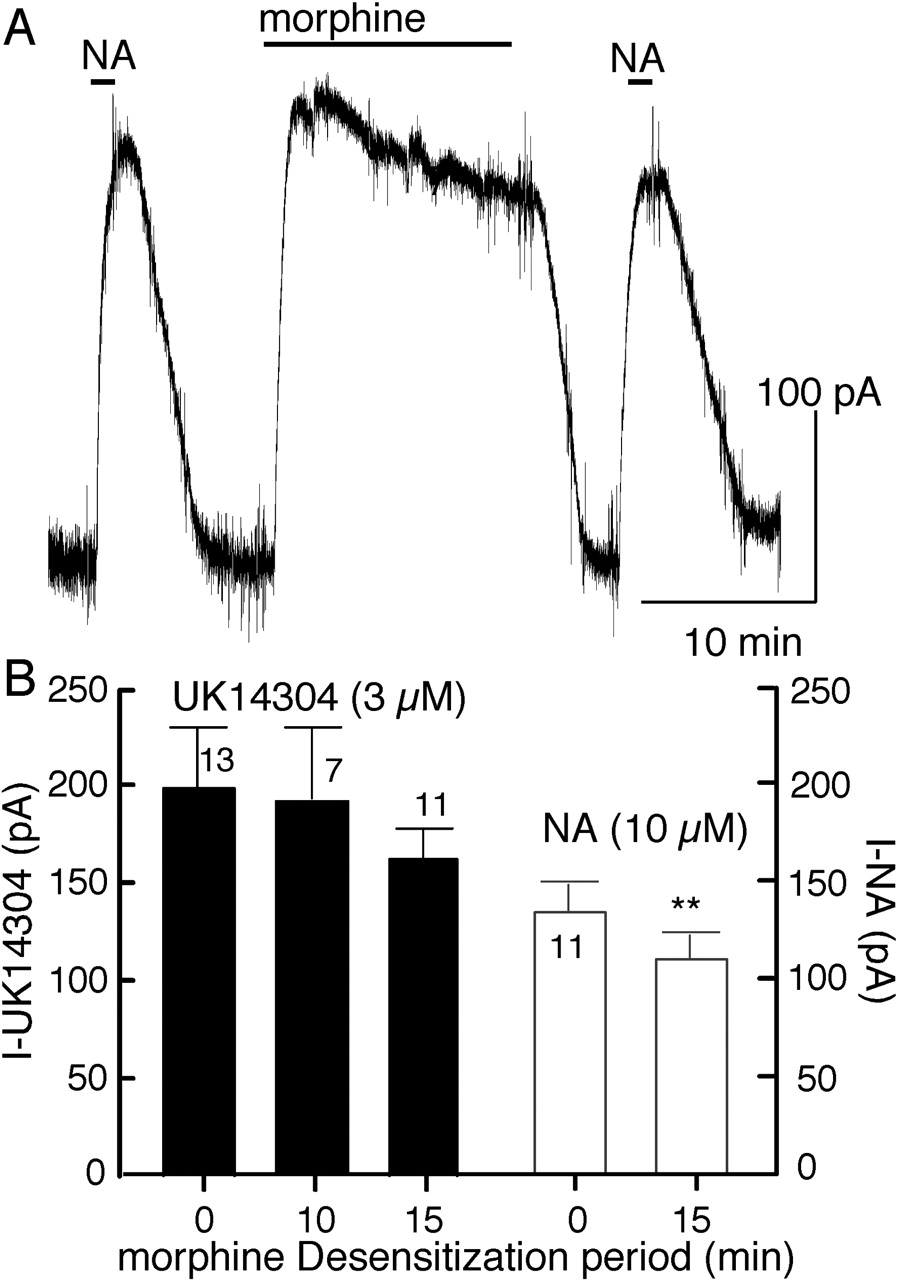
To determine whether the morphine-induced desensitization was homologous or heterologous, the current induced by noradrenalin (NA; 10 μM) was measured before (131 ± 17 pA; n = 11) and after (115 ± 14 pA) treatment with morphine (15 μM, 15 min, Fig. 4A). In addition, the current caused by a saturating concentration of UK14304 (3 μM) was tested in cells with and without prior application of morphine (15 μM, 10 and 15 min, Fig. 4B). The results show that morphine induced desensitization was primarily homologous with a small heterologous component. A small amount of heterologous desensitization was also caused by desensitization with ME (Harris and Williams, 1991; Fiorillo and Williams, 1996).

A prior application of ME (10 μM) inhibits morphine-induced desensitization. A, representative trace showing desensitization induced by morphine (15 μM, 15 min). B, the outward current induced by morphine did not decline after a brief test with ME (10 μM). C, the peak morphine-induced current, normalized to the maximum UK14304 (3 μM) evoked current, is significantly reduced by the test application of ME (10 μM).
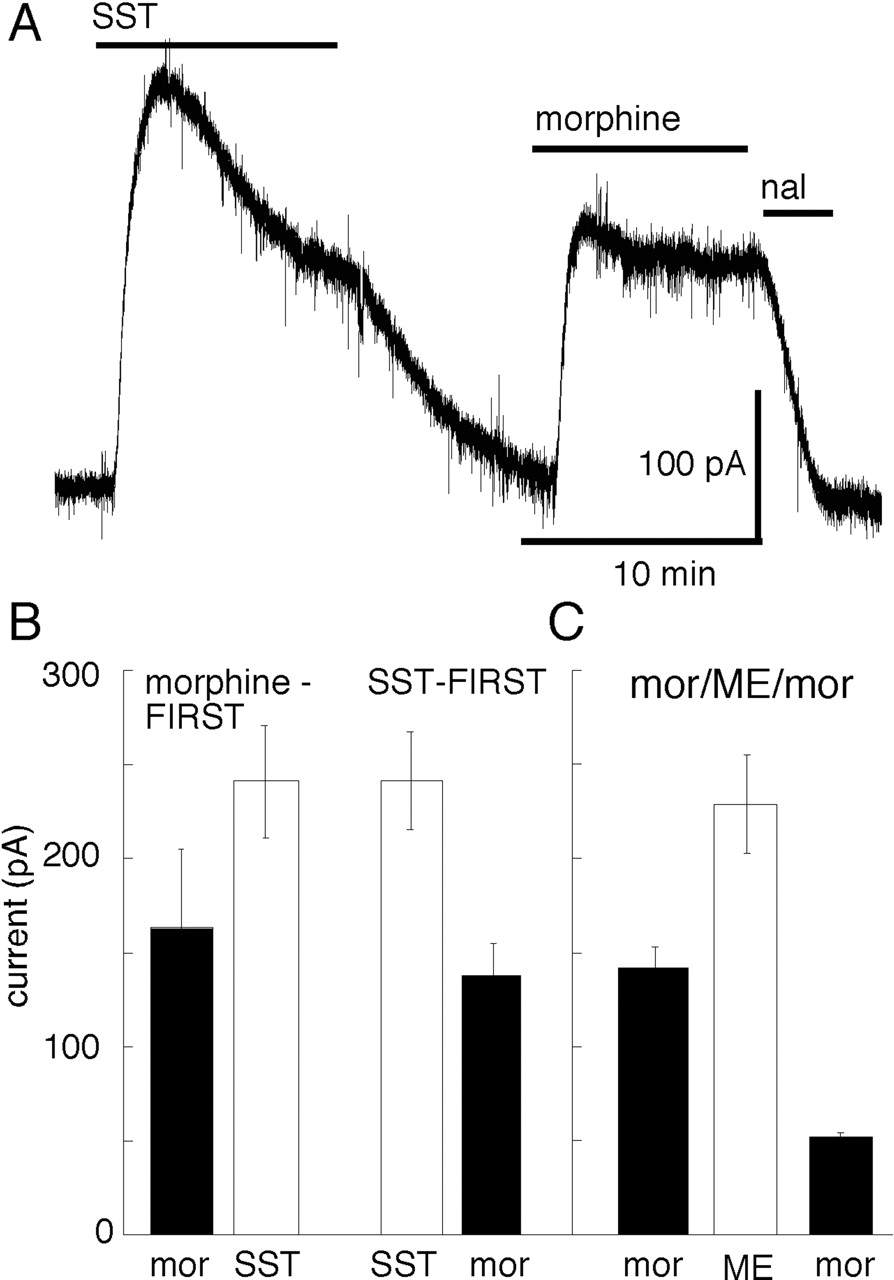
Given the small amount of heterologous desensitization induced by opioids, the question of heterologous desensitization of opioid currents induced by other G-protein-linked receptors was examined. The current induced by somatostatin (1 μM) declined to 54 ± 3% (n = 10) of the peak during an application period of 10 min (Fig. 5). After the slow washout of somatostatin, the current induced by morphine was not significantly different from that measured in another group of cells before the administration of somatostatin (Fig. 5B).
Previous work has found that the maximum morphine-induced potassium current was decreased by 40% when tested in tissues taken from animals undergoing long-term treatment with morphine (Christie et al., 1987). Similar results were obtained in experiments measuring the inhibition of calcium conductance in rapidly dissociated LC neurons, where the maximum effect of morphine was reduced in cells from morphine-treated animals compared with control animals (Connor et al., 1999). The decreased effect of morphine was interpreted to result from a reduction of receptor reserve. In the experiments measuring the potassium current (but not the inhibition of calcium current), the maximum opioid effect was determined using a short application of a saturating concentration of ME before the application of morphine. In the present study, animals underwent long-term treatment with morphine, and the outward current induced by morphine (15 μM) was determined using a protocol that did not include a prior test with a saturating concentration of ME. Using this protocol, the peak current induced by morphine was not significantly different in cells from untreated control animals (113 ± 9 pA, n = 22, Fig. 6) and morphine-treated animals (106 ± 14 pA, unpaired t test, P > 0.703). When the maximum current induced by morphine was measured after a brief treatment with ME (10 μM, 1 min), the peak current induced by morphine was reduced to 44 ± 12 pA (Fig. 6D). This result demonstrates that a brief application of ME caused a profound reduction of the current induced by morphine in slices taken from morphine-treated animals (Fig. 6D). A decrease in receptor reserve or an increase in the kinetics of short-term desensitization or both could account for this observation.

Recovery of morphine-induced desensitization is slow. A, an experiment illustrating the inability of morphine to cause desensitization 10 min after a test application of ME. B, a summary of results showing the recovery of ability for morphine to cause desensitization after a brief application of ME. The open bar (control) indicates the decline in current induced by morphine during a 15-min application without a prior application of ME. Solid bars indicate the decline in morphine current during a 15-min application period at various times after the test application of ME (10 μM, 1 min). Although the peak morphine-induced current is the same, the decline in current during the 15-min application is reduced. Full recovery was not observed after even 30 min.
The desensitization induced by morphine was facilitated in tissues taken from morphine-treated animals. The peak current induced by morphine (15 μM) declined by 17 ± 7% (n = 6) after 5 min (n = 9, Fig. 6) compared with 2 ± 3% in untreated control animals. The rate of morphine-induced desensitization was also faster than that from control animals (t1/2 = 2.9 ± 0.5 versus 5.3 ± 0.3 min from controls, Fig. 6E). Thus, long-term morphine treatment did not affect the peak current induced by morphine but increased the speed at which morphine caused desensitization. This result is consistent with previous work and suggests that the increase in desensitization caused by long-term morphine treatment can contribute to receptor-mediated tolerance (Dang and Williams, 2004).

Morphine-induced desensitization is primarily homologous. A, an experiment in which noradrenalin (10 μM, NA) was tested before and after morphine (15 μM, 15 min). There was a small but significant decrease in the amplitude of the NA current in response to the second application (control, 131 ± 17 pA; after morphine, 115 ± 14 pA, n = 11; percentage decline, 12 ± 2.5%). The decrease in the current induced by noradrenalin was significantly smaller than that induced by morphine (P < 0.015). B, summarized results showing the amplitude of the outward current induced by UK14304 (3 μM) before application of morphine (control) and after treatment with morphine (15 μM) for 10 and 15 min, and the summarized results with noradrenalin using the protocol illustrated in A. There was no significant change in the current induced by UK14304 caused by prior treatment of the preparation with morphine. There was a small and significant decrease in the current induced by noradrenalin.
Discussion
Morphine has always been considered unique among opioid agonists in that it does not cause desensitization or receptor trafficking resulting in sustained signaling. This property of morphine has been suggested to result in downstream adaptations, leading to the development of tolerance and dependence (Finn and Whistler, 2001; He et al., 2002). This study demonstrates that morphine does cause desensitization and may not be qualitatively different from other agonists, including endogenous opioids such as ME. Morphine-induced desensitization is quantitatively different from that induced by ME in that it is slower and the magnitude of decline is less. Once desensitization has reached steady state, the outward potassium current that remains after morphine (100 ± 11 pA; n = 19) and ME (98 ± 11 pA; n = 17) desensitization are the same.

Prior desensitization with somatostatin does not significantly decrease the current induced by morphine. A, somatostatin (SST, 1 μM) caused an outward current that declined during 10 min. After the washout of SST, the current induced by morphine (15 μM) was similar to that found in experiments in which morphine was applied before somatostatin. B, summary of data showing the current induced by morphine (solid bars) and somatostatin (open bars). The left bars (morphine-FIRST, n = 7) show the currents induced in the two agonists when added in that order. The bars to the right (SST-FIRST, n = 10) show results where the application of somatostatin preceded the test with morphine. C, for comparison with the experiments shown in B is a summary of results showing that ME (30 μM, 5 min) caused a reduction of the current induced by morphine.
The possibility that morphine and ME share a common desensitization mechanism is based on the observation that the current induced by morphine is decreased and does not desensitize after a short application of ME (10 μM). The decrease in the current induced by morphine probably contributes to the inability to observe desensitization and suggests that desensitization is a labile and sensitive measure of receptor-dependent processes. The slow onset of morphine-induced desensitization could be because morphine is not as efficient as other agonists at activating protein kinases and/or other steps in the initiation of desensitization. This suggestion is based on two observations. First, overexpression of GRK2 results in the increased ability for morphine to cause MOR trafficking (Kovoor et al., 1998; Zhang et al., 1998). Second, long-term morphine treatment increases the expression of GRK2 and β-arrestin2 (Terwilliger et al., 1994). This up-regulation after long-term treatment of animals with morphine could account for the increase in the rate of morphine-induced desensitization.

Long-term morphine treatment facilitates morphine-induced desensitization. Shown are current traces in slices from control (A) and morphine-treated (B) animals. Morphine (15 μM, 5 min) caused an outward current that did not decline in control animals (A) but did in the experiment using a slice from a morphine-treated animal (MTA, B). C, summarized results showing the decline in morphine-induced current during a 5-min application. D, the amplitude of the morphine-induced current in slices from morphine-treated animals before application of ME (-ME) but was reduced after a brief test with ME (10 μM, 1 min, +ME). E, the time course of morphine-induced desensitization was increased in slices from morphine-treated animals. Summary of the t1/2 of morphine- and ME-induced desensitization. After long-term morphine treatment, rate of morphine-induced desensitization was similar to that of ME.
There are indications that opioid receptor desensitization can result from multiple mechanisms. Rapid morphine-induced desensitization in LC neurons was recently reported to occur after the activation of protein kinase C (PKC) (Bailey et al., 2004). The PKC-dependent desensitization was significantly faster than that found in the present study (t1/2 ∼2 min compared with ∼5 min). It is unlikely, therefore, that the PKC-dependent mechanism underlies the desensitization described in the present work. Morphine (30 μM) has also been reported to mediate apparent desensitization through an interaction with the potassium channel (Blanchet et al., 2003). This mechanism is unlikely to account for the present observations because morphine-induced desensitization was primarily homologous. That is, there was only a small decrease in the outward current induced by a submaximal concentration noradrenalin and no significant change in the current induced by a saturating concentration of UK14304. The facilitated desensitization resulting from the activation of PKC was also homologous (Bailey et al., 2004).
After long-term morphine treatment, the maximum current induced by morphine was unchanged. This observation is different from a previous study reporting a reduction of maximum morphine current in morphine-treated animals (Christie et al., 1987). The explanation for this difference is that in previous work, the current induced by morphine was measured after a brief test with a maximum concentration of ME. This study shows that prior treatment with ME caused a dramatic reduction of the current induced by morphine (Fig. 6). After treatment with ME, the results of the present study are the same as those reported previously. That is, the morphine-induced current was dramatically reduced in slices from morphine-treated animals. Given that morphine is a partial agonist, any reduction in receptor reserve or receptor function or both can have a dramatic affect on morphine-induced signaling (Williams and North, 1984; Christie et al., 1987; Dang and Williams, 2004). The interpretation of the present and previous results is that long-term morphine treatment decreases receptor reserve such that signaling by a partial agonist, such as morphine, is more affected than full agonists. The difference between the present work and previous publications is the realization that desensitization after long-term morphine treatment plays a significant role in receptor-mediated opioid tolerance.
A role of desensitization in the long-term actions of morphine has also been suggested in studies using mice that lacked β-arrestin2. Those animals had both an increased sensitivity to morphine and reduced tolerance after long-term treatment with morphine (Bohn et al., 2000). At the cellular level, the postsynaptic sensitivity to morphine was not different in these knockout animals compared with wild-type control animals (Bradaia et al., 2005). However, a cAMP-dependent increase in synaptic transmission in the knockout animals was sensitive to morphine. It is possible that the increased presynaptic inhibition caused by morphine results in the increased behavioral response to morphine. This may be a synapse-specific observation, because no such difference in transmission between knockout and wild-type control animals was observed in the peri-aqueductal gray (Hack et al., 2005). The role that β-arrestin2 plays in postsynaptic desensitization remains to be explored. The results of the present study suggest that morphine-induced desensitization would be disrupted in the knockout animals.
Footnotes
-
This work was supported by National Institute on Drug Abuse grant DA08163.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.013185.
-
ABBREVIATIONS: MOR, μ-opioid receptor; LC, locus ceruleus; GRK2, G-protein receptor kinase 2; MES, 2-[morpholino]-ethane-sulfonic acid; UK14304, 5-bromo-6-(2-imidazolin-2-ylamino)quinoxaline; ME, [MET]5-enkephalin; NA, noradrenalin; PKC, protein kinase C.
- Received March 23, 2005.
- Accepted July 14, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}