


Abstract
We have examined the ligand regulation and G protein selectivity of the human cannabinoid CB1 and CB2 receptors by an in situ reconstitution technique directly measuring G protein activation. Membranes from Spodoptera frugiperda cells expressing CB1 and CB2 receptors were chaotrope extracted to denature endogenous GTP-binding proteins. The ability of the receptors to catalyze the GDP-GTP exchange of each G protein was then examined with purified bovine brain Gi and Go. Activation of CB1 receptors produced a high-affinity saturable interaction for both Gi and Go. Agonist stimulation of CB2 receptors also resulted in a high-affinity saturable interaction with Gi. In contrast, CB2 receptors did not interact efficiently with Go. G protein activation was then examined with a diverse group of ligands. For the interaction of CB2receptors with Gi, HU210 was the only compound tested that demonstrated maximal activation. In contrast, WIN55,212 (64%), anandamide (42%), and Δ9-tetrahydrocannabinol (Δ9-THC) (44%) all initiated submaximal levels of G protein activation. For CB1 receptor-catalyzed activation of Gi, HU210, WIN55,212, and anandamide all elicited maximal activation, whereas Δ9-THC (56 ± 6%) caused only partial Gi activation. In contrast, only HU210 effected maximal CB1 stimulation of Go, with anandamide, WIN55,212, and Δ9-THC all stimulating between 60 and 75% compared with HU210. These data demonstrate that different agonists induce different conformations of the CB1receptor, which in turn can distinguish between different G proteins. Our data thus demonstrate agonist-selective G protein signaling by the CB1 receptor and suggest that therapeutic agents may be designed to regulate individual G protein-signaling pathways selectively.
Cannabinoid receptors belong to the G protein-coupled receptor superfamily. To date, two subclasses of cannabinoid receptors have been isolated, CB1found primarily in the central nervous system and testis (Matsuda et al., 1990), and CB2 located predominantly in the immune system (Munro et al., 1993). The CB1 and CB2 receptors exhibit a low overall amino acid sequence identity (44%, with 68% in the transmembrane domains) but they share a common pharmacology and few of the available ligands distinguish between them. Like several other G protein-coupled receptors, the cannabinoid receptors activate multiple intracellular signal transduction pathways. CB1 and CB2 receptor agonists inhibit forskolin-stimulated adenylyl cyclase by activation of a pertussis toxin-sensitive G protein (Felder et al., 1995). However in heterologous cells, CB1 but not CB2 receptors inhibit N-, P-, and Q-type calcium channels and activate inwardly rectifying potassium channels (Caulfield and Brown, 1992; Mackie and Hille, 1992; Felder et al., 1995; Mackie et al., 1995; Pan et al., 1996). Inhibition of calcium channels and enhancement of inwardly rectifying potassium currents is pertussis toxin-sensitive but independent of cAMP inhibition, suggestive of a direct G protein mechanism (Mackie and Hille, 1992; Mackie et al., 1995). An additional layer of complexity for the signaling of CB1 receptors derives from their ability to stimulate cAMP formation under certain conditions, consistent with a possible Gs linkage of this receptor (Glass and Felder, 1997). Given the complexity of the cannabinoid receptor-mediated signaling, it is likely that the diverse range of behavioral effects of cannabinoids arise from the activation of several distinct intracellular processes. Data from cannabinoid ligand binding (Houston and Howlett, 1998; Kearn et al., 1999) and regulation of GTPγS binding (Burkey et al., 1997a; Breivogel et al., 1998; Griffin et al., 1998; Kearn et al., 1999) in membrane fractions has led to the suggestion of agonist-selective G protein coupling. However, these experiments cannot directly examine this proposal because these methods fail to distinguish among different G proteins. Furthermore, these approaches have not been used with CB2 receptors, probably due to lower receptor levels. Thus, although the hypothesis of agonist-selective cannabinoid stimulation of multiple G protein-coupled pathways is attractive, it remains untested.
We have recently developed an approach to the investigation of receptor-G protein coupling that enables precise characterization of the coupling properties of the receptors to individual G protein subtypes (Hartman and Northup, 1996). Our technique uses recombinant expressed receptors in situ in membrane fractions from which extrinsic membrane proteins have been removed or inactivated by urea extraction. Although depleted of G protein, the uncoupled receptors remain fully functional for reconstitution with purified G protein subunits. The depletion of endogenous G proteins from the membrane enables the controlled addition of isolated G proteins for analysis. In addition to depleting endogenous G proteins, the urea washing removes or destroys nonintegral membrane proteins, including small GTP-binding proteins, greatly reducing the GTP-binding capacity of the membranes and enhancing the signal from reconstituted G proteins. This technique has previously been successfully used to characterize receptor-G protein interactions of the 5-hydroxytryptamine (5-HT)2c(serotonin) receptor (Hartman and Northup, 1996), and the bombesin family of receptors (Jian et al., 1999) with Gq. However, the high intrinsic rate of GDP-GTP exchange has inhibited its application for receptors coupled to Gi/o. To overcome this technical obstacle, we have adapted our procedures to suppress the spontaneous binding signal and to require the exchange to be receptor catalyzed.
In this study, we have examined the ability of the human CB1 and CB2 receptors expressed in Spodoptera frugiperda cells (Sf9) to catalyze the activation of the pertussis toxin-sensitive G proteins Gi and Go. In situ reconstitution of CB1 receptors reveals G protein-selective agonist efficacies of cannabinoid ligands.
Experimental Procedures
Materials
(−)-11-hydroxy-Δ8-tetrahydrocannabinol-dimethylheptyl (HU210) was purchased from Tocris Cookson (Ballwin, MO). (R)-(+)-(2,3-Dihydro-5-methyl-3-[(morphonolinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl)(1-napthalenyl)methanone mesylate (WIN55,212–2) was purchased from Research Biochemicals (Natick, MA).N-(Peperidino-1-yl)-5-(4-chloropheyl)-1-(2,4-dichlorophenyl)-4-methyl-pyrazole-3-carboxamide, hydrochloride (SR141716A) was provided by Research Biochemicals as part of the Chemical Synthesis Program of the National Institute of Mental Health, Contract NO1 MH30003. Anandamide was purchased from Avanti Polar Lipids (Alabaster, AL). Tritiated antagonists SR141716A (18.8Ci/mmol) andN-[(1S)-endo-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl]5-(4-chloro-3-methyl-phenyl)-1-(4-methylbenzyl)pyrazole-3-carboxamide (SR155528; 30Ci/mmol) were provided by Research Triangle Institute (Research Triangle Park, NC) as part of the Chemical Synthesis Program of the National Institute of Drug Abuse, Contract NO1DA-6–7054. Δ9-Tetrahydrocannabinol (Δ9THC) was provided to Dr. Miles Herkenham by the National Institute of Drug Abuse. [35S]GTPγS (specific activity, 1100–1200 Ci/mmol) was purchased from NEN (Boston, MA). cDNA for the human CB1 receptor was received from Dr. T. Bonner (National Institute of Mental Health, Bethesda, MD), and for the human CB2 receptor was from Dr. S. Munro (Cambridge, UK). All other compounds were purchased from Sigma Chemical Co. (St. Louis, MO).
Methods
Formation of Recombinant CB1 and CB2Cannabinoid Receptors.
A cDNA fragment encoding the open reading frame of the human CB1 receptor was produced withBpu102 and XbaI and ligated between theSmaI and XbaI sites of the transfer vector pBacPAK9 (Clontech, Palo Alto, CA). A cDNA fragment encoding the open reading frame of the human CB2 receptor was produced with ApaI and DraI, and ligated into theSmaI site of the transfer vector pBacPAK 8 after ligation of an ApaI linker. Sf9 insect cell culture, transfection, plaque purification, and virus amplification of CB1 and CB2 receptors were carried out according to manufacturer's protocol (Clontech).
Membranes Preparation and Urea Extraction.
Sf9 cells in suspension were infected with either CB1 receptor or CB2 receptor virus at a multiplicity of infection of ∼4. The cells were harvested 48 h postinfection by sedimentation at 500 rpm for 5 min in a Hereaus megafuge 2.0. After one wash in PBS, the cells were resuspended into solution A [10 mM 4-morpholinepropanesulfonic acid (MOPS), pH 7.5, 1 mM EGTA,100 μM 4-(2-aminoethyl)benzenesulfonyl fluoride] and left at 4°C for 30 min before cell lysis with a Dounce homogenizer. Nuclei and cell debris were removed by centrifugation at 1600g for 10 min in a Hereaus megafuge 2.0, and the postnuclear fraction (P2) was collected at 40,000g for 30 min in a Beckman JA-20 rotor and J2–21 centrifuge. The P2 membrane pellet was suspended in ice-cold solution A containing 7 M urea. After incubation with urea for 30 min on ice, the membrane solution was diluted to <4 M urea with solution A and then sedimented at 142,000g for 30 min at 4°C in a Beckman 45Ti rotor and L8–70 M ultracentrifuge. The membrane pellet was then washed once with solution A and collected by sedimentation as before. The final pellet was suspended in solution A with 200 mM sucrose, and aliquots were snap frozen and stored at −80°C.
Quantification of Receptor Sites.
Binding site abundance was determined by saturation-binding assay with 0.1 to 30 nM [3H]SR141716A (CB1) or [3H]SR144528 (CB2). Binding reactions were performed for 1 h at 30°C in solution B (50 mM Tris, pH 7.5, 3 mM MgCl2, 1 mM EDTA) and 5 mg/ml fraction V BSA in a final reaction volume of 300 μl containing 10 to 40 μg of membrane protein. The incubation was terminated by addition of 2.5 ml of ice-cold solution B, and samples were filtered through Whatman GF-C filters and washed with 2.5 ml of solution B. Filters were then soaked in 400 μl of 2% SDS for 2 h before the addition of 10 ml of Cytoscint (ICN Pharmaceuticals, Costa Mesa, CA) scintillation fluid. The filters were soaked in Cytoscint for at least 5 h before analysis by liquid scintillation spectometry in a Wallac 1219 beta counter.
Purification of G Protein Subunits.
G proteins Gi, Go, and βγ-subunits were isolated from bovine cortex following a previously published protocol (Sternweis and Robinshaw, 1984; Mumby et al., 1988). Due to the apparent instability of Gαi in the absence of βγ, Gi was maintained as a trimer. Immunological analysis by Western blot indicated that the Gi sample contained Gαi-1-, Gαi-2-, and Gαi-3-subunits. Squid Gαq (Hartman and Northup, 1996) and bovine retinal Gαt (Fawzi and Northup, 1990) were purified following previously published protocols. After purification, all subunits were eluted over a G50 column to ensure the final solution concentration for α-subunits was 10 mM MOPS, pH 7.5, 1 mM MgCl2, and 4 mM 3-[(3-cholamidopropyl)dimethylamminio]propanesulfonate, and for βγ was 10 mM MOPS, pH 7.5, 100 mM NaCl2, and 8 mM 3-[(3-cholamidopropyl)dimethylamminio]propanesulfonate. Gα-subunit concentration assay was assessed by [35S]GTPγS binding in the presence of 1 μM GTPγS for up to 2 h in a solution containing 50 mM Tris-HCl, pH 7.5, 1 mM EDTA, 11 mM MgCl2, and 0.1% lubrol (Northup et al., 1982). Reactions were terminated by the addition of 2.5 ml of solution D (20 mM Tris-HCl, pH 8.0, 25 mM MgCl2, and 100 mM NaCl) and filtered over nitrocellulose membranes on a Millipore vacuum manifold. The filters were washed four times with 2.5 ml of ice-cold solution D, dried, and the bound radioactivity was counted by liquid scintillation in a Wallac 1219 beta counter.
Reconstitution of Cannabinoid Receptors with G Protein Subunits.
The receptor catalyzed GDP-[35S]GTPγS exchange was determined by incubation of 5 to 10 nM CB1 or CB2 receptor (10–30 μg of membrane), with varying concentrations of Gα-subunits in the presence of a previously determined saturating concentration of βγ (100 nM). [35S]GTPγS binding proceeded linearly beyond 10 min and this time was used to estimate rates in all experiments. The assays were carried out at 30°C in a final reaction mixture (50 μl) containing, 10 mM MOPS, pH 7.5, 2 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 0.5% (w/v) BSA, 4 μM GDP, and [35S]GTPγS (0.4–0.8 nM; 2–5 × 105 cpm) (Solution C). G protein-binding activity was measured alone or with urea washed membrane in the presence and absence of cannabinoid ligands. Reactions were terminated by the addition of 2.5 ml of solution, and filtered over nitrocellulose membranes on a Millipore vacuum manifold as described above.K m and V maxvalues for the agonist catalyzed GDP-[35S]GTPγS exchange were calculated with nonlinear regression analysis for a single site Michaelis-Menton interaction with GraphPad Prism software. All experiments were carried out in siliconized test tubes.
Agonist Saturation Analysis.
Agonist saturation analysis were determined for all receptor-G protein high-affinity interactions (CB1 with Gi and Go; CB2 with Gi only). Experiments were performed at approximate K m values (20 nM Gαi or 80 nM Gαo) and saturating βγ (100 nM), with 3 to 10 nM CB1or CB2 receptor. Membranes were preincubated with agonist for 10 min at 30°C before the addition of [35S]GTPγS containing reaction mixture. The reaction was allowed to proceed for an additional 10 min before termination and filtration as described above. Affinity constants for the agonist catalyzed GDP-[35S]GTPγS exchange were calculated with nonlinear regression analysis for a sigmoidal dose response with GraphPad Prism.
Inverse Agonism of SR141716A at CB1 Receptor.
To test for inverse agonism of the CB1 receptor, 10 μg of urea-washed membranes was incubated with 80 nM Gαo or 20 nM Gαi and saturating βγ in a modified solution C (10 mM MOPS, pH 7.5, 100 mM NaCl, 6 mM MgCl2, 1 mM EDTA, 5 mg/ml BSA, 4 μM GDP, and ∼0.4 nM [35S]GTPγS) in the presence and absence of 1 μM SR141716A. The reaction proceeded for 10 min at 30°C before termination and filtration as described above.
Results
Reconstitution of Cannabinoid Receptors with G Protein Subunits.
Sf9 cells infected with either the human CB1 or CB2receptor-encoding baculoviruses were harvested 48 h after infection, and were urea-washed. Binding assays with the antagonists SR141716A (CB1) and SR144528 (CB2) demonstrated high-affinity binding to a single site in both cases. Baculovirus expression of CB1 or CB2 receptors resulted in membranes expressing ∼15 and 33 pmol/mg, respectively. The conditions for the reconstitution experiments were optimized to decrease the rates of the noncatalyzed [35S]GTPγS G protein binding by the addition of 4 μM GDP. Because the concentration of GDP is more than three orders of magnitude greater than that of the tracer, the binding of [35S]GTPγS to Gα is not stoichiometric and each binding event represents multiple receptor-catalyzed activation events. Kinetic analyses of the [35S]GTPγS-binding reactions performed in this way were consistent with a ligand-regulated rate constant proportional to the added CB1 or CB2 receptor (data not shown). Furthermore, no cannabinoid ligand regulation of the rate of binding was observed in the absence of expressed CB1 or CB2 receptor (data not shown).
As demonstrated in Fig. 1, a and c, urea-washed Sf9-CB1 and CB2membranes displayed a very low rate of GDP-[35S]GTPγS exchange in the absence of agonist. The addition of the agonist HU210 dramatically increased the rate of GDP-[35S]GTPγS exchange of Gαi for both receptors. Gαi demonstrated a saturable high-affinity interaction with both CB1 and CB2 receptor-agonist complexes. The contribution of the endogenous membrane [35S]GTPγS binding and the basal G protein activity could be removed from the total binding signal to assess the specific agonist-stimulated [35S]GTPγS exchange rates as shown in Fig. 1, b and d. These curves were fitted with a single-site saturation isotherm. Both the CB1 and CB2 cannabinoid receptors demonstrated similar apparent affinity for Gαi withK m values of 28.3 ± 2.2 nM (CB1) and 39.7 ± 3.6 nM (CB2) (mean ± S.E.; n = 3).
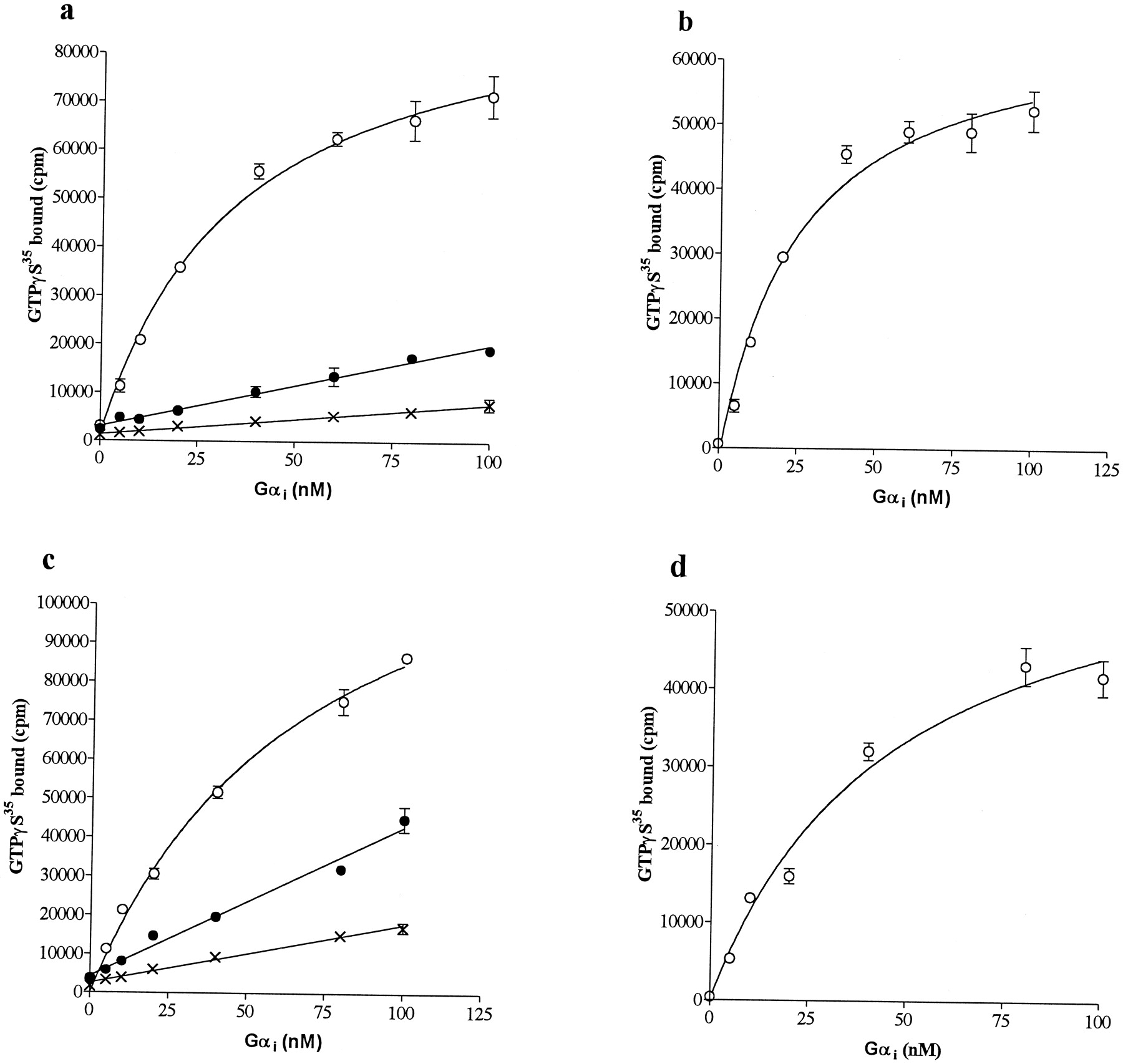
Reconstitution of CB1 or CB2receptors with Gαi. Urea-extracted Sf9-CB1 (a and b) and Sf9-CB2 (c and d) membranes were assessed for agonist stimulated [35S]GTPγS binding to Gαi-subunits at the indicated concentrations in the presence of 100 nM βγ. ×, Sf9 membrane; ●, Sf9 membrane with Gαi ; ○, Sf9 membrane with Gαi and 1 μM HU210. Specific HU210-stimulated [35S]GTPγS binding (b and d) was calculated by subtracting binding in the presence of agonist from binding in the absence of agonist at each Gαi-concentration. The data presented are the averages and errors of duplicate values from a representative experiment. The experiment was performed three times.
The activation of Gαo by CB1 and CB2 receptors is shown in Fig. 2, a and c. The CB1 and CB2 receptors demonstrate low catalysis of Gαo activation in the absence of agonist. Addition of HU210 resulted in a significant increase in G protein activation for both CB1 and CB2 receptors. In contrast to the nearly identical interaction of Gαi with these receptors, Gαo clearly distinguishes between them. As seen in Fig. 2, b and d, only the interaction of CB1 was saturable, whereas the CB2 interactions did not saturate within this range of Gαo concentrations. The CB1 receptor agonist-mediated GDP-[35S]GTPγS exchange saturated with aK m of 81 ± 9 (mean ± S.E.;n = 4) for Gαo. In contrast, we estimate the K m of CB2 for Gαo at 290 ± 10 nM (mean ± S.E.; n = 3). Both cannabinoid receptors appear selective for G protein because neither CB1 nor CB2 receptor significantly catalyzed the activation of Gαqor Gαt (data not shown).

Reconstitution of CB1 or CB2receptors with Gαo. Urea-extracted Sf9-CB1 (a and b) and Sf9-CB2 (c and d) membranes were assessed for agonist-stimulated [35S]GTPγS binding to isolated Gαo-subunits at the indicated concentrations in the presence of 100 nM βγ. ×, Sf9 membrane; ●, Sf9 membrane with Gαo; ○, Sf9 membrane with Gαo and 1 μM HU210. Specific HU210-stimulated [35S]GTPγS binding (b and d) was calculated by subtracting binding in the presence of agonist from binding in the absence of agonist at each Gαo-concentration. The data presented are the averages and errors of duplicate values from a representative experiment. The experiment was performed three times.
Agonist Saturation of CB1 and CB2Receptor-Stimulated [35S]GTPγS
Binding. Because the above-mentioned experiments confirmed the expectation that cannabinoid receptors can couple to multiple G proteins, we performed the experiments shown in Fig. 3 to address the issue of agonist-selective G protein regulation. These experiments examine the ligand saturation of CB1and CB2 receptor activation of Gαi or Gαo with several distinct cannabinoid ligands. As shown in Fig. 3, the efficacies of cannabinoid ligands are not only an intrinsic property of the cannabinoid receptor structure but also are dependent upon the G protein. Figure 3a presents data for the saturation of Gαi activation by CB1receptors. Although displaying varying apparent affinities (Table1), HU210, WIN55,212, and anandamide were all equally efficacious for CB1 catalyzed GDP-[35S]GTPγS exchange on Gαi. Δ9-THC displayed an efficacy of 57% compared with these three. In contrast to the data for Gαi, anandamide, WIN55,212, and Δ9-THC were all partial agonists and only HU210 stimulated maximal GDP-[35S]GTPγS exchange for Gαo. The observed efficacies for these agonist ligands were as follows: anandamide (71%), WIN55,212 (72%), and Δ9-THC (64%) compared with HU210 (Fig. 3b; Table 1). For all the agonists tested with both G proteins the Hill coefficients for the interaction approximated 1 (data not shown) and the saturation data were well fit by a single-site model.

Agonist saturation binding analysis for cannabinoid agonist stimulated [35S]GTPγS binding to G proteins. Urea-washed Sf9-CB1 (a and b) and Sf9-CB2 (c) membranes were incubated at 20 nM Gαi (a and c) or 80 nM Gαo (b) with 100 nM added βγ and the indicated concentrations of cannabinoid agonists. ×, WIN55,212; , ●, HU210; ○, anandamide ♦, Δ9-THC. The data presented are the averages and errors of duplicate values from a representative experiment. The experiment was performed four (b and c) or five (a) times.
Agonist saturation analysis of Gi or Go at the CB1 or CB2 receptor
The ligand saturation of CB2 receptor activation of Gαi is shown in Fig. 3c. Because of the low apparent affinity and a doubtful physiological relevance of Gαo for CB2, this receptor was examined only with Gαi. Consistent with published K d values (Pertwee, 1997), EC50 values for Δ9-THC, anandamide, and HU210 were similar to those seen for CB1 receptors, whereas WIN55,212 demonstrated a significantly higher potency at CB2 receptors than at CB1 receptors. As for Gαi activation by CB1receptors, Δ9-THC was again a partial agonist for CB2 (42%). Distinct from the CB1 receptor, anandamide efficacy was equivalent to Δ9-THC for CB2 (45%), whereas WIN55,212 induced a rate of Gαiactivation that was intermediate between Δ9-THC and HU210 (Fig. 3c; Table 1). The Hill coefficient of all the agonist response curves approximated 1 (data not shown).
Several lines of evidence argue that these differences in agonist efficacies are accurately measured in our studies. No difference was observed in the ability of anandamide to stimulate receptor-G protein coupling in the presence or absence of 4-(2-aminoethyl)benzenesulfonyl fluoride (data not shown), suggesting that amido-hydrolase enzymes do not survive the urea washing of the membranes. For both CB1 and CB2 receptors the activation of Gαi and Gαo increased with higher receptor concentrations (data not shown), indicating that these assays are limited by receptor. These results argue that the maximal stimulation of GDP-[35S]GTPγS exchange produced by each ligand must be directly proportional to its intrinsic efficacy.
Inverse Agonism of SR141716A at CB1 Receptor.
The inhibition of the CB1 receptor by SR141716A has been reported for a number of intact cell models (Bouaboula et al., 1997; MacLennan et al., 1998; Pan et al., 1998). Our in situ reconstitution allows the elimination of alternative cellular pathways or the existence of autocoid ligands as possible explanations for this phenomenon. To test for inverse agonism of SR141716A at the CB1 receptor assay conditions were altered from those used for agonist stimulation to enable a greater rate of spontaneous activity of the receptor-G protein complex. In the presence of 5 mM Mg2+ CB1 receptors catalyzed a higher level of [35S]GTPγS exchange for both Gαi and Gαo in the absence of added ligand than found at lower Mg2+ concentrations. This activity is receptor catalyzed because it is significantly greater than the sum of the binding of [35S]GTPγS for unreconstituted membranes or G protein alone, and it requires the expression of CB1 receptors (data not shown). Addition of 1 μM SR141716A reduced the [35S]GTPγS binding to levels equivalent to the additive total of membrane and G protein activity (Fig. 4), thus confirming that this competitive antagonist is indeed an inverse agonist of the CB1 receptor.

Inverse agonism of 1 μM SR141716A at CB1 receptors. [35S]GTPγS binding in the presence and absence of SR141716A was measured with 20 nM Gi or 80 nM Go and 100 nM added βγ. The [35S]GTPγS binding of unreconstituted membranes and Gαβγ alone also were tested. The data presented are the averages and errors of duplicate values from a representative experiment. The experiment was performed three times.
Discussion
A variety of important receptor types is thought to signal through pertussis toxin-sensitive pathways, and physiological studies of ion channel and cAMP regulation have suggested that a single receptor type may couple to multiple distinct pertussis toxin-substrate-G protein α-subunits (Gudermann et al., 1997). The cannabinoid receptors are of particular interest in this regard because the various pharmacological actions of cannabinoids may be linked to distinct intracellular pathways and the development of therapeutically effective agents devoid of intoxicating properties would be welcome. Furthermore, synthetic cannabinoid ligands may bind to distinct surfaces of the CB1 receptor (for review, see Howlett, 1998). This has led to the speculation that these chemically disparate agonists may direct receptor activation of selective G proteins. We therefore modified an in situ reconstitution approach previously used to characterize the Gq coupling of 5-HT2C receptors (Hartman and Northup, 1996), and the bombesin family of receptors (Jian et al., 1999) to examine the G protein coupling of recombinant CB1 and CB2 receptors. Our data demonstrate that this technique can be applied successfully to other families of G proteins, including Gi and Goproteins that have high rates of spontaneous GDP dissociation.
As predicted by signal transduction, studies CB1and CB2 receptors did indeed show different abilities to activate the G proteins. Furthermore, the ability to recognize G proteins was selective because nonappropriate G proteins such as Gq and Gt were not recognized by either receptor. Both CB1 and CB2 receptors could activate Gi with similar apparent affinity. In contrast to Gi, the abilities of CB1and CB2 receptors to activate Go protein were substantially different. Both receptors showed a lower apparent affinity for Gothan for Gi. However, the apparent affinity for the CB2 receptor was too low to be measured accurately within the detergent constraints of this assay and was estimated to be at least 3- to 4-fold lower than that measured for CB1 receptor activation of Go. This finding is consistent with the regional distribution of the cannabinoid receptors and G proteins. CB1 receptors are localized to the brain and a few peripheral organs (Pertwee, 1997), whereas CB2 receptor are localized primarily to immune cells. Conversely, although Gi and Go are both neuronally localized, Gi has been demonstrated to be present in immune cells, whereas Go is not abundant peripherally, suggesting that physiologically Gi is likely to be the G protein encountered by CB2. Furthermore, this finding helps to explain the differences in signal transduction pathways observed for these receptors. The failure of CB2 receptors to modulate ion channels may be explained by their low affinity for Go. Inhibition of voltage gated Ca2+ channels is probably mediated via Gαo, whereas activation of K+ channels is probably via βγ-subunits derived from either Gi or Go (Gudermann et al., 1997). It is also possible that βγ-subunits of differing composition have higher affinity for Go versus Gi (or CB1 versus CB2), and that these subunits differentiate the ability of cannabinoid receptors to activate K+ channels. This data would suggest that CB1 receptor coupling to ion channels is Go mediated, and that the lower apparent affinity of CB2 for this G protein is sufficient to prevent regulation of ion channels.
The efficacy of a range of agonists to stimulate Gi via the CB2 receptors observed in this study correlated well with the existing signal transduction data for these receptors. In this study, HU210 was a full agonist, whereas WIN55,212, Δ9-THC, and anandamide were only partial agonists, although the degree of stimulation differed between the agonists with WIN55,212 producing an intermediate level of activation. Previous studies on the ability of cannabinoids to inhibit cAMP formation in cells transfected with CB2 receptors have produced inconsistent results. Although Felder et al. (1995) found both Δ9-THC and anandamide to be inhibitory in CB2-transfected Chinese hamster ovary (CHO) cells, other investigators have reported either or both of these agents to have little or no inhibitory effect on cAMP production by CHO or COS-7 cells (Bayewitch et al., 1995, 1996; Slipetz et al., 1995). Only one study appears to have compared the abilities of HU210 and WIN55,212 to inhibit cAMP formation in CB2-transfected CHO cells (Slipetz et al., 1995). That study found the maximal inhibition induced by these two compounds to be equivalent. This finding suggests that the receptor number was sufficient to overcome the slight differences we observed for the efficacies of these agonists to activate G proteins, or that submaximal activation of Gi is sufficient to produce maximal inhibition of cAMP. Given that the K i and IC50 values in this study appeared to be similar, the latter explanation seems more likely.
Our analysis of the efficacies for CB1 suggest that different cannabinoid agonists can indeed direct the interaction of CB1 receptors with Gαior Gαo. We found that WIN55,212 and anandamide were full agonists in the activation of Gαi but only partial agonists in the activation of Gαo. Consistent with our finding of partial agonism at both Gi and Go proteins, Δ9-THC has been demonstrated previously to produce either no activation or only partial activation of [35S]GTPγS binding in rat and mouse brain cerebellar membranes and slices (Sim et al., 1995; Burkey et al., 1997a,b; Griffin et al., 1998). Anandamide also produced partial agonism at Go in our study and has previously demonstrated submaximal activation of [35S]GTPγS in cerebellar and whole-brain homogenates (Burkey et al., 1997a; Griffin et al., 1998; Kearn et al., 1999). In contrast, WIN55,212 has been demonstrated to produce maximal GTPγS35 stimulation in the former studies. This difference most likely reflects the inability of the homogenate [35S]GTPγS-binding assays to detect differences between different G proteins. Our data clearly demonstrate that one ligand can induce a receptor conformation that is maximally active in stimulating one G protein, whereas only partially active in its ability to activate a different G protein. Thus, HU210 stabilizes a conformation of the CB1 receptor that can fully activate both Gi and Go. However, WIN55,212 must induce a different conformation, as it was a full agonist at the CB1 receptor for activation of Gi, but was only a partial for the activation of Go. This finding clearly suggests that ligands may be designed that are fully selective for one G protein pathway over another. Therapeutically, this could provide a powerful mechanism for selecting for particular actions of cannabinoids, while avoiding some of the unwanted effects.
That different agonists might induce different conformations of the CB1 receptor is not entirely unpredicted given that the agonists are known to bind differentially to the receptor. WIN55,212 belongs to the nonclassical class of cannabinoids, the aminoalkylindoles. Although classical cannabinoids and aminoalkylindoles agonists show competitive binding interactions at the CB1 receptor and appear to exhibit some pharmacophoric elements in common (for review, see Howlett, 1998), it appears that their interaction with the cannabinoid receptor is not identical. Mutation studies of the CB1 receptor have identified at least one point of receptor interaction with cannabinoid ligands, a predicted helix III lysine, that inhibits binding of anandamide and CP55,940 [(−)-cis-3-[2-hydroxy-4-(1-1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol], but has no effect on the binding of aminoalkylindole ligands (Song and Bonner, 1996; Chin et al., 1998). It is possible that the different points of ligand-receptor interaction promote different receptor conformations, which in turn result in selective interaction with different G proteins. Although much work has been carried out on the sites of interaction of receptors with G proteins (for review, seeGudermann et al., 1997), it is still unclear whether contact sites for different G proteins on cytoplasmic receptor parts can be differentiated or are identical. Previous studies on a range of receptor types have suggested that this “agonist trafficking” of signaling pathways is possible (Tucek, 1997). However, these studies have focused on the activation of second messenger pathways, rather than directly measuring G protein activation. A recent study has demonstrated WIN55,212 to be more efficacious than other agonists tested in stimulating cAMP accumulation in CHO cells expressing the CB1 receptor following pertussis toxin treatment (Bonhaus et al., 1998). Previous studies have suggested that this pathway may be mediated by Gαs (Glass and Felder, 1997), suggesting that agonist-receptor complexes may differ in their recognition of Gs in addition to Gi and Go.
Several previous reports have suggested that the CB1 receptor antagonists SR141716A may exhibit inverse agonist properties (Bouaboula et al., 1997; MacLennan et al., 1998; Pan et al., 1998). Inverse agonism differs from conventional antagonism, in that rather than possessing equivalent affinity for both the active and inactive receptor states, inverse agonists have a higher affinity for the inactive state, thereby inhibiting any spontaneous activity of the receptor. SR141716A also has been reported to reduce basal GTPγS binding in membranes from cells with CB1 receptors (Landsman et al., 1997). However, other studies have failed to observe this effect (Breivogel et al., 1998; Kearn et al., 1999). Recently, Pan et al. (1998) demonstrated constitutive activity of CB1 receptors in inhibiting Ca2+ currents that was not due to endogenous agonist, confirming that CB1 receptors can be tonically active. In our study, CB1receptors exhibited spontaneous activation of both Gi and Go that could be enhanced by additional magnesium. This spontaneous activity was completely blocked by SR141716A, indicative of strong inverse agonism. It is tempting to speculate based on our findings with cannabinoid agonists that inverse agonists also may be capable of distinguishing between G proteins.
The physiological relevance of the difference in the ability of ligands to regulate G protein signaling will depend on a combination of the number of receptors in the cell, and the saturation properties of the effector molecules. Thus, if saturation of the second messenger response (e.g., inhibition of adenylate cyclase, enhancement of potassium conductance) requires full stimulation of G protein, then partial efficacy would be visible if receptor number was limited. If, however, the maximal response can be generated by submaximal G protein activation, then the difference between agonists may not be readily discernable. This model, therefore, provides a mechanism for explaining the differences observed in potency of agonists in different tissues or cells. For example, Mackie et al., (1993) demonstrated that anandamide was a partial agonist in the inhibition of calcium channels in N18 neuroblastoma cells, but they observed full agonism of this effect in AtT20 cells that express higher receptor number (Mackie et al., 1995). Our studies have demonstrated that anandamide is a partial agonist in the activation of Gαo, the G protein thought to mediate calcium channel activation. Thus, these findings are consistent that when receptor is limited, the differences in response to particular agonists become detectable. This finding emphasizes the importance of future studies focused on designing agonists that more fully distinguish between different G proteins. The ability of the cannabinoid receptors to activate G proteins in the absence of agonist confirms that the cannabinoid signaling may have an intrinsic tone independent of cell activity. Previous studies with 5-HT receptors have demonstrated that not all antagonists can act as inverse agonists (Hartman and Northup, 1996). This would suggest that the physiological response of a pure antagonist will differ from an inverse agonist, again increasing the range of potential therapeutic outcomes mediated through the cannabinoid receptors. Furthermore, this study demonstrates reconstitution as an ideal system for screening potential antagonists, inverse agonists, and subclasses of agonists.
Acknowledgments
We gratefully acknowledge the assistance of Haya Laufer in Sf9 culture and baculovirus production, and Joanne Guiterrez and Loren Chen in G protein production. We thank Drs. Paul Randazzo, Michael Brownstein, and James Battey for critical reading of this manuscript.
Footnotes
- Received June 15, 1999.
- Accepted September 14, 1999.
-
Send reprint requests to: Dr. John K. Northup, Section on Signal Transduction, National Institute on Deafness and Other Communication Disorders, 5 Research Court, Rockville, MD 20850. E-mail:drjohn{at}codon.nih.gov
Abbreviations
- GTPγS
- guanosine-5′-O-(3-thio)-triphosphate
- Sf9
- Spodoptera frugiperda cells
- 5-HT
- 5-hydroxytryptamine
- HU210
- (−)-11-hydroxy-Δ8-tetrahydrocannabinol-dimethylheptyl
- WIN55,212
- (R)-(+)-(2,3-dihydro-5-methyl-3-[(morphonolinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl)(1-napthalenyl)methanone mesylate
- SR141716A
- N-(peperidino-1-yl)-5-(4-chloropheyl)-1-(2,4-dichlorophenyl)-4-methyl-pyrazole-3-carboxamide, hydrochloride
- SR144528
- N-[(1S)-endo-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl]5-(4-chloro-3-methyl-phenyl)-1-(4-methylbenzyl)pyrazole-3-carboxamide
- Δ9-THC
- Δ9-tetrahydrocannabinol
- MOPS
- 4-morpholinepropanesulfonic acid
- P2
- postnuclear fraction
- CHO
- Chinese hamster ovary
- U.S. Government




{kind=link}
{kind=link}
{kind=link}
{kind=link}