


With at least 1000 family members encoded by mammalian genomes, the G protein-coupled receptors (GPCRs) represent the most diverse group of signaling proteins known (Bockaert and Pin, 1999). GPCRs are involved in the regulation of a wide variety of physiological processes including, but not limited to, the sensory perceptions of pain, light, odors and tastes, cognition, muscle contraction, endocrine and exocrine secretion, metabolism, inflammation, and immunity.
The classic paradigm of the GPCR signal transduction process is that upon ligand binding, conformational changes in the receptor arise that allow it to couple to the heterotrimeric G proteins. This coupling stimulates the G protein to alter the activity of a variety of downstream effector molecules (Neer, 1995). In addition to G protein coupling, activation of a GPCR by its ligand also initiates the process of receptor desensitization, an adaptive response used by cells to arrest G protein signaling, therefore preventing the potentially harmful effects that can result from persistent receptor stimulation.
Almost every GPCR that has been studied undergoes desensitization and, despite their diversity, all cells use a universal mechanism for desensitizing GPCRs. This involves the coordinated actions of two families of proteins, the G protein-coupled receptor serine/threonine kinases (GRKs) and the arrestins (Freedman and Lefkowitz, 1996;Krupnick and Benovic, 1998; Pitcher et al., 1998; Ferguson, 2001). After binding to its agonist, a GPCR assumes a conformation that allows it to bind one or more of the GRKs (of which there are seven) and, in doing so, becomes phosphorylated at residues on its intracellular loops and carboxyl terminus. Phosphorylation of the receptor promotes the high-affinity binding of the arrestin family of proteins (of which there are four) to the receptor, which physically interdicts further coupling to G proteins. This hindrance of coupling can result in as much as an 80% diminution of receptor signaling (Attramadal et al., 1992; Lohse et al., 1992).
It is also known that phosphorylation of GPCRs by several other kinases, such as protein kinases A and C (PKA and PKC) (Benovic et al., 1985; Roth et al., 1991; Pitcher et al., 1992a) and c-Src (Fan et al., 2001), can result in receptor desensitization. This process of desensitization involves a feedback mechanism in which the second messenger generated by the agonist-stimulated GPCR activates a kinase that decreases the activity of the receptor and ultimately attenuates production of the second messenger. Despite contributions of a feedback mechanism to the desensitization of GPCRs, desensitization can be fully reconstituted in vitro using highly purified receptors, GRKs, and arrestins (Benovic et al., 1987; Attramadal et al., 1992; Lohse et al., 1992). Receptor desensitization is, in fact, highly regulated both through differences in activity exhibited by each of the individual GRK and arrestin subtypes and through their modulation by the activities of many accessory proteins. Herein, we review our current understanding of how GPCR desensitization is regulated by GRKs and arrestins.
Receptor Phosphorylation Requires Recruitment of GRKs to the Plasma Membrane
For an agonist-occupied receptor to become phosphorylated by a GRK, the kinase first must be recruited to the plasma membrane and into a complex with the receptor. Of the seven GRK types identified, three are known to be constitutively associated with the plasma membrane through covalent attachment of either fatty acids or isoprenes to their carboxyl termini: GRK4 (Premont et al., 1996) and GRK6 (Stoffel et al., 1994) are palmitoylated, whereas GRK1 (Inglese et al., 1992) is farnesylated. Furthermore, GRK7 (Hisatomi et al., 1998; Weiss et al., 1998) has a CAAX sequence at its carboxyl terminus that predicts geranylgeranyl modification. GRK5 is also predominantly associated with the membrane through interactions between a positively charged domain located near its carboxyl terminus and the negatively charged head groups of membrane lipids, including phosphatidylinositol-4,5-bisphosphate (PIP2) (Pitcher et al., 1996). The binding of phospholipids also enhances the activity of GRK5 by promoting its autophosphorylation (Kunapuli et al., 1994). Thus, five GRKs (1, 4, 5, 6, and 7) are located at the membrane and near the activated receptors, which they bind and phosphorylate.
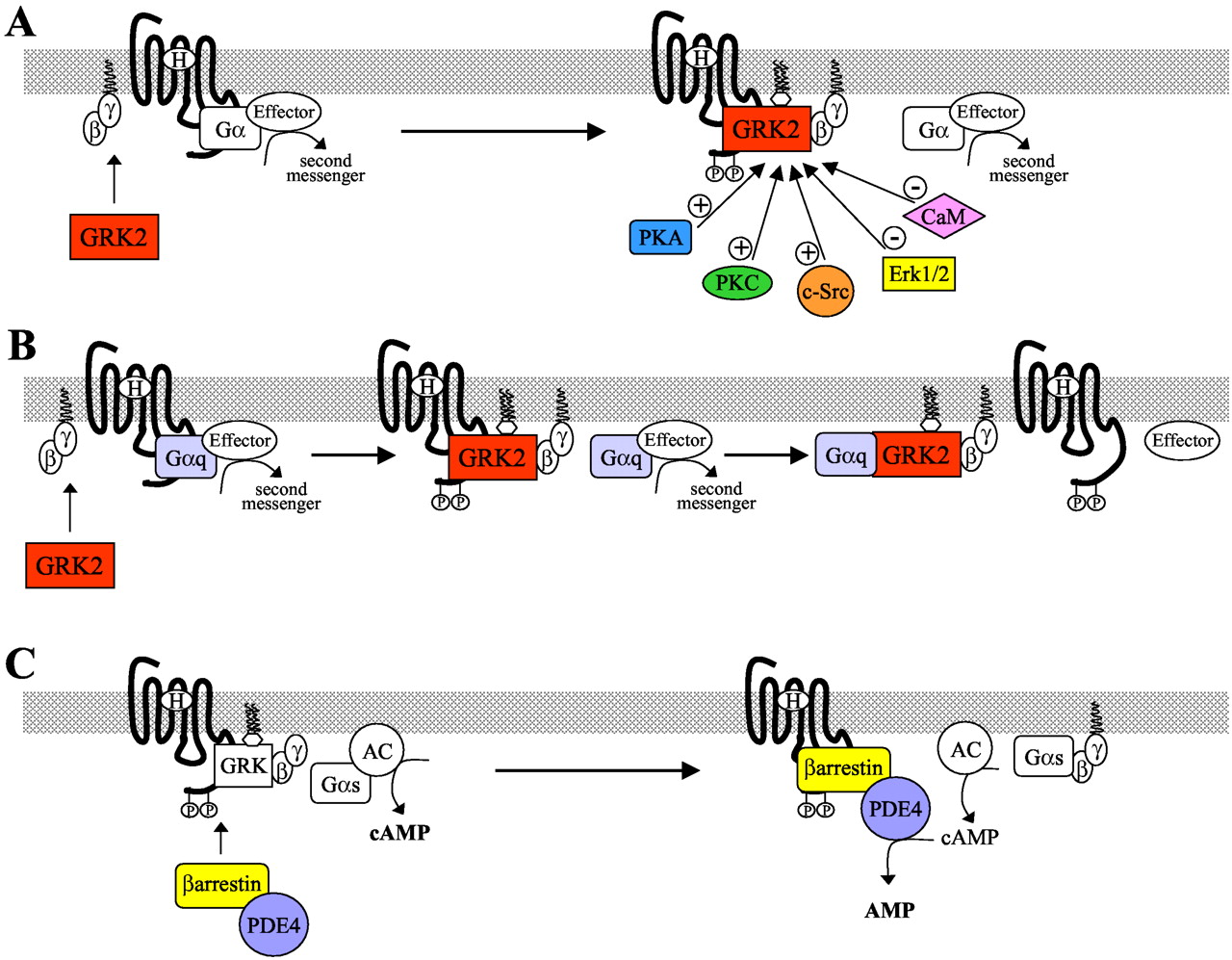
The situation is more complex with regard to GRK2 and GRK3 because they do not undergo permanent lipid or isoprene modification and are not constitutively associated with membranes. Rather, most of the cellular complement of these kinases is located in the cytosol, and they undergo only transient recruitment to the plasma membrane after GPCR activation. Both these kinases possess pleckstrin homology domains, through which they bind to PIP2 in the plasma membrane (Fig. 1A) (Pitcher et al., 1995). However, translocation also requires G protein activation because GRK2 and GRK3 must bind liberated Gβγdimers to be recruited to the membrane (Pitcher et al., 1992b). The importance of recruitment of GRK2 in GPCR desensitization has been clearly demonstrated in vitro (Koch et al., 1993) and in vivo (Koch et al., 1995) using the isolated pleckstrin homology/Gβγ-binding domain of GRK2 as an inhibitor to prevent the kinase from interacting with Gβγ subunits. This results in the receptors becoming poorly phosphorylated, and thus desensitization is greatly diminished.

The regulation of GPCR desensitization by GRK2 and β-arrestins. A, receptor activation is followed by the recruitment of GRK2 by Gβγ to the receptor where it is anchored by phosphatidylinositol-4,5-bisphosphate and positioned to phosphorylate the C terminus of the receptor. GRK2 activity can be enhanced by its phosphorylation by protein kinase A (PKA), protein kinase C (PKC), and c-Src. Alternatively, GRK2 activity can be reduced by its phosphorylation by Erk1/2 or its binding to calcium/calmodulin (CaM). B, GRK2 can also inhibit receptor signaling by sequestering Gαq to prevent its coupling to its effectors, such as PLCβ. C. Gs-coupled receptors activate the effector adenylyl cyclase (AC) to generate the second messenger cAMP. After phosphorylation of the receptor by GRK, β-arrestin is recruited to the receptor, translocating PDE4 with it. This places the PDE4 on the membrane, the site of cAMP generation, thus allowing it to efficiently degrade cAMP to AMP and reduce signaling.
GRK2 and GRK3 have also been demonstrated to bind to the activated, GTP-bound form of the Gαq subunit through domains located near the amino termini of the kinases, domains that show significant homology to the regulator of G protein signaling (RGS) family of proteins. Although RGS proteins are known to act as potent GTPase accelerating proteins for a number of G protein α subunits (Berman and Gilman, 1998), the RGS-like domains of GRK2 and GRK3 enhance Gαq GTPase activity only weakly (Carman et al., 1999). However, this interaction greatly reduces Gαq activation of phospholipase Cβ after Gq-coupled receptor stimulation by a mechanism that is independent of desensitization of the receptors, because both a catalytically inactive mutant and an amino terminal fragment of GRK2 can inhibit inositol polyphosphate production in cells (Carman et al., 1999; Sallese et al., 2000b). Thus, in addition to their ability to phosphorylate GPCRs and mediate desensitization, GRK2 and GRK3 also are able to limit the extent of Gq-coupled receptor signaling by sequestering Gαq and preventing its coupling to downstream effectors (Fig. 1B). It has also been hypothesized that the mere interaction of GRK2 and -3 with the GPCR, without the requirement for receptor phosphorylation, could be sufficient to suppress signaling (Dhami et al., 2002; Freedman et al., 1997).
Regulation of GRKs through Phosphorylation by Other Kinases
Several recent studies have shown that receptor phosphorylation by GRKs is modulated by the activity of other kinases that directly phosphorylate the GRKs and alter a variety of their properties (Fig.1A). The effects of a specific phosphorylation event are determined by what site becomes phosphorylated, which is dependent on the identity of the phosphorylating kinase. These effects include changes in catalytic activity, protein binding affinity, and stability of the GRK protein. Most of these studies have focused on the phosphorylation of GRK2, but some have investigated the regulation of GRK5.
Most of the cellular complement of GRK2 exists in a basally phosphorylated state that maintains it in an inactive conformation in the cytosol. This was first demonstrated when two chemically distinct fractions of GRK2 protein were purified by gel filtration from Sf9 cell lysates (Pitcher et al., 1999). The more abundant fraction was shown to be phosphorylated at Ser670, within a confirmed phosphorylation consensus sequence for the extracellular signal-regulated kinases, Erk1 and Erk2. Phosphorylation of Ser670 in GRK2 causes a dramatic reduction in both the activity of the kinase and its ability to bind Gβγ subunits (Fig. 1A) (Pitcher et al., 1999). In doing so, the ability of GRK2 to be targeted to the plasma membrane and to phosphorylate receptors is directly regulated by the Erk1/2 mitogen-activated protein kinase (MAPK) cascade (Pitcher et al., 1999). Indeed, constitutively active or dominant-negative mutants of the Erk1/2 activating kinase MEK1, alter the ability of GRK2 to phosphorylate ligand-occupied receptors in the expected manner: inhibition of Erk1/2 activation with dominant negative MEK1 enhances receptor phosphorylation, whereas activation of Erk1/2 with constitutively active MEK1 reduces it (Pitcher et al., 1999).
Furthermore, it has been demonstrated that GRK2 and Erk1 rapidly associate in cells after activation of the β2adrenergic receptor. In vitro assays have shown that this is enhanced by the presence of both agonist-occupied receptor molecules and Gβγ subunits (Elorza et al., 2000). Taken together, these observations suggest that phosphorylation of GPCRs by GRK2 is a tightly regulated mechanism in which dephosphorylated GRK2 is recruited to its site of action at the plasma membrane after GPCR activation. Once in complex with the receptor and Gβγ subunits, GRK2 binds Erk1 or Erk2 (Elorza et al., 2000) and becomes phosphorylated on Ser670 (Pitcher et al., 1999), deactivating the GRK2 and promoting its release from the plasma membrane back into to the cytosol. Thus, phosphorylation by Erk1/2 maintains the majority of the cellular pool of GRK2 in an inhibited or “off” state, which becomes active only during agonist activation of the GPCR but is then rapidly returned to the inactive state after binding to receptors and Gβγ subunits. However, for such a mechanism to work, it would also require a phosphatase to become activated shortly after GPCR activation to remove the inhibitory phosphate moiety from GRK2 and allow it to be recruited to the receptors and to phosphorylate them. The identity of this phosphatase has yet to be elucidated.
GPCR-mediated production of the second messengers cAMP, diacylglycerol, and inositol-1,4,5-trisphosphate (IP3), leads to the activation of the second messenger-activated kinases: PKA by cAMP, PKC by diacylglycerol, and calcium/calmodulin-activated kinases by IP3-induced calcium mobilization. It has long been appreciated that two of these kinases, PKA and PKC, directly induce receptor desensitization by phosphorylating GPCRs (Benovic et al., 1985; Roth et al., 1991; Pitcher et al., 1992a). More recently, it has been demonstrated that PKA and PKC can also affect the desensitization of GPCRs by phosphorylating GRK2 and altering its activity (Fig. 1A) (Winstel et al., 1996; Cong et al., 2001). The best studied of these systems involves the specific enhancement by PKA of GRK2 activity toward the Gs/adenylyl cyclase-coupled β2 adrenergic receptor. The β2 adrenergic receptor indirectly binds to PKA by virtue of its interaction with the PKA scaffolding protein A-kinase anchoring protein 79 (AKAP79) (Fraser et al., 2000). When the β2 adrenergic receptor is stimulated, cellular levels of cyclic AMP increase and PKA becomes activated, resulting in enhanced receptor phosphorylation. This is sensitive to both chemical and transfectable inhibitors of PKA activity, and is also dependent on PKA being tethered to the receptor by AKAP79, because disruption of this interaction also inhibits receptor phosphorylation (Fraser et al., 2000). However, the enhancement is not the direct result of increased phosphorylation of the receptor by PKA, because the same effect is observed with a β2 adrenergic receptor mutant that is not a PKA substrate. Instead, PKA phosphorylates GRK2 at serine 685, which increases its binding affinity for Gβγ dimers and thus promotes the recruitment of GRK2 to the plasma membrane and into a complex with its activated receptor substrates (Cong et al., 2001). This mechanism seems to be specific for GPCRs that bind AKAP79, although alternative mechanisms for tethering PKA to other GPCRs may exist. Furthermore, it cannot be assumed that tethering is necessary for PKA to phosphorylate GRK2 under all circumstances.
A similar mechanism may also exist after the activation of PKC by Gq-coupled receptors. Stimulation of the Gq-coupled α1B adrenergic receptor causes GRK2 to become phosphorylated at a site within its carboxyl terminus, which may be the same site phosphorylated by PKA. Similarly, GRK2 phosphorylation can also be induced when cells are treated with either calcium ionophores or with phorbol esters, both of which potently activate PKC. As is the case with phosphorylation by PKA, PKC phosphorylation increases the activity of GRK2 toward receptors but not soluble substrates. Moreover, treating cells with phorbol esters causes GRK2 to redistribute from the cytosol to the plasma membrane (Chuang et al., 1995; Winstel et al., 1996), indicating that phosphorylation of GRK2 by PKC enhances recruitment to the membrane rather than increasing the specific activity of the kinase.
Like GRK2, GRK5 also undergoes phosphorylation by PKC at an undetermined site in the carboxyl terminal region. However, rather than translocating it to the plasma membrane, phosphorylation of GRK5 inhibits its activity toward both receptors and soluble substrates (Pronin and Benovic, 1997). Thus the PKC-mediated phosphorylation of GRK2 and GRK5 result in opposite effects on their activity. In fact, GRK5 phosphorylation by PKC functions in a manner similar to that of GRK2 phosphorylation by Erk1/2: to inactivate the GRK in response to specific cell-signaling events after receptor activation. It will be interesting to see whether other mechanisms known to regulate GRK2 activity are also applicable to GRK5 and the other GRKs and whether similarities or differences in their regulation help illuminate specific functions for each of these kinases.
After stimulation of the β2 adrenergic receptor, GRK2 has also been shown to undergo phosphorylation at several tyrosine residues by the nonreceptor tyrosine kinase c-Src (Sarnago et al., 1999). This phosphorylation is dependent on the ability of β-arrestin to bind to and recruit c-Src to the receptor, because mutants of β-arrestin that fail to bind to c-Src or to the receptor inhibit the phosphorylation of GRK2 (Penela et al., 2001). Furthermore, a catalytically inactive mutant of GRK2 fails to be phosphorylated by c-Src (Penela et al., 2001), presumably because it blocks phosphorylation of the receptor and so inhibits β-arrestin/c-Src recruitment.
Tyrosine phosphorylation affects GRK2 activity in two ways. First, it rapidly and transiently increases GRK2 activity, enhancing phosphorylation of GPCR substrates and promoting more rapid receptor desensitization (Fig. 1A) (Sarnago et al., 1999). Second, the tyrosine phosphorylation of GRK2 promotes its degradation by the ubiquitin/proteosome pathway (Penela et al., 1998, 2001). It should be noted that phosphorylation is a vital initial step in the ubiquitination and degradation of a number of proteins. Although it is not known which of the many E3 ubiquitin ligases that are expressed in cells is responsible for GRK2 ubiquitination, it should also be noted that the β-arrestins are known to bind to one such ligase, Mdm2 (Shenoy et al., 2001). Thus, it is tempting to speculate that β-arrestin may recruit both c-Src and an ubiquitin ligase into complex with GRK2 and the receptor and so supply both the proteins necessary to target GRK2 for degradation. Phosphorylation of GRK2 by c-Src, then, seems to act as the initial event in a long-term feedback loop that regulates GRK2 activity within the cell by reducing GRK2 protein levels. This supplies the cell with a mechanism that allows it to alter its responsiveness to a wide variety of receptor ligands and is of particular interest because cellular levels of GRK2 protein are positively and negatively regulated in a number of pathologies (Ungerer et al., 1994; Lombardi et al., 2001). Whether this represents a normal mechanism used by cells to respond to a disease state or it is somehow pathogenic, possibly through dysregulation of receptors, remains to be clarified.
Regulation of GRKs by Calcium Sensing Proteins
Calcium signaling is a common feature in all cell types and impacts many aspects of cell physiology, including GPCR signaling. Increases in the cytosolic concentration of calcium ions, either because of its release from intracellular stores or influx from outside the cell through calcium channels in the plasma membrane, results in the activation of a group of calcium-binding proteins known collectively as calcium sensor proteins (CSPs). A number of CSPs are known to interact with and alter the activities of GRKs (Sallese et al., 2000a). The best studied of these is recoverin, a CSP found predominantly in photoreceptor cells, where it functions by binding to GRK1 and inhibiting its ability to phosphorylate rhodopsin (Chen et al., 1995; Klenchin et al., 1995). Because basal calcium levels in the cytosol of photoreceptors are high, it is believed that recoverin acts to “silence” GRK1 activity in the dark when the cells are not signaling and to relieve the inhibition after light stimulation when calcium levels are lowered, allowing GRK1 to phosphorylate rhodopsin and initiate desensitization. Several other recoverin-like CSPs also bind to and inhibit GRK1 (De Castro et al., 1995; Sallese et al., 2000a). The specific functions of these interactions have yet to be determined.
The ubiquitously expressed calcium sensor calmodulin (CaM) inhibits the activity of all the GRKs except GRK1 (Fig. 1A) (Chuang et al., 1996). Calcium-bound calmodulin (Ca/CaM) binds to an amino-terminal region common to GRK2, -3, -4, -5, and -6 (Pronin et al., 1997). In addition, Ca/CaM also binds to GRK2 and 5 through sites in their carboxyl termini (Levay et al., 1998; Pronin et al., 1998). The relative sensitivity of the GRKs to CaM inhibition varies greatly: GRK2 is the least sensitive (IC50, ∼2 μM), and GRK5 is the most sensitive (IC50, 40–50 nM) (Chuang et al., 1996; Pronin et al., 1997). The mechanism of inhibition also varies between GRKs, because Ca/CaM directly inhibits GRK2 catalytic activity but acts on GRK5 by inducing inhibitory autophosphorylation as well as blocking membrane association and possibly also by interfering with receptor recognition (Pronin et al., 1998). The functional significance of these differences is not well studied, but it is likely that they allow cells to regulate GPCRs in a highly specific manner in response to fluctuations in intracellular calcium levels.
GRK Specificity for GPCRs: Lessons from Genetically Altered Mice
It has been suggested that functional redundancy might exist between the seven isoforms of GRK, with there being little or no specificity, or that there may be some subtype specificity in phosphorylating different GPCR substrates. In fact, numerous studies using heterologous expression of the different GRKs to identify which GPCRs can act as their substrates have revealed examples of both redundancy and specificity of GRK function. For example, GRKs exhibit redundancy with regard to the β2 adrenergic receptor because it can be phosphorylated and desensitized by GRKs 2 through 6 (Benovic et al., 1989, 1991; Benovic and Gomez, 1993; Premont et al., 1994, 1996). In contrast, the secretin and the parathyroid hormone receptors can be phosphorylated and desensitized only by GRKs 2, 3, and 5 (Shetzline et al., 1998; Flannery and Spurney, 2001). Furthermore, although overexpression of either GRK2, -3, -4, -5, or -6 results in phosphorylation of the vasoactive intestinal polypeptide type-1 receptor, only phosphorylation by GRKs 2, 3, or 5 actually leads to the receptors becoming desensitized (Shetzline et al., 2002).
With the recent generation of GRK transgenic and knockout mouse models, the true extent of specificity of function for the GRKs has now begun to be elucidated. Although these studies are still in their infancy, with only a few receptors having been examined, the results so far show that defined roles for each of the GRKs do exist (Tables1 and2). The following is a review of the results of the GRK transgenic and knockout models investigated so far.
GRK and arrestin knockout mice phenotypes and target GPCRs
In vivo specificity of GPCR regulation in GRK-overexpressing transgenic mice
Genetically modified animals bearing targeted deletions of GRK1, GRK2, GRK3, GRK5, and GRK6 have been constructed to study the roles of individual GRKs in various pathways of receptor signaling. In addition, mice with cardiac-specific overexpression of GRK2, GRK3, and GRK5 have been generated, and a mouse overexpressing GRK4 in all tissues has also been developed.
GRK1 (rhodopsin kinase) knockout animals display an almost complete lack of light-induced phosphorylation of rhodopsin, resulting in a larger and more prolonged response of the retinal rod cells to light (Table 1) (Chen et al., 1999). Similarly, the response of the cone cells is also impaired, showing a recovery rate 30 to 50 times slower than in wild-type mice (Lyubarsky et al., 2000). Furthermore, the absence of GRK1 resulted in morphological changes in the retina from light-dependent apoptosis of the rod cells and the degeneration of vision.
A GRK2 knockout animal has not been studied because they displayed embryonic lethality by gestational day 15.5. The embryos showed pronounced hypoplasia of the ventricular myocardium, suggesting that GRK2 plays an important role during embryogenesis and is essential in cardiac development (Jaber et al., 1996). Because ablation of GRK2 is embryonically lethal, in vivo studies have been performed using the heterozygous GRK2 knockout mice [GRK2(−/+)] instead. In studies of β-adrenergic signaling in the heart, GRK2(−/+) mice showed enhanced cardiac contractile function compared with the wild-type mice demonstrating that cardiac function can be modulated by GRK2 activity (Table 1) (Rockman et al., 1998). To further decrease GRK2 activity in vivo, the GRK2(−/+) mice were crossed with a transgenic mouse with targeted myocardial overexpression of a GRK2 inhibitor fragment, βARKct. The result of this cross is a further lowering of GRK2 activity in cardiac tissues and a subsequent enhancement of cardiac contractility in comparison with the GRK2(−/+) mouse (Rockman et al., 1998). The biochemical analysis of the hearts of the hybrid mice showed decreased receptor phosphorylation and enhanced β-adrenergic signaling. Conversely, the targeted overexpression of GRK2 in the myocardium (Koch et al., 1995) or in vascular smooth muscle (Eckhart et al., 2002) resulted in an attenuation of agonist-stimulated cardiac contractility or vasodilation, respectively (Table 2). This series of experimental data clearly demonstrate in vivo that GRK2 serves to modulate β-adrenergic signaling in the heart and vasculature.
Overexpression of either GRK2 or -3 in cultured cells results in an agonist-stimulated increase in both α1B-adrenergic (α1B-AR) (Diviani et al., 1996) and β-AR (Benovic et al., 1991) phosphorylation and desensitization. To investigate whether both GRK2 and -3 affect in vivo function of the α1B-AR, transgenic mice were generated with concomitant cardiac overexpression of α1B-AR and either GRK2 or -3. Interestingly, overexpression of GRK2 had no effect on the α1B-AR–mediated cardiac phenotype; however, overexpression of GRK3 completely attenuated cardiac α1B-AR signaling (Table 2) (Eckhart et al., 2000). Thus, although α1B-AR shows no in vitro specificity for either GRK2 or -3, GRK3 is the relevant GRK for desensitizing the α1B-AR in the heart. As described previously, myocardial overexpression of GRK2 leads to attenuation of β-AR signaling. In contrast, overexpression of GRK3 resulted in completely normal β-AR signaling, having no effect on the β-AR–mediated cardiac responses (Iaccarino et al., 1998). Analysis of the in vivo function of other GPCRs in the GRK3 cardiac overexpression model revealed that angiotensin II type 1 receptor function is unaltered, whereas thrombin signaling is attenuated (Table2) (Iaccarino et al., 1998), again indicating that GRKs show substrate specificity even when expressed within the same tissue type.
In olfactory epithelium, GRK2 is virtually absent but GRK3 is expressed to a high degree. Consistent with in vitro data suggesting the involvement of GRK3 in olfactory signal transduction, deletion of the GRK3 gene results in the complete lack of fast odorant-induced desensitization of second messenger production in cilia preparations (Table 1) (Peppel et al., 1997). Interestingly, despite the extensive similarity to GRK2, GRK3 gene deletion does not alter embryonic and postnatal development. Further physiological studies on the GRK3 knockout mouse [GRK3(−/−)] have determined a role for GRK3 in airway responses. Agonist stimulation of cholinergic muscarinic receptors in GRK3(−/−) animals resulted in an enhanced airway response compared with wild-type mice, as well as a baroreflex potentiation of heart rate (Table 1) (Walker et al., 1999). Thus, in vivo, GRK3 displays a physiological specificity for the desensitization of odorant and muscarinic receptors.
Because the GRK4 gene locus has been linked to hypertension, a transgenic mouse overexpressing GRK4 in all tissues was constructed to explore the in vivo role of GRK4 in hypertension and in desensitization of the D1 dopaminergic receptor (D1R) (Felder et al., 2002). In essential hypertension there is a defective coupling of the renal D1R with its effector that results in impaired urinary sodium excretion. The naturally occurring polymorphism A142V in GRK4 has increased kinase activity toward the D1R relative to wild-type GRK4. Transgenic mice carrying this polymorphism displayed a hypertensive phenotype, whereas wild-type GRK4 transgenic mice did not (Table 2). Furthermore, whereas D1R agonists normally increase urinary sodium excretion, this effect was impaired in GRK4(A142V) transgenic mice (Felder et al., 2002). Thus, the increased phosphorylation of the D1R by GRK4 results in impaired signal transduction that may ultimately lead to the pathogenesis of hypertension.
In addition to GRK2, GRK5 is also highly expressed in heart supporting a role for this kinase in the regulation of cardiac functions. Transgenic mice overexpressing GRK5 in a cardiac specific manner show a marked enhancement of β-AR desensitization (Table 2) (Rockman et al., 1996). Furthermore, contractility in response to β-agonists is attenuated, yet, the response to angiotensin II remains unchanged (Rockman et al., 1996). To further investigate the role of GRK5 in vivo, GRK5 gene deletion mice were generated. The GRK5 knockout mice [GRK5(−/−)] displayed no differences in dopamine agonist-induced locomotor responses compared with wild-type animals, as well as in serotonin 5-HT1A induced hyperthermic responses (Gainetdinov et al., 1999). However, classic cholinergic muscarinic receptor responses such as hypothermia and hypoactivity were enhanced in GRK5(−/−) mice (Table 1) (Gainetdinov et al., 1999). Biochemical assays also demonstrated a lack of muscarinic receptor desensitization in the knockout animals. Thus, in vivo, GRK5 targets the β-adrenergic receptors and muscarinic receptors but not the angiotensin II, dopamine, and serotonin receptors.
GRK-mediated phosphorylation of the chemokine receptor CXCR4 has been shown to be important in the regulation of the CXCL12 (stromal cell-derived factor 1)-stimulated CXCR4 signaling (Orsini et al., 1999). To understand the role and specificity of GRKs in the chemotactic response generated by the activation of CXCR4, lymphocytes derived from GRK5 and GRK6 knockout animals were stimulated with CXCL12 and their ability to undergo chemotaxis was measured (Fong et al., 2002). Whereas there was no difference in the chemotactic activity between GRK5 knockout lymphocytes and those derived from wild-type mice, lymphocytes from GRK6-deficient animals were strikingly impaired in their ability to respond to CXCL12 (Table 1). Thus, GRK6 is specifically required for the in vivo phosphorylation of the CXCR4, which allows lymphocytes to respond correctly to the chemotactic agent (Fong et al., 2002).
The evidence collected to date from cellular studies and genetically altered mice suggests that there is GRK specificity for particular GPCRs. That is, all GPCRs are not regulated equivalently by all the GRKs expressed in a cell, and many GPCRs seem to be functionally paired with a particular GRK. Further research in cellular and mouse models will be required to delineate fully the GRK specificity for GPCRs.
Regulation of Arrestins during Desensitization
Several reports have described how the activities of the arrestins are regulated through post-translational modification and interactions with accessory proteins. However, the majority of these interactions are concerned with the regulation of arrestins while functioning as adaptors during receptor internalization (for reviews, see Krupnick and Benovic, 1998; Ferguson, 2001; Claing et al., 2002; Perry and Lefkowitz, 2002) and have little or no effect on their ability to desensitize receptors. For instance, phosphorylation of β-arrestin 1 (arrestin 2) on serine 412 by the Erk1/2 kinases does not affect its ability to bind to and desensitize the β2adrenergic receptor (Lin et al., 1999). However, phosphorylation at this site does considerably reduce the internalization of the receptor, because it inhibits the binding of clathrin to β-arrestin 1, which prevents the accumulation of receptors in clathrin-coated pits (Lin et al., 1997). However, one example has been described in which phosphorylation of an arrestin does alter its ability to desensitize a receptor. In the Drosophila melanogaster visual system, arrestin 2 (the major arrestin found in D. melanogasterphotoreceptor cells and most homologous to mammalian β-arrestin 2) binds to and desensitizes light-activated rhodopsin molecules. Only unphosphorylated D. melanogaster arrestin 2 is capable of binding rhodopsin, and its subsequent phosphorylation by calcium/calmodulin-dependent kinase II releases arrestin 2 from rhodopsin (Alloway et al., 2000; Kiselev et al., 2000). BecauseD. melanogaster rhodopsin is a Gq-coupled receptor, activation by light stimulates the production of the second messengers IP3 and diacylglycerol and the release of calcium ions from intracellular stores. This results in a rapid activation of Ca/CaM-dependent kinase II and phosphorylation of arrestin 2. Thus, very soon after rhodopsin becomes activated and desensitized by binding arrestin 2, Ca/CaM-dependent kinase II phosphorylates the arrestin 2 and it dissociates from rhodopsin, allowing the photoreceptors to resensitize. This tightly regulated mechanism of rapid activation, desensitization, and resensitization ensures that the photoreceptors can be repeatedly stimulated with light and not become permanently desensitized, a necessary adaptation for the correct function of any highly responsive sensory system.
To allow a resensitized receptor to instigate further signaling, second messenger molecules that were synthesized (or mobilized, in the case of calcium ions) during previous rounds of signaling must be removed from the cell. In the D. melanogaster visual system, this requires the return of calcium ions to the endoplasmic reticulum or to the extracellular milieu and the degradation of IP3 and diacylglycerol. In the case of Gs-coupled receptors, such as the β2AR, cAMP must be degraded to AMP by phosphodiesterase enzymes (Houslay, 2001). Until recently, the mechanisms that are responsible for degrading second messengers were thought to function independently of receptor desensitization. However, it has now been shown that, at least for the Gs-coupled β2AR, the β-arrestins form a link between the two processes because they interact with the PDE4 family of phosphodiesterases (Fig. 1C) (Perry et al., 2002). After β2 adrenergic receptor activation, the β-arrestins are recruited to and desensitize the receptor molecules, translocating PDE4s with them to the plasma membrane. Because the plasma membrane is the site of cyclic AMP production by adenylyl cyclase enzymes, this results in both a reduction in the rate of synthesis of cyclic AMP (because the desensitized receptors can no longer couple as well to Gs) and an increase in the rate of its degradation by the higher levels of PDE4 activity present on the membrane (Perry et al., 2002). In this manner, the β-arrestins both desensitize the receptors and facilitate the quenching of signaling by aiding in the degradation of second messenger molecules. It will be very interesting to see whether this paradigm extends beyond PDEs and Gs-coupled receptors to other second messenger degrading enzymes, such as those that eliminate diacylglycerol and IP3 after Gq-coupled receptor stimulation.
Of Mice and MEFs: Revealing Specificity for GPCRs and the Arrestins
Until genetic knockout mouse strains lacking expression of specific arrestins became available the first, and best, evidence that the different arrestin isoforms might be used preferentially by different receptors came from a study that compared in vitro the ability of purified visual arrestin, β-arrestin 1, and β-arrestin 2 (arrestins 1, 2, and 3) to desensitize rhodopsin and the β2 adrenergic receptor (Attramadal et al., 1992). This study confirmed a high degree of specificity for visual arrestin by rhodopsin that had already been suspected based on the highly restricted and overlapping expression patterns of the two molecules (Wilden et al., 1986). However, it also suggested that the two β-arrestins might be functionally redundant because they showed identical desensitizing activity toward the β2adrenergic receptor (Attramadal et al., 1992). Later efforts that used heterologous expression and antisense RNA “knock-down” methods also failed to identify any receptor preferences between β-arrestin 1 and 2 (Mundell et al., 1999).
Unlike arrestin, β-arrestin 1 and 2 are ubiquitous; hence, their GPCR specificities cannot be inferred from their expression patterns (Attramadal et al., 1992). To define the physiological roles of β-arrestin 1 and 2 in the regulation of GPCRs, knockout mouse models were generated. Both knockout animals have neither gross abnormalities nor an overt phenotype (Conner et al., 1997; Bohn et al., 1999). When challenged with various stimuli, however, physiological differences with their wild-type littermates become apparent. When increasing concentrations of the β-agonist isoproterenol were infused into the heart of the β-arrestin 1 knockout mouse, the resulting cardiac ejection fraction was significantly greater than in the wild-type mouse (Table 1) (Conner et al., 1997). This result suggested that there is enhanced β-adrenergic signaling in mice lacking β-arrestin 1 and consequently that β-arrestin 1 is important for in vivo β-AR desensitization.
To date, the β-arrestin 2 knockout animals have been used to study the in vivo desensitization of the opioid and chemokine CXCR4 receptors (Table 1). In the β-arrestin 2 knockout mice, the analgesic effects of morphine were potentiated and prolonged compared with the wild type. Using specific antagonists for the different isoforms of the opioid receptor, the effect was localized to the impaired desensitization of the μ-opioid receptor (Bohn et al., 1999). Interestingly, the β-arrestin 2 knockout mice did not develop tolerance to morphine but did develop dependence (Bohn et al., 2000). This suggests that the phenomena of desensitization and tolerance are closely linked, whereas dependence occurs by a different mechanism. The chemotactic responses to CXCL12 mediated through the CXCR4 receptors were markedly impaired in lymphocytes derived from β-arrestin 2 deficient mice, similar to the phenotype observed in GRK6 knockout lymphocytes (Fong et al., 2002). Thus, for desensitization of the CXCR4 receptor to occur in vivo, it would seem to need to be first phosphorylated by GRK6 and then to bind β-arrestin 2. It will be of great interest in future studies to compare the physiological responses of the β-arrestin 1 and 2 knockout mice to the same stimulus and to thus determine the selectivity of either β-arrestin for a specific GPCR.
There is a limitation in studying the overall effects of β-arrestin in the knockout mice generated, because only one of the two ubiquitous β-arrestins was eliminated. The ideal experimental model in which to analyze the roles of β-arrestin and the selectivity of β-arrestin 1 and 2 for particular GPCRs would be one that lacks both β-arrestins. Unfortunately, β-arrestin 1 and 2 double knockout mice could not be obtained because they displayed embryonic lethality. This hurdle was overcome by generating mouse embryonic fibroblasts from embryos that lack β-arrestin 1 or 2, or both (Kohout et al., 2001). These cells were then used to carefully dissect the roles of β-arrestin 1 and 2 in the desensitization of various GPCRs. Experiments showed that desensitization of the β2-adrenergic and the AT1A receptors is impaired in cells lacking one or the other of the β-arrestins and severely impaired in cells lacking both β-arrestins. However, there is no appreciable difference between β-arrestins 1 and 2 in their ability to desensitize these receptors (Kohout et al., 2001). This is in sharp contrast to the individual abilities of β-arrestins 1 and 2 to mediate sequestration of the β2 adrenergic receptor, because β-arrestin 2 is dramatically more efficient than β-arrestin 1 (Kohout et al., 2001). However, similar experiments analyzing the sequestration of the AT1A receptor showed that both β-arrestins are equally capable of mediating its internalization. Interestingly, the protease-activated receptor 1 (PAR1) does not desensitize in the absence of β-arrestin 1 but has a normal desensitization profile in the absence of β-arrestin 2 (Paing et al., 2002). Hence, β-arrestin 1 seems to be the major regulator of PAR1 desensitization. This is the first example of a β-arrestin differentially regulating GPCR desensitization. These data suggest that the ability of a particular β-arrestin to either desensitize or sequester a GPCR is receptor-specific.
Summary and Perspectives
Our understanding of the mechanisms involved in the regulation of GPCR desensitization has developed considerably over the past decade. The complexity of regulation of GPCR desensitization is now known to far exceed the simple model of GPCR phosphorylation by GRKs followed by arrestin binding and uncoupling of G protein signaling. GRK activities are not simply triggered by agonist-occupied receptors; rather, they are extensively regulated by a plethora of interactions with and modifications by other proteins. Similarly, β-arrestins not only serve to physically interdict signaling to the G protein but also further enhance GPCR desensitization by translocating cytosolic proteins such as PDEs and c-Src to the receptor. Once at the membrane, PDEs and c-Src can turn off signaling at its source by degradating cAMP or by phosphorylating GRK2 to enhance its activity toward the receptor, respectively.
One of the most intriguing questions in GPCR desensitization is why the family of GRKs comprises seven members and the family of arrestins has four members. Are the enzymatic activities of the various GRKs redundant or is there substrate specificity for each one? Do the arrestins translocate to and subserve desensitization equally well for all GPCRs or do they have specialized functions? Recent observations in transgenic mouse models, as well as knockout mice and cells derived from knockout mice, have clearly indicated that such specificity does exist for both GRKs and arrestins. For example, in vivo signaling of the α1B-AR in the heart was attenuated by the overexpression of GRK3 but not GRK2. In contrast, in the same cardiac-specific transgenic mouse models, GRK2 overexpression attenuates AT1R function, whereas GRK3 overexpression is ineffective. An even more illustrative example of differential regulation by GRK2 and 3 is their different roles in embryonic development. Whereas GRK2 knockout mice display embryonic lethality by gestational day 15, GRK3 knockout mice live normally to maturity. It is evident that despite their structural and regulatory similarities, GRK2 and GRK3 cannot substitute for one another. Indeed, it is likely that GRK4, -5, -6, and -7, which are even more divergent in structure and in vitro substrate specificity from GRK2 and -3, have very different in vivo modes of action and receptor specificity. The effects of β-arrestins 1 and 2 on the function of a specific GPCR have not been compared in an in vivo setting. However, it is quite possible that such differences will be found, as has been the case with the GRKs, because studies in mouse embryonic fibroblasts derived from the β-arrestin knockout animals have shown, for example, that the PAR1 receptor can only be desensitized by β-arrestin 1 and that β-arrestin 2 cannot substitute for this function.
The regulation of GRKs and arrestins in GPCR desensitization process remains an exciting and vibrant field of investigation. Because more research is carried out in genetically altered mice, our appreciation of the distinctions between the members of the GRK and arrestin families will certainly increase.
Acknowledgments
We thank Dr. Stephen J. Perry for valuable discussions and D. Addison and J. Turnbough for excellent secretarial assistance.
Footnotes
- Received September 9, 2002.
- Accepted October 16, 2002.
-
This work was supported in part by National Institutes of Health Grant HL16037. R.J.L. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- PKA
- protein kinases A
- PKC
- protein kinase C
- PIP2
- phosphatidylinositol-4,5-bisphosphate
- RGS
- regulator of G protein signaling
- Erk
- extracellular signal-regulated kinase
- IP3
- inositol-1,4,5-trisphosphate
- CSP
- calcium sensor protein
- CaM
- calmodulin
- AR
- adrenergic receptor
- PDE
- phosphodiesterase
- PAR1
- protease-activated receptor 1
- The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
Jump to section
- Article
- Receptor Phosphorylation Requires Recruitment of GRKs to the Plasma Membrane
- Regulation of GRKs through Phosphorylation by Other Kinases
- Regulation of GRKs by Calcium Sensing Proteins
- GRK Specificity for GPCRs: Lessons from Genetically Altered Mice
- Regulation of Arrestins during Desensitization
- Of Mice and MEFs: Revealing Specificity for GPCRs and the Arrestins
- Summary and Perspectives
- Acknowledgments
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters