


Abstract
Recent studies have shown that morphine, in contrast to other agonists at the μ-opioid receptor, causes very little rapid μ-opioid receptor desensitization or internalization in adult rat mammalian neurons, raising important questions about how morphine tolerance is induced. Here we show that morphine can indeed cause marked rapid desensitization of μ-opioid receptors in mature rat locus ceruleus neurons when protein kinase C is also activated. Thus, activation of Gq-coupled M3 muscarinic receptors or application of a phorbol ester enhanced the desensitization of the μ-opioid receptor-evoked potassium current in rat locus ceruleus neurons. The enhancement of desensitization was reversible by the protein kinase C inhibitors chelerythrine and 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)-maleimide (GF109203X) and resulted from an effect at the level of the μ-opioid receptor rather than the potassium channel. This is the first finding that morphine can induce rapid μ-opioid receptor desensitization in adult rat neurons, and because reduced protein kinase C activity in vivo attenuates morphine tolerance, we propose that G-protein coupled receptor cross-talk and the level of protein kinase C activity may play critical roles in the desensitization of the μ-opioid receptor and could underlie the development of morphine tolerance.
The rewarding and analgesic effects of morphine occur through activation of μ-opioid receptors (MORs) (Matthes et al., 1996). In the whole animal, tolerance to morphine develops rapidly, even after a single dose (Inoue and Ueda, 2000), compromising the clinical efficacy of morphine as well as contributing to the social problems inherent in recreational opioid abuse. Much effort has been spent trying to uncover the cellular mechanisms underlying the development of morphine tolerance, but they remain poorly understood. It was long held that tolerance to morphine occurs, at least in part, by MOR desensitization (Nestler and Aghajanian, 1997). However, recent studies examining rapid MOR desensitization and internalization by morphine have cast doubt over these processes contributing to morphine tolerance, because it has recently been observed that morphine, in direct contrast to other agonists, causes very little rapid MOR desensitization or internalization in adult rat locus ceruleus (LC) neurons or HEK293 cells (Whistler et al., 1999; Finn and Whistler, 2001; Alvarez et al., 2002; Bailey et al., 2003). This finding has resulted in a re-examination of the fundamental assumptions concerning opioid action and how tolerance to morphine occurs (Whistler et al., 1999; Finn and Whistler, 2001). Finn and Whistler (2001) speculated that morphine produces tolerance because it does not induce rapid receptor desensitization but continues to activate the receptors and thus evokes other adaptive mechanisms that result in the development of tolerance.
In the present study, we examined the role of protein kinase C (PKC) in morphine-induced desensitization of native MORs in mature mammalian LC neurons at the single neuron level. LC neurons have been studied because they are a homogeneous population of neurons that express the MOR in the absence of other opioid receptor types (North et al., 1987). We observed that concomitant PKC activation through either M3 muscarinic receptor activation or by exposure to a phorbol ester resulted in a marked increase in the amount of MOR desensitization induced by morphine and enhanced MOR desensitization induced by the endogenous opioid peptide Met-enkephalin (ME). At present, it is unclear how PKC enhances morphine-induced desensitization. This could be by direct phosphorylation of the MOR or by activation and potentiation of other cellular mechanisms involved in G-protein-coupled receptor desensitization such as G-protein-coupled receptor kinases (GRKs), arrestins, and mitogen-activated protein kinases, which could also lead to an increase in MOR desensitization (Liu and Anand, 2001).
A number of previous in vivo studies have shown that reduced PKC activity attenuates morphine tolerance (Watkins et al., 1984; Trujillo and Akil, 1991; Granados-Soto et al., 2000; Inoue and Ueda, 2000; Popik et al., 2000; Zeitz et al., 2001; Bohn et al., 2002; Hua et al., 2002; Powell et al., 2003). This study constitutes the first observation of morphine causing significant rapid MOR desensitization in mature rat neurons and highlights the crucial requirement for PKC in this phenomenon. We therefore propose that morphine-induced MOR desensitization may contribute to morphine tolerance and that PKC plays a crucial role in this process.
Materials and Methods
Brain Slice Preparation. Male Wistar rats (130-170 g) were killed by cervical dislocation, and the brains were removed and rapidly submerged in ice-cold cutting solution containing 20 mM NaCl, 2.5 mM KCl, 0.5 mM CaCl2, 7 mM MgCl2, 1.25 mM NaH2PO4, 85 mM sucrose, 25 mM d-glucose, and 60 mM NaHCO3, saturated with 95% O2/5% CO2. Horizontal slices (250 μm thick) containing the LC were prepared using a vibratome (Agar Scientific, Essex, England, UK) (Fiorillo and Williams, 1996). Immediately upon cutting, slices were submerged in an artificial cerebrospinal fluid (aCSF) containing 126 mM NaCl, 2.5 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 11.1 mM d-glucose, 21.4 mM NaHCO3, and 0.1 mM ascorbic acid, saturated with 95% O2/5% CO2 at 34°C, and were left to equilibrate for at least 1 h. All experiments were performed in accordance with the U.K. Animals (Scientific Procedures) Act of 1986, the European Communities Council Directive of 1986 (86/609/EEC), and the University of Bristol ethical review document.
Whole-Cell Patch-Clamp Recordings. Slices were submerged in a slice chamber (0.5 ml) mounted on the microscope stage and superfused (2.5-3 ml/min) with aCSF at 33 to 34°C. LC neurons were visualized by Nomarski optics (Nomarski, Edgewater, NJ), and individual cell somata were cleaned by gentle flow of aCSF from a pipette. Whole-cell voltage-clamp recordings (Vh = -60 mV) were made using electrodes (3-6 MΩ) filled with 115 mM potassium gluconate, 10 mM HEPES, 11 mM EGTA, 2 mM MgCl2, 10 mM NaCl, 2 mM MgATP, and 0.25 mM Na2GTP, pH 7.3 (osmolarity, 270 mOsM). Recordings of whole-cell current were filtered at 2 kHz using an Axopatch 200B amplifier (Axon Instruments Inc., Union City, CA) and analyzed offline using pClamp (Axon Instruments). All drugs were applied in the superfusing solution in known concentrations. Naloxone (1 μM) was applied at the end of each morphine application to antagonize the response completely, and ME was allowed to wash out. In those experiments in which α2-adrenoceptor desensitization was studied, noradrenaline (NA) was applied in the presence of prazosin (1 μM) and cocaine (3 μM). Desensitization of evoked currents was quantified by expressing the magnitude of decline in current as a percentage of the initial peak current. Cells in which the maximum opioid-evoked currents were <50 pA were excluded from the data analysis.
Statistical Analysis. All data are presented as mean ± S.E.M. Two-way ANOVA was used for statistical analysis of desensitization of opioid-induced currents. Student's t test was used for analysis of lines of best fit of desensitization kinetics.
Results
Activation of PKC by a Phorbol Ester Enhances Morphine Desensitization. Activation of MORs on LC neurons evoked a transmembrane current caused by the opening of G-protein-coupled inwardly rectifying K+ channels (GIRKs), and by performing whole-cell patch-clamp recordings, a real-time index of MOR activation could be recorded. When a receptor-saturating concentration of morphine (30 μM) was applied, an outward current (122 ± 7 pA; n = 12) was rapidly induced, and over the course of a 7-min drug application, only a small degree of desensitization occurred (Fig. 1, a, c, and g). In contrast, ME applied at the receptor-saturating concentration of 30 μM evoked an outward current (peak current, 232 ± 18 pA; n = 14) that desensitized rapidly in the continued presence of the drug (Fig. 1, e, f, and g). This desensitization did not involve PKC because it was not inhibited by chelerythrine (5 μM) (Fig. 1, h-i).

Direct activation of PKC by a phorbol ester enhances MOR desensitization. a and b, in the presence of PMA (1 μM), morphine (30 μM) caused MOR desensitization. Drugs were applied for the durations indicated by the bars. Each trace is from a separate cell. Scale bars, 50 pA for 5 min. c, superimposed traces, scaled to give equal peak responses, evoked by morphine in the absence (▪) and presence (\#9641;) of PMA. Scale bar, 5 min. d, pooled data (n = 4-12) showing the time course of morphine-induced desensitization in control cells (▪) and in cells exposed to PMA (○; ★, P < 0.01 versus control). The effect of PMA was inhibited by chelerythrine (5 μM; ▴;†, P < 0.01 versus PMA alone). e, ME-induced desensitization was increased after PMA incubation (gray trace, in the presence of PMA; black trace, control; scale bar, 5 min). f, pooled data showing ME-induced desensitization in control slices (▪) in the presence of PMA (○) and in the presence of PMA + chelerythrine (▴, n = 4-14; ★, P < 0.01 versus control; †, P < 0.01 versus PMA alone). g, desensitization kinetics of data displayed in b and d. All data from a single treatment group were pooled, and a single line of best-fit was constructed from a single exponential decay (all lines of best-fit R2 > 0.999). Maximum desensitization (%) was obtained from the plateau of the line of best-fit and are displayed as mean ± S.E.M. t1/2 Values for decay (in seconds) are displayed as means with 95% confidence intervals in parentheses. ★, P < 0.01 versus morphine alone; †, P < 0.01 versus ME alone, Student's t test. h and i, administration of chelerythrine (5 μM) alone had no effect on agonist-induced MOR desensitization. Pooled data from 3 to 12 cells (h) showing no effect of chelerythrine (○) on morphine-induced MOR desensitization; ▪, control. Likewise, chelerythrine did not affect ME-induced MOR desensitization (i, ▪, control; ○, + chelerythrine). All data were analyzed by two-way ANOVA.
PKC can be directly activated by phorbol esters. Figure 1, b-d, shows that in slices preincubated for 20 min with phorbol-12-myristate-13-acetate (PMA; 1 μM), morphine-induced desensitization was dramatically increased. The effect of PMA was blocked by the PKC inhibitor chelerythrine (5 μM) (Fig. 1d).
PMA treatment also increased ME-induced desensitization. Preincubation with PMA (1 μM; 20 min) increased ME-induced desensitization (Fig. 1, e-f), an effect that was blocked by chelerythrine (5 μM) (Fig. 1f). The apparent increased “noise” level in the presence of PMA (see current traces in Fig. 1) is caused by the increased frequency of spontaneous excitatory postsynaptic currents and inhibitory postsynaptic currents produced by PMA. This has been reported previously to occur at other central nervous system synapses (Malenka et al., 1986).
All desensitization traces could be fitted well to single exponential decays (Fig. 1g) (R2 > 0.999 for each condition). These data show that the extent of PMA-induced morphine desensitization is 36.5 ± 2.1%. The extent of decay but not the t1/2 was significantly different in the presence of PMA compared with control slices. Likewise, when the desensitization kinetics of ME were fitted to single exponential decays, only the maximum extent but not the 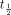 , was significantly altered by the presence of PMA. The maximum extents and rates of desensitization induced by morphine or ME in the combined presence of PMA and chelerythrine were not significantly different from those of control slices (data not shown).
, was significantly altered by the presence of PMA. The maximum extents and rates of desensitization induced by morphine or ME in the combined presence of PMA and chelerythrine were not significantly different from those of control slices (data not shown).
Further evidence that the enhancing effect of PMA on morphine and ME-induced MOR desensitization was mediated by activation of PKC is that the phorbol ester 4α-phorbol-12,13-didecanoate, which does not activate PKC, had no effect on MOR desensitization when ME or morphine was applied (data not shown). We then examined the effects of GF109203X, a bisindolylmaleimide inhibitor of PKC that binds to the active site of the enzyme rather than to the phorbol ester-responsive C1 domain common to a number of phorbol ester-responsive proteins (Yang and Kazanietz, 2003). GF109203X (1 μM), like chelerythrine, abolished the enhancement of MOR desensitization by PMA without affecting the desensitization induced by ME alone (data not shown).
M3 Receptor Activation Enhances Morphine Desensitization. LC neurons also express Gq-coupled M3 muscarinic receptors (Egan and North, 1985). We next tested whether increasing PKC activity by activating M3 muscarinic receptors can convert morphine into a desensitizing MOR agonist. Addition of the muscarinic agonist oxotremorine-M (oxo-M; 10 μM) resulted in a small inward current (77 ± 11 pA; n = 6) that decayed to a steady level within 10 min (57 ± 12 pA). When morphine was applied after 10 min of oxo-M treatment, the level of MOR desensitization was dramatically increased (Fig. 2, a-c). The facilitatory effect of oxo-M on morphine-induced desensitization was abolished in the presence of the PKC inhibitor chelerythrine (preincubation of 5 μM for 20 min) (Fig. 2c), indicating that it resulted from the activation of PKC.

MOR desensitization in LC neurons is enhanced by M3 muscarinic receptor activation. a and b, morphine-evoked currents in single LC neurons. Morphine (30 μM) alone caused little MOR desensitization. In the presence of oxo-M (10 μM), morphine induced a current that showed significant desensitization. Oxo-M was present for 10 min before and during morphine application. The oxo-M-induced current had declined to steady state before morphine application. c, pooled data from 4 to 12 cells showing the time course of morphine-induced desensitization in control (▪) and oxo-M-treated cells (○); ★, P < 0.01 versus control. The effect of oxo-M was inhibited by the PKC inhibitor chelerythrine (5 μM, ▴; n = 3; †, P < 0.01 versus oxo-M alone). d and e, in control cells, the current evoked by ME (30 μM) rapidly desensitized. Pre-exposure to oxo-M (10 μM for 10 min) increased the ME-induced desensitization. f, pooled data from 3 to 14 cells showing the time course of the oxo-M enhancement (○) of the ME-induced desensitization (▪), an effect that was inhibited by chelerythrine (5 μM, ▴; n = 4; ★, P < 0.01 versus control; †, P < 0.01 versus oxo-M alone). For current traces, scale bars represent 50 pA and 5 min. All data were analyzed by two-way ANOVA. g, desensitization kinetics of c and f, analyzed as described for Fig. 1g. All lines of best-fit R2 > 0.999. ★, P < 0.01 versus morphine alone; †, P < 0.01 versus ME alone, Student's t test.
We then determined whether the effect of oxo-M on the desensitization was induced by ME. ME-induced MOR desensitization was increased by the activation of M3 muscarinic receptors (10 μM oxo-M for 10 min) (Fig. 2, e and f). The enhancement of desensitization by oxo-M was blocked by preincubation of chelerythrine (5 μM) (Fig. 1f). No enhancement of spontaneous synaptic events was observed after administration of oxo-M, unlike with PMA.
All desensitization kinetics fitted well to single exponential decays (R2 > 0.999). As with PMA, the maximum extents but not the rates of morphine or ME-induced desensitization were significantly enhanced by the presence of oxo-M (Fig. 2g), an effect that was abolished by the presence of chelerythrine (data not shown).
Increased MOR Desensitization after PKC Activation Occurs at the Level of the Receptor and RequiresMOR Activation. Other researchers have reported that in some cell types (Mao et al., 2004) but not in others (Lesage et al., 1995) PKC has been shown to inhibit GIRK channels. Because our functional readout of MOR activation was the opening of GIRKs, it was possible that the apparent increase in MOR desensitization produced by oxo-M or PMA was caused by a direct effect of PKC on the channel rather than on the receptor itself. However, we have several pieces of evidence that demonstrate this is not the case and confirm that the enhancement of MOR desensitization by activation of PKC is mediated at the level of the μ-opioid receptor.
Morphine is a partial agonist at MORs in the LC (Bailey et al., 2003). If PKC was acting to inhibit the GIRK channels, then PMA should cause a decrease in the initial peak of the morphine-evoked current. Figure 3a shows that there was no decrease in the peak of the morphine-evoked current after 20-min PMA application. In addition, we performed experiments whereby two brief applications of a submaximal concentration of ME were applied 7 min apart after preincubation with PMA for 20 min. Figure 3b shows that PMA did not reduce the amplitude of the second exposure to ME (second ME response was 101 ± 2% of the first, n = 3), which is in direct contrast to the enhancement of desensitization seen over the same time scale (Fig. 1, e and f).
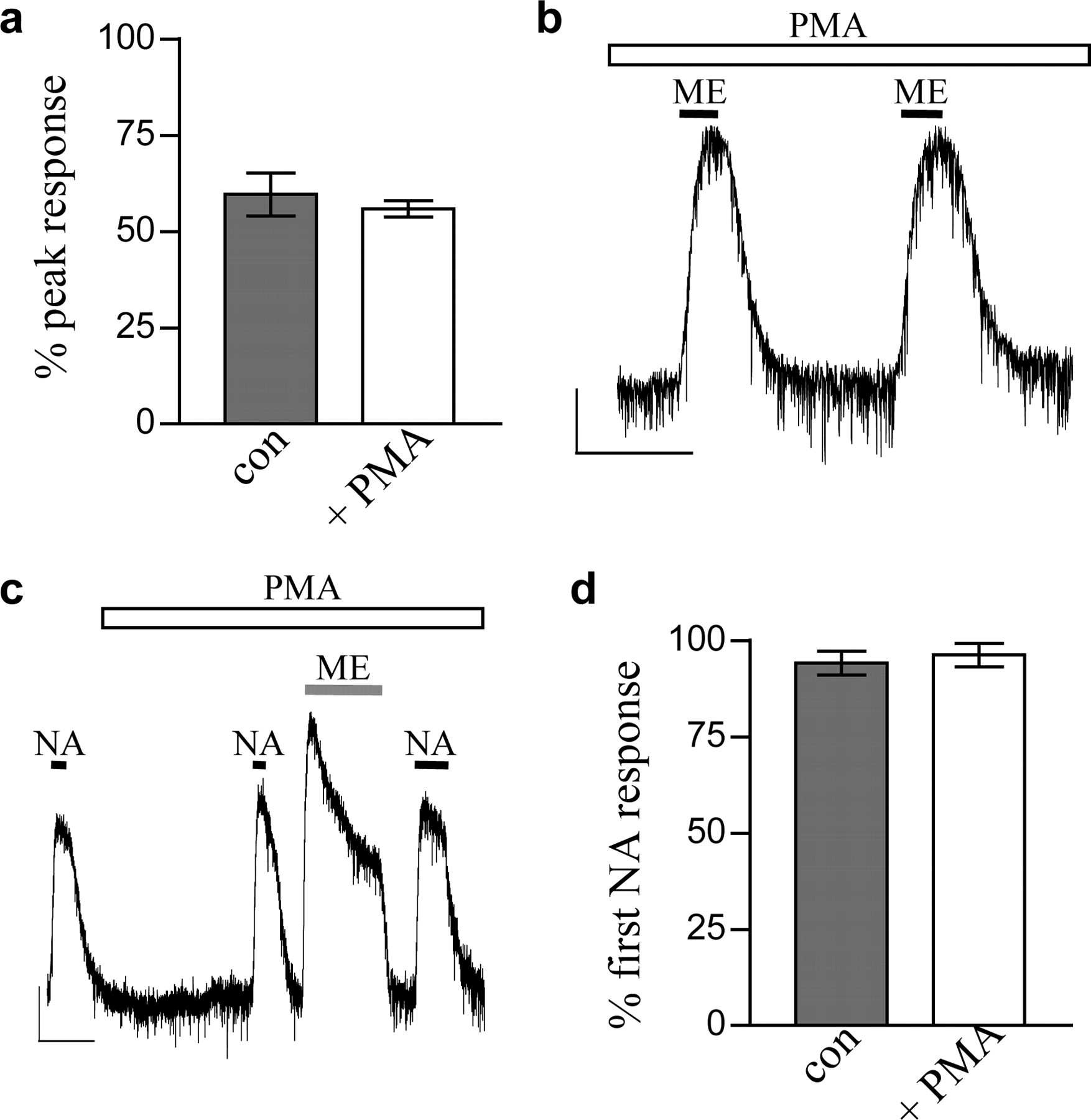
Activation of PKC increases MOR desensitization at the level of the receptor. a, the peak current evoked by morphine (30 μM), expressed as a percentage of maximal MOR current (derived by application of 10 μM ME), was unaffected by 20-min preincubation of PMA (1 μM). \#9641;, control; □, in the presence of PMA (n = 5). b, responses to brief applications of ME (1 μM) in the presence of PMA (1 μM) did not decrease. This sample trace is typical of three experiments in the presence of PMA in which the second response was 101 ± 2% of the first. Scale bars, 50 pA and 5 min. c and d, NA current amplitude was unaffected by PMA (1 μM) or by PMA-enhanced 30 μM ME-induced MOR desensitization. Scale bars, 50 pA and 5 min. d, pooled data (n = 5-12 cells) showing the NA current amplitude after ME-induced MOR desensitization as a percentage of the initial NA response. Con, control; + PMA, in the presence of 1 μM PMA.
To exclude the possibility that PKC activation was in some way producing a use-dependent block of the GIRK channels, we made use of the fact that MORs and α2-adrenoceptors couple to the same set of GIRK channels in LC neurons (North and Williams, 1985; C.P. Bailey and G. Henderson, unpublished observations). First, we applied NA (100 μM) before and after ME-induced desensitization. Figure 3, c and d, shows that in the presence of PMA, the amplitudes of the current evoked by NA before and after ME-induced desensitization were identical in control and PMA-treated slices. Second, we examined the effect of PMA on the rate of desensitization of the noradrenaline-evoked GIRK current. PMA did not significantly enhance the desensitization of the NA-induced response, although it did enhance the desensitization of the morphine response (Fig. 4). Because these two receptors couple to the same set of GIRK channels, the enhancement of MOR desensitization by PMA cannot therefore be caused by GIRK channel block.

Activation of PKC does not increase NA-induced desensitization of α2 receptors. a, a saturating concentration of NA (100 μM) caused a small degree of desensitization over the course of a 7-min exposure to the drug. The level of desensitization was not increased in the presence of PMA (1 μM). Sample traces, scale bars represent 50 pA and 5 min. b, traces shown in a are scaled and superimposed. Black trace, control; gray trace, + PMA. Scale bar, 5 min. c, pooled data from 5 to 11 cells show that slices incubated with PMA (○) exhibit a similar degree of NA-induced desensitization to control cells (▪). d, PMA increased morphine-induced MOR desensitization, measured after 7 min of drug exposure, to a significantly greater extent than noradrenaline-induced α2-adrenoceptor desensitization. Columns represent the percentage enhancement of desensitization after 7 min of drug exposure to morphine (30 μM) or NA (100 μM) in the presence of PMA (1 μM). n = 5; ★, P < 0.05 morphine versus NA, Student's t test.
In addition to causing a small inward current, oxo-M also reduced the peak current evoked by both ME and morphine by approximately 40%. The oxo-M-evoked inward current results from the opening of nonselective cation channels and the closing of GIRK channels (Shen and North, 1992), possibly caused by phosphatidylinositol 4,5-bisphosphate depletion (Huang et al., 1998). An action to close GIRK channels would reduce the MOR current and, if ongoing during the exposure to morphine, might result in a gradual decay of the morphine-evoked current. However, the oxo-M enhancement of desensitization was reversed by PKC inhibition, whereas the reduction of peak morphine- or ME-evoked current in the presence of oxo-M was not reversed by PKC inhibition (data not shown). Thus, oxo-M enhancement of desensitization cannot be caused by phosphatidylinositol 4,5-bisphosphate depletion and subsequent GIRK channel closure, but it is mediated by PKC.
Discussion
There has been much recent controversy concerning the ability of morphine to induced rapid MOR desensitization, a finding that has given rise to new theories concerning the mechanisms underlying morphine tolerance. In LC neurones (Alvarez et al., 2002; Bailey et al., 2003) and HEK293 cells (Whistler et al., 1999; Finn and Whistler, 2001), morphine has been reported to produce little desensitization of MORs, whereas other opioid agonists such as ME, [D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin, and methadone induce significant rapid MOR desensitization. Furthermore, in a number of cell systems, morphine has been shown to cause negligible rapid MOR internalization (Whistler et al., 1999; Finn and Whistler, 2001; Alvarez et al., 2002; Bailey et al., 2003; Borgland et al., 2003; Celver et al., 2004), although a recent study examining trafficking of recombinant MORs in nucleus accumbens neurons showed dendritic redistribution of receptors after morphine administration (Haberstock-Debic et al., 2003). We now report that in LC neurons, morphine can, in fact, cause rapid MOR desensitization, but only when PKC is activated. PKC-mediated enhancement of MOR desensitization was evoked in two ways: by directly activating PKC with a phorbol ester (PMA), or by indirectly activating PKC through endogenous Gq-coupled M3 muscarinic receptors. Unlike morphine, ME, in the absence of PKC activation, did induce MOR desensitization. This ME-induced desensitization was not reduced by the inhibition of PKC and is likely to be mediated by a G-protein receptor kinase (Kovoor et al., 1998; Zhang et al., 1998). However, increasing PKC activity with either PMA or an M3 muscarinic agonist did enhance ME-induced MOR desensitization (Fiorillo and Williams, 1996). The enhancement of morphine- or ME-induced desensitization by PMA or an M3 muscarinic agonist was completely reversed by inhibitors of PKC, indicating that these effects are mediated entirely by PKC activation.
The enhancement of MOR desensitization by PKC activation occurs at the level of the receptor, not at the level of the K+ channel. This was demonstrated by the fact that the desensitization of α2 receptors, which are coupled to the same population of GIRK channels, was not significantly enhanced by PMA. Furthermore, the PKC enhancement of desensitization requires agonist activation of the receptor because PMA did not affect the initial amplitude of MOR-evoked currents. We did observe that exposure of slices to PMA resulted in an increase in spontaneous synaptic currents, presumably through a presynaptic action to enhance transmitter release (Malenka et al., 1986). It is possible, therefore, that PMA could enhance MOR desensitization indirectly by enhancing the release of various neurotransmitters that subsequently act on the MOR-expressing neuron to somehow increase MOR desensitization. However, we believe that this is unlikely because enhanced transmitter release was not seen after activation of M3 muscarinic receptors, whereas PKC-mediated enhanced MOR desensitization was still observed. The ability of PKC to enhance MOR desensitization is unlikely to be mediated by direct phosphorylation of the G-protein, because this has been shown, if anything, to enhance the activity of the Gβγ subunit (Chakrabarti and Gintzler, 2003).
Although we and others working on LC neurones (Alvarez et al., 2002; Bailey et al., 2003) and HEK293 cells (Whistler et al., 1999; Finn and Whistler, 2001) have reported that morphine alone does not desensitize MORs, other groups studying MORs transfected into ATt20 cells have found that morphine does induce MOR desensitization (Borgland et al., 2003; Celver et al., 2004). Although at first glance this seems to be a contradiction, one possibility is that in ATt20 cells, a high level of basal PKC activity is responsible for desensitizing the morphine-activated MOR.
After 3-day in vivo morphine treatments, LC neurons show marked cellular tolerance to morphine (Christie et al., 1987). This study found that the maximum response to morphine was decreased by approximately 40%. This correlates well with our finding of a decrease in maximum response to morphine of 35 to 37% in the presence of PMA or oxo-M. Because morphine is a partial agonist, this corresponds to approximately 37% loss of functional receptor in the presence of PMA.
MOR desensitization to opioid agonists other than morphine is believed to occur largely by phosphorylation of the activated receptor by GRKs (Kovoor et al., 1998; Zhang et al., 1998), with subsequent binding of arrestins to the phosphorylated receptor uncoupling it from its G proteins (Krupnick and Benovic, 1998). Therefore, PKC activation could enhance agonist-induced MOR desensitization by 1) direct phosphorylation of the receptor; 2) enhancing the activation of other regulatory proteins such as GRKs, arrestins, and mitogen-activated protein kinases; 3) a combination of both; or 4) inhibiting receptor recycling and reactivation. Putative consensus sequences for PKC phosphorylation exist on the third intracellular loop and the C terminal tail of the MOR, and PKC has been shown to directly phosphorylate MORs (Zhang et al., 1996), although this was seen even in the absence of opioid agonists, whereas in our experiments, agonist activation of the receptor was required before PKC enhancement of desensitization was observed. Therefore, any PKC phosphorylation of the MOR in the absence of an opioid agonist in itself does not cause desensitization of the receptor but may permit some other interaction to take place (e.g., arrestin binding) once the agonist is bound. The specific GRK isoforms most heavily implicated in MOR desensitization are GRK2 and GRK3 (Kovoor et al., 1998; Zhang et al., 1998). The abilities of GRK2 and GRK3 to phosphorylate MOR are enhanced by PKC, either by direct GRK phosphorylation by PKC or by inducing translocation of GRK2 to the membrane (Chuang et al., 1995; Winstel et al., 1996; Mandyam et al., 2002; Thakker and Standifer, 2002). Therefore, PKC could enhance agonist-induced MOR desensitization indirectly by increasing the activity of GRKs. Indeed Zhang et al. (1998) showed that morphine could cause MOR desensitization in oocytes only when GRK2 was overexpressed.
In arrestin knockout mice, morphine tolerance was attenuated (Bohn et al., 2000), suggesting that arrestin-dependent MOR desensitization plays a critical role in morphine tolerance. Arrestin can bind not only to GRK-phosphorylated receptors but also to second-messenger kinase-phosphorylated receptors (Kohout and Lefkowitz, 2003). Thus, arrestin may bind to and desensitize PKC-phosphorylated MORs. It is interesting to note that the residual component of morphine tolerance present in arrestin knockout mice was removed by administration of a PKC inhibitor (Bohn et al., 2002). Thus, PKC may induce MOR desensitization in an arrestin-dependent and -independent manner. Indeed, some groups have reported that the regulation of GPCR-mediated signaling by PKC and arrestin are separable phenomena (Krupnick and Benovic, 1998).
A further potential explanation for our findings is that PKC may inhibit MOR trafficking, reinsertion, and therefore resensitization. Indeed, Ueda et al. (2001) demonstrated that inhibition of PKC could cause enhanced morphine-bound MOR internalization and reinsertion in Chinese hamster ovary cells. However, a recent study (Dang and Williams, 2004) showed that PKC inhibition attenuated the rate of recovery from ME-induced desensitization. Therefore, if anything, PKC activation may increase receptor resensitization in these neurons.
The original observation that morphine caused very little rapid MOR desensitization (Whistler et al., 1999) led to the suggestion that morphine tolerance occurs not because of receptor desensitization, but precisely because the lack of MOR desensitization gives rise to prolonged signaling from the morphine-activated MOR, and this results in the development of other compensatory cellular changes that act to diminish that signaling and thus result in the development of tolerance (Finn and Whistler, 2001). Our data demonstrate that morphine can indeed cause significant rapid MOR desensitization in adult rat LC neurons when PKC is activated. We suggest that PKC may therefore be involved in the development of morphine tolerance. In the intact nervous system, there is likely to be ongoing activation of Gq-coupled receptors; thus, the levels of PKC activity within individual neurons are likely to be increased compared with those in quiescent neurons in brain slices. Therefore, in vivo, G-protein-coupled receptor cross-talk between the MOR and Gq-coupled receptors and enhanced PKC activity may play significant roles in morphine-induced desensitization of MORs and thus may be responsible for the development of morphine tolerance.
Evidence to support our hypothesis comes from behavioral studies showing that morphine tolerance can be reduced by PKC inhibitors (Granados-Soto et al., 2000; Inoue and Ueda, 2000; Bohn et al., 2002; Hua et al., 2002), in PKC knockout mice (Zeitz et al., 2001), or by blockade of various Gq-coupled receptors (Watkins et al., 1984; Popik et al., 2000; Powell et al., 2003) or can be enhanced by activation of PKC (Narita et al., 1997). In addition, N-methyl-d-aspartate receptor antagonism has been shown to reduce morphine tolerance (Trujillo and Akil, 1991). N-methyl-d-aspartate receptor activation would result in calcium influx and thus PKC activation (Mao et al., 1995). Indeed, the facilitatory effect of PKC on morphine-induced rapid MOR desensitization can also contribute to acute morphine tolerance seen during a single dose (Watkins et al., 1984; Bohn et al., 1999), meaning that agents which inhibit PKC can be used clinically as adjuncts to morphine (Caruso, 2000).
Footnotes
-
This work was supported in part by the Wellcome Trust.
-
ABBREVIATIONS: MOR, μ-opioid receptor; GF109203X, 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)-maleimide; GIRK, G-protein-coupled inwardly rectifying K+ channel; GRK, G-protein coupled receptor kinase; PMA, phorbol-12-myristate-13-acetate; HEK, human embryonic kidney; LC, locus ceruleus; PKC, protein kinase C; ME, Met-enkephalin; aCSF, artificial cerebrospinal fluid; NA, noradrenaline; ANOVA, analysis of variance; oxo-M, oxotremorine-M.
- Received July 8, 2004.
- Accepted September 9, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}