


Abstract
R214127 was shown to be a potent and noncompetitive metabotropic glutamate 1 (mGlu1) receptor-selective antagonist. The kinetics and pharmacology of [3H]1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone (R214127) binding to rat mGlu1a receptor Chinese hamster ovary (CHO)-dhfr− membranes was investigated, as well as the distribution of [3H]R214127 binding in rat brain tissue and sections. Specific binding to rat mGlu1a receptor CHO-dhfr− membranes was ∼92% of total and was optimal at 4°C. Full association was reached within 5 min, and [3H]R214127 bound to a single binding site with an apparent KD of 0.90 ± 0.14 nM and aBmax of 6512 ± 1501 fmol/mg of protein. Inhibition experiments showed that [3H]R214127 binding was completely blocked by 2-quinoxaline-carboxamide-N-adamantan-1-yl (NPS 2390), (3aS,6aS)-6a-naphtalan-2-ylmethyl-5-methyliden-hexahydro-cyclopenta[c]furan-1-on (BAY 36-7620), and 7-(hydroxyimino)cyclo-propa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt), but was not displaced by competitive mGlu1 receptor ligands such as glutamate and quisqualate, suggesting that R214127, NPS 2390, BAY 36-7620, and CPCCOEt bind to the same site or mutually exclusive sites. Experiments using rat cortex, striatum, hippocampus and cerebellum revealed that [3H]R214127 labeled a single high-affinity binding site (KD ∼ 1 nM).Bmax values were highest in the cerebellum (4302 ± 2042 fmol/mg of protein) and were 741 ± 48, 688 ± 125, and 471 ± 68 fmol/mg of protein in the striatum, hippocampus, and cortex, respectively. The distribution of [3H]R214127 binding in rat brain was investigated in more detail by radioligand autoradiography. A high density of binding sites was detected in the molecular layer of the cerebellum. Moderate labeling was seen in the CA3 and dentate gyrus of the hippocampus, thalamus, olfactory tubercle, amygdala, and substantia nigra reticulata. The cerebral cortex, caudate putamen, ventral pallidum, and nucleus accumbens showed lower labeling. The high affinity and selectivity of [3H]R214127 for mGlu1 receptors renders this compound the ligand of choice to study the native mGlu1 receptor in brain.
Eight metabotropic glutamate (mGlu) receptors have been cloned to date. These G protein-coupled glutamate receptors have been divided into three groups based on sequence homology, second-messenger coupling, and pharmacology (Pin et al., 1999). Numerous studies have been performed to elucidate the role of these receptors in physiological and pathological conditions of the central nervous system. Group I mGlu receptors, which comprise the mGlu1 and mGlu5 receptors and several splice variants thereof, in particular have received quite some attention. Several studies have provided clues that group I receptors play a physiological role in regulating ion channels and synaptic transmission and in synaptic plasticity that underlies learning and memory (Pin and Duvoisin, 1995). Overactivation of these mGlu receptors could be involved in psychiatric and neurological diseases (Bordi and Ugolini, 1999). The suggested role of mGlu1 and mGlu5 receptors was based on pharmacology (Schoepp et al., 1999), immunohistochemistry (Martin et al., 1992; Shigemoto et al., 1993, 1997; Grandes et al., 1994; Lujan et al., 1996), in situ hybridization (Shigemoto et al., 1992; Mutel et al., 2000), studies using knock-out animals (Aiba et al., 1994; Conquet et al., 1994), or antibody ablation studies (Shigemoto et al., 1994). A selective radioligand with high affinity for either the mGlu1 or mGlu5 receptor, an obvious tool in the study of the physiology and pathology of group I mGlu receptors, had been missing for a long time. Recently, however, the selective mGlu5 receptor radioligand [3H]2-methyl-6-((3-methoxyphenyl)ethynyl)-pyridine was developed (Gasparini et al., 2002). Until now, there has been no radioligand available to selectively label the mGlu1 receptor.
In recent years, [3H]glutamate and [3H]quisqualate have been frequently used to study group I receptor binding. [3H]Glutamate has been used on rat (Thomsen et al., 1993) and human (Kingston et al., 1998) mGlu1a receptors expressed in baby hamster kidney cells and in an RGT cell line, respectively. In both cases, low binding affinities (KD of 296 and 158 nM for the rat mGlu1a receptor and the human mGlu1a receptor, respectively) were found. [3H]Quisqualate has been used on the human mGlu1a receptor, expressed in baculovirus (Ohashi et al., 1997); a KD value of 53 nM was found. Recently, the binding of [3H]quisqualate to membranes from human embryonic kidney 293 cells transfected with the rat mGlu1a and mGlu5a receptor was described (Mutel et al., 2000).KD values of 37 and 81 nM for the mGlu1a and the mGlu5a receptor were measured, respectively. Besides their relatively low affinity, the lack of selectivity limits the usefulness of [3H]glutamate and [3H]quisqualate for studying mGlu receptor subtypes in rat or human brain. Previous attempts to examine mGlu receptor binding in brain tissue using [3H]glutamate (Schoepp and True, 1992; Wright et al., 1994) or [3H]quisqualate (Mutel et al., 2000) were performed under saturating concentrations of ionotropic glutamate receptor antagonists. Under these conditions, either mGlu1 and mGlu5 receptors (for [3H]quisqualate) or several mGlu receptors (for [3H]glutamate) were labeled; thus, no distinction between these subtypes could be made. Consequently, the lack of a selective mGlu1 receptor radioligand has hampered studies of mGlu1 receptor binding to brain homogenates or of the distribution of the mGlu1 receptor in brain slices.
In this article, we describe the characteristics of a new mGlu1 receptor antagonist radioligand, [3H]R214127. We show the receptor binding profile of R214127 and illustrate that it selectively binds to the mGlu1 receptor. We further demonstrate that R214127 blocks mGlu1 receptor function in a potent and noncompetitive manner, leaving the mGlu2, -3, -4, -5, and -6 receptors unaffected. We next describe the properties of the radiolabeled [3H]R214127. We show highly specific [3H]R214127 binding to membranes prepared from CHO-dhfr− cells expressing the cloned rat mGlu1a receptor (rat mGlu1a receptor CHO-dhfr−membranes), and we demonstrate the usefulness of [3H]R214127 in radioligand binding of mGlu1 receptors in rat brain membranes and in mGlu1 receptor autoradiography in brain sections. Our studies show that [3H]R214127 is the first high-affinity antagonist radioligand that selectively labels the mGlu1 receptor.
Materials and Methods
Materials
All cell culture reagents were obtained from Invitrogen (Carlsbad, CA). Glutamate was from Aldrich Chemical Company (Milwaukee, WI). R214127 and R193845 (see Fig. 1 for structures) are original products from Johnson and Johnson Pharmaceutical Research and Development, a division of Janssen Pharmaceutica N.V. (Mabire et al., 2002). [3H]R214127 (28.11 Ci/mmol) was labeled by Johnson and Johnson Pharmaceutical Research and Development (for detailed description of its synthesis, see[3H]R214127 Synthesis, below). [3H]quisqualate (29 Ci/mmol), [3H]Ro 48-8587 (53 Ci/mmol), [myo-2-3H (N)]inositol (22 Ci/mmol), and [35S]GTPγS (1030 Ci/mmol) were obtained from Amersham Biosciences (Paisley, UK). [3H]MK-801 (22.5 Ci/mmol) and [3H]CGP39653 (20–50 Ci/mmol) were obtained from PerkinElmer (Zaventem, Belgium). GDP was from Roche Molecular Biochemicals (Basel, Switzerland) and glycine was from Bio-Rad (Hercules, CA). [3H]L689560 (10–30 Ci/mmol), [3H]LY341495 (34.61 Ci/mmol), [3H]MPEP (50.2 Ci/mmol), (S)-4C3HPG, (1S,3R)-ACPD, (S)-3,5-DHPG, (S)-4CPG, AIDA, MCPG, MPEP, CPCCOEt (see Fig. 1 for structure) l-serine-O-phosphate, andl-quisqualic acid were purchased from Tocris Cookson (Essex, UK). BAY 36-7620, NPS 2390 (see Fig. 1 for structures), and phencyclidine were synthesized for in-house use as reference compounds by Johnson and Johnson Pharmaceutical Research and Development. Fluo-3-acetoxymethyl ester and pluronic acid were from Molecular Probes (Leiden, The Netherlands). Probenecid, strychnine,d-serine, and Triton X-100 were purchased from Sigma-Aldrich (Steinheim, Germany). All other reagents were from Merck (Darmstadt, Germany).

Chemical structure of [3H]R214127 (T, tritium), R193845, BAY 36-7620, NPS 2390, and CPCCOEt.
[3H]R214127 Synthesis
2-(4-Bromophenyl)-1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)- 1-ethanone, a p-bromo derivative of R214127 (0.872 mg), together with triethylamine (0.872 μl) in sodium-dried tetrahydrofuran (0.174 μl), was added to a carefully measured amount of palladium on carbon. The reaction flask was connected to a tritiation manifold, and the reaction mixture was carefully degassed and exposed to tritium gas (19.5 Ci at a pressure of 105 Pascals, generated from uranium tritide) for 30 min. The reaction mixture was then frozen using liquid nitrogen, and the excess of tritium gas was captured onto uranium sponge. Solvent was lyophilized from the reaction mixture. Methanol was added to the mixture and lyophilized to remove labile tritium. This procedure was repeated twice. The residue was taken up in ethanol, filtered over a GHP Acrodisk 13-mm syringe filter, and depleted with ethanol. From this, the compound was purified by quantitative high-performance liquid chromatography (Kromasil KR 100-10, column dimensions 4.6 × 300 mm). UV detection took place at 265 nm and elution was performed isocratically with water/methanol/acetonitrile/diisopropylamine (47:26.5:26.5:0.2; v/v/v/v) at a flow rate of 2 ml/min. The product containing fractions were combined and concentrated under vacuum at 30°C. The residue was dissolved in ethanol and concentrated again. This procedure was repeated twice and the remaining residue was finally dissolved in ethanol and stored at −20°C. The resulting [3H]R214127 had a radiochemical purity of 98.5% and a specific activity of 28.11 Ci/mmol.
Cell Transfection and Culture
L929sA cells stably expressing the human mGlu1a receptor were obtained as described previously (Lavreysen et al., 2002) and were cultured in GlutaMAX I medium supplemented with 10% heat-inactivated dialyzed fetal calf serum, 0.1 mg/ml streptomycin sulfate, and 100 units/ml penicillin. CHO-dhfr− cells stably expressing the rat mGlu1a, -2, -3, -4, -5, and -6 receptor were a kind gift from S. Nakanishi (Tokyo University, Tokyo, Japan) and were grown in Dulbecco's modified Eagle's medium with GlutaMAX I with 10% heat-inactivated dialyzed fetal calf serum, 0.4 mMl-proline, 0.2 mg/ml streptomycin sulfate, and 200 units/ml penicillin. Cells were kept in an atmosphere of 37°C and 5% CO2.
Intracellular Ca2+ Response in Rat and Human mGlu1a Receptor Expressing Cells and in Rat mGlu5 Receptor-Expressing Cells
Intracellular calcium ion levels ([Ca2+]i) in human mGlu1a receptor-expressing L929sA cells were measured using the fluorometric imaging plate reader (Molecular Devices, Sunnyvale, CA), as described previously (Lavreysen et al., 2002). The same procedure was followed for CHO-dhfr− cells expressing the rat mGlu1a receptor. For the rat mGlu5 receptor, cells were seeded at 30,000 cells/well 2 days before the experiment.
IP Response in Rat mGlu1a Receptor Expressing CHO-dhfr−Cells
IP accumulation was measured as described previously (Lavreysen et al., 2002). Briefly, cells were seeded at 30,000 cells/well in 24-well plates and were labeled with 2.5 μCi/ml [myo-2-3H (N)]inositol overnight. On the day of the experiment, cells were washed and incubated for 10 min with 10 mM LiCl. After 30 min of incubation with increasing concentrations of R214127, 1 N HClO4 was added and plates were put at 4°C. KOH/phosphate solution and a solution containing 30 mM Na2B4O7·10H2O and 3 mM EDTA were added before application to ion exchange chromatography.
Membrane Preparation from CHO-dhfr− Cells Expressing the Rat mGlu1a, -2, -3, -4, -5, and -6 Receptors
Confluent cells were washed in ice-cold phosphate-buffered saline and stored at −20°C until membrane preparation. After thawing, cells were suspended in 50 mM Tris-HCl, pH 7.4, and collected through centrifugation for 10 min at 23,500g at 4°C. The cells were lysed in 10 mM hypotonic Tris-HCl, pH 7.4. After recentrifugation for 20 min at 30,000g at 4°C, the pellet was homogenized with an Ultra Turrax homogenizer in 50 mM Tris-HCl, pH 7.4. Protein concentrations were measured by the Bio-Rad protein assay using bovine serum albumin as standard.
[35S]GTPγS Binding to Membranes from CHO-dhfr− Cells Expressing the Rat mGlu2, -3, -4, and -6 Receptors
Membranes were thawed on ice and diluted in 10 mM HEPES acid, 10 mM HEPES salt, pH 7.4, containing 100 mM NaCl, 3 mM MgCl2, 3 μM GDP, and 10 μg/ml saponine. Assay mixtures contained 10 μg of membrane protein and were preincubated with compounds or buffer for 5 min at 37°C. Then, glutamate was added and the assay mixtures were further incubated for 30 min at 37°C. [35S]GTPγS was added to a final concentration of 0.1 nM for another 30 min at 37°C. Reactions were terminated by rapid filtration through Unifilter-96 GF/B filter plates (PerkinElmer Life Sciences, Boston, MA) using a 96-well Packard filtermate harvester. Filters were washed 2 times with ice-cold 10 mM NaH2PO4/10 mM Na2HPO4 buffer, pH 7.4. Filter-bound radioactivity was counted in a Microplate scintillation and luminescence counter from Packard.
Radioligand Binding to Rat mGlu1a Receptor CHO-dhfr−Membranes
[3H]R214127 Binding.
After thawing, the membranes were homogenized using an Ultra Turrax homogenizer and suspended in ice-cold binding buffer containing 50 mM Tris-HCl, pH 7.4, 1.2 mM MgCl2, and 2 mM CaCl2, unless otherwise indicated. Ligand saturation experiments were performed at apparent binding equilibrium (30 min of incubation) with 20 μg of membrane protein and 10 concentrations (0.1, 0.2, 0.3, 0.4, 0.5, 1, 2, 2.5, 5, and 10 nM) of radioligand. Nonspecific binding was estimated in the presence of 1 μM R193845. The incubation was stopped by rapid filtration under suction over GF/C glass-fiber filters using a manual 40-well filtration manifold. To measure association kinetics, membranes were incubated at 4°C, 25°C or 37°C in the presence of 2.5 nM [3H]R214127 for 2, 5, 10, 15, 20, 30, 45, 60, 90 or 120 min, then terminated by rapid filtration using a manual 40-well filtration unit. Dissociation kinetics were measured by adding, at different times before filtration 1 μM R193845 to membranes preincubated for 30 min at 4°C or 25°C in the presence of 2.5 nM [3H]R214127. The filters were transferred to scintillation vials and, after the addition of Ultima-Gold MV, the radioactivity collected on the filters was counted in a Packard scintillation counter. For inhibition experiments, assay mixtures were incubated for 30 min at 4°C in a volume of 0.5 ml containing 10 to 20 μg membrane protein, appropriate concentrations of test compounds and 2.5 nM [3H]R214127. Nonspecific binding was defined as above. Filtration was performed using Unifilter-96 GF/C filter plates and a 96-well PerkinElmer FilterMate harvester. After the addition of microscint-O, radioactivity on the filters was counted in the Microplate scintillation and luminescence counter.
[3H]Quisqualate Binding.
Thawed membranes were homogenized and suspended in ice-cold binding buffer. For saturation experiments, 30 μg of membrane protein was incubated for 1 h at 25°C with 10 concentrations (1, 2, 5, 10, 20, 40, 60, 90, 120, and 150 nM) of [3H]quisqualate. Nonspecific binding was determined in the presence of 1 mM l-glutamate. Bound and free radioligand was separated by rapid filtration over GF/C glass-fiber filters using a manual 40-well filtration manifold. For inhibition experiments, 30 μg of membrane protein was incubated for 1 h at 25°C in a volume of 0.5 ml containing appropriate concentrations of test compounds and a final concentration of 10 nM [3H]quisqualate. Filtration was performed using Unifilter-96 GF/C filter plates and FilterMate harvester. Radioactivity trapped on the filters was counted as above.
Radioligand Binding to Membranes from CHO-dhfr− Cells Expressing the Rat mGlu2, -3, -4, -5, and -6 Receptors
After thawing, the membranes were homogenized using an Ultra Turrax homogenizer and suspended in ice-cold binding buffer containing 50 mM Tris-HCl, pH 7.4, 1.2 mM MgCl2, and 2 mM CaCl2. For [3H]R214127 binding, 20 to 160 μg of membrane protein and a final concentration of 20 nM [3H]R214127 was used. As indicated under Results, different blanks were used to define nonspecific binding. Incubation time and temperature as well as filtration were as described for rat mGlu1a receptor CHO-dhfr− membranes. Expression of rat mGlu2, -3, -5, and -6 receptors was confirmed by specific binding of [3H]LY341495 (mGlu2, -3, and -6) or [3H]MPEP (mGlu5). For [3H]LY341495 binding, 1 nM (mGlu2 and mGlu3) or 10 nM (mGlu6) [3H]LY341495 was used. Nonspecific binding was determined using 1 mM glutamate. Assay mixtures were incubated for 30 min (mGlu2 and mGlu3) or 60 min (mGlu6) at 4°C. Incubation was stopped by filtration over GF/B glass-fiber filters (Whatman, England) using a manual 40-well filtration manifold. For rat mGlu5 receptor CHO-dhfr− membranes, 10 nM [3H]MPEP and 10 μM MPEP were used to reveal nonspecific binding. Incubation was performed at 4°C for 30 min. Bound and free radioligand were separated over GF/C glass-fiber filters (Whatman, England) using a 40-well filtration unit.
[3H]R214127 Binding to Rat Brain Membranes
Tissue Preparation.
Male Wistar rats (∼200 g) were sacrificed by decapitation. The brains were rapidly removed and cortex, hippocampus, striatum and cerebellum were immediately dissected. The fresh tissue was homogenized with an Ultra Turrax homogenizer in 20 volumes of 50 mM Tris-HCl, pH 7.4, and tissue was centrifuged at 23,500g for 10 min. After homogenization using a DUALL homogenizer, membranes were washed twice by centrifugation at 23,500g for 10 min. The final pellet was suspended in 10 volumes of 50 mM Tris-HCl, pH 7.4, and frozen at −80°C.
In Vitro Binding Assay.
After thawing, membranes from rat cortex, cerebellum, striatum, and hippocampus were rehomogenized using a DUALL and suspended in ice-cold binding buffer containing 50 mM Tris-HCl, 1.2 mM MgCl2, and 2 mM CaCl2, pH 7.4. The binding assay was carried out in a total volume of 0.5 ml containing 2.5 nM [3H]R214127 and a membrane aliquot corresponding to 40 μg for cerebellar membranes, 60 μg for hippocampal membranes, 80 μg for striatal membranes, or 150 μg for cortical membranes. Specific binding was calculated as the difference between the total binding and the binding measured in the presence of 1 μM R193845. After incubation for 30 min at 4°C, the labeled membranes were washed and harvested by rapid vacuum filtration over Whatman GF/C glass-fiber filters using a 40-well filtration manifold, and radioactivity collected on the filters was counted as above.
[3H]Ro 48-8587, [3H]L689560, [3H]CGP39653, and [3H]MK-801 Binding to Rat Brain Membranes
Tissue Preparation.
Male Wistar rats (∼200 g) were sacrificed by decapitation. The brains were rapidly removed and forebrain was dissected. The tissue was homogenized with an Ultra Turrax homogenizer in 20 volumes of ice-cold H2O and was centrifuged at 48,000g for 20 min. After homogenization using a DUAL homogenizer, membranes were washed by centrifugation at 48,000g for 10 min. The pellet was then suspended in 20 volumes of 50 mM Tris-HCl, pH 7.4, containing 0.04% Triton X-100 and again centrifuged at 48,000g for 20 min. The final pellet was frozen at −80°C.
In Vitro Binding Assay.
On the day of the experiment, the pellet was thawed, washed, and rehomogenized using a DUAL in ice-cold 50 mM Tris-acetate, pH 7.4. Assay conditions for the different radioligands were as follows. The final concentration of membrane in the assay was 20 mg/ml (wet weight) for [3H]Ro 48-8587 and [3H]L689560 and 10 mg/ml (wet weight) for [3H]CGP39653 and [3H]MK-801. Radioligand concentrations of 2 nM [3H]Ro 48-8587, 2 nM [3H]L689560, 2 nM [3H]CGP39653, and 3 nM [3H]MK-801 were used. Incubation was performed in the presence of 1 mM KSCN for [3H]Ro 48-8587, 100 μM strychnine for [3H]L689560, and 1 μM glycine + 1 μM glutamate for [3H]MK-801 binding. Nonspecific binding was determined in the presence of 1 mM glutamate for [3H]Ro 48-8587 and [3H]CGP39653 binding. For [3H]L689560 and [3H]MK-801 binding, 100 μMd-serine or 10 μM phencyclidine was used to define nonspecific binding, respectively. Assays were incubated for 1 h at 37°C, 2 h at 4°C, 30 min at 25°C, and 1 h at 4°C for [3H]Ro 48-8587, [3H]L689560, [3H]CGP39653, and [3H]MK-801 binding, respectively. After incubation, bound and free radioligand was separated using a 40-well filtration manifold. Radioactivity collected on the filters was counted as above.
[3H]R214127 Binding and Autoradiography on Rat Brain Sections
Tissue Preparation.
Male Wistar rats (200 g) were sacrificed by decapitation. Brains were immediately removed from the skull and were rapidly frozen in dry-ice-cooled 2-methylbutane (−40°C). Brains were then stored at −70°C until sectioning. Sagittal sections (20 μm) were cut using a Leica C3050 cryostat microtome (Leica Microsystems, Wetzlar, Germany) and thaw-mounted on SuperFrost Plus microscope slides (Menzel-Glaser GmbH, Braunschweig, Germany). The sections were then kept at −70°C until use.
Receptor Autoradiography.
Sections were thawed and dried under a stream of cold air, preincubated (3 × 5 min) in 50 mM Tris-HCl, 1.2 mM MgCl2, 2 mM CaCl2, 0.1% bovine serum albumin, pH 7.4, at room temperature. Sections were then incubated for 60 min at room temperature, in buffer containing 50 mM Tris-HCl, 1.2 mM MgCl2, 2 mM CaCl2, 0.1% bovine serum albumin, pH 7.4, and 1.5 nM [3H]R214127. Nonspecific binding was determined by addition of 1 μM R193845 in the incubation buffer. After the incubation, the excess of radioligand was washed off (3 × 5 min) in ice-cold buffer containing 50 mM Tris-HCl, 1.2 mM MgCl2, and 2 mM CaCl2, followed by a rapid dip in cold distilled water. The sections were dried under a stream of cold air and then exposed to [3H]Hyperfilm (Amersham Biosciences, Buckinghamshire, UK) for 6 weeks at room temperature. The films were developed manually in Kodak D19 and fixed with Kodak Readymatic (Eastman Kodak, Rochester, NY). Some sections were exposed to a Fuji Imaging Plate for 2 days at room temperature and scanned using a Fujix BAS 2000 Bio Imaging Analyzer (Fuji, Tokyo, Japan).
Data Analysis and Statistics
Data analysis was performed using the Prism program (GraphPad Software, Inc., San Diego, CA). Saturation binding experiments were analyzed using a nonlinear regression analysis. Inhibition curves were fitted using nonlinear regression analysis fitting the one-site competition equation: Y = Bottom + ((Top − Bottom)/1 + 10X−LogIC50).Ki values were calculated using the Cheng-Prusoff equation: Ki = IC50/[1 + ([C]/KD)], where C is the concentration of radioligand and KD is the dissociation constant of the radioligand (Cheng and Prusoff, 1973). The observed on (kob) and off (koff) rates were calculated from association-dissociation curves using the one-phase-exponential association and decay equations in the Prism program, respectively.kon was calculated by subtractingkoff fromkob and dividing by the radioligand concentration.
The two-tailed Student's t test was used for statistical evaluation of the binding data. The Dunnett's t test after a two-way analysis of variance (with compound concentration and experiment as factors) was used to analyze the data from the IP experiments.
Results
Selectivity and Mode of Antagonism of R214127 for the mGlu1 Receptor.
In CHO-dhfr− cells expressing the rat mGlu1a receptor, R214127 inhibited the glutamate-induced increase in [Ca2+]i with an IC50 value of 21.6 ± 5.0 nM (n = 4; Fig. 2A) and seemed to be about 8-fold more potent than the recently described specific mGlu1 receptor antagonist BAY 36-7620 (IC50 = 161 ± 38 nM, n = 3) and 500-fold more potent than CPCCOEt (IC50 = 10.3 ± 0.8 μM, n = 3), tested in the same assay. For the human mGlu1a receptor, R214127 had an IC50 value of 10.4 ± 4.7 nM (n = 3). R214127 did not inhibit glutamate-induced Ca2+ signaling of the rat mGlu5 receptor expressed in CHO-dhfr− cells, tested up to a concentration of 10 μM. IC50 values for inhibition of glutamate (30 μM)-induced [35S]GTPγS activation were above 30 μM at the recombinant rat mGlu2, -3, -4, or -6 receptor. In [35S]GTPγS assays, R214127 did not exhibit agonist activity toward any of the mGlu receptors up to a concentration of 30 μM. In addition, we investigated whether R214127 could act as a positive allosteric modulator on one of these mGlu receptor types. For this, we performed glutamate concentration-response curves by adding glutamate alone or together with 10 μM R214127. [35S]GTPγS assays on recombinant rat mGlu2, -3, -4, or -6 receptors showed that the glutamate EC50 was not altered and that the glutamateEmax value was not increased upon addition of R214127. The EC50 andEmax value of glutamate-induced intracellular Ca2+ mobilization also did not change in cells expressing the rat mGlu5 receptor when R214127 was added together with glutamate (data not shown). Together, these data exclude agonist, antagonist, or positive allosteric action on mGlu2, -3, -4, -5, and -6 receptors. Radioligand binding studies on rat forebrain using [3H]Ro48-8587, [3H]L689560, [3H]CGP39653 and [3H]MK-801 revealed that R214127 did not bind to the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor or to the glycine, glutamate, or channel pore site of theN-methyl-d-aspartate receptor (tested up to a concentration of 10 μM). To analyze how R214127 inhibits glutamate activation of the mGlu1a receptor, mobilization of Ca2+ in response to glutamate was compared in the absence and presence of R214127 (Fig. 2B). The presence of R214127 not only caused a rightward shift in the concentration-response curve of glutamate but also resulted in a dramatic decrease in the maximal response evoked by the agonist, revealing that antagonism by R214127 was noncompetitive. Complete inhibition of mGlu1a receptor-mediated signaling was observed in the presence of 100 nM-1 μM R214127. To investigate whether R214127 could act as an inverse agonist, we measured basal IP accumulation in rat mGlu1a receptor containing CHO-dhfr− cells in the presence of R214127. Figure 2C shows that there is a clear reduction in basal IP production with increasing concentration of R214127. This reduction was statistically significant (p < 0.05) as of 1 μM R214127, at which basal IP accumulation decreased by 24 ± 4%. A maximal decrease of 33 ± 3% was found when using 100 μM R214127. These data indicate that R214127 can indeed act as an inverse agonist toward the mGlu1a receptor.

Antagonist profile of R214127. A, inhibition of glutamate (30 μM)-induced Ca2+ mobilization in CHO-dhfr− cells expressing the rat mGlu1a receptor. Data are expressed as mean percentage ± S.D. of the signal obtained using 30 μM glutamate, which was set at 100% (three experiments). B, concentration-response curve of glutamate alone or together with 20 nM, 30 nM, 60 nM, 100 nM, and 1 μM R214127. Values are mean ± S.D. of triplicate determinations within one experiment. An additional experiment showed the same results. C, basal IP accumulation in the presence of increasing concentrations of R214127. Values are expressed as mean percentage ± S.D. of basal IP production in the presence of solvent, which was set as 100% (three experiments performed in quadruplicate; *, p < 0.05).
Characterization of [3H]R214127 Binding to Rat mGlu1a Receptor CHO-dhfr− Membranes.
The specific binding of 2.5 nM [3H]R214127 at 4°C to rat mGlu1a receptor CHO-dhfr− membranes was proportional to the amount of membrane protein and increased linearly between 10 and 50 μg of membrane protein per assay (Fig.3). Nonspecific binding was defined using 1 μM R193845 as inhibitor. R193845 was identified as a specific mGlu1 receptor antagonist with a potency of 7.2 ± 1.2 nM (n = 3) for reversal of glutamate-induced [Ca2+]i mobilization. Using 20 μg of protein per assay, specific binding of [3H]R214127 was ∼92% of the total binding; in typical assay conditions, total and nonspecific binding were in the range of 3800 and 300 dpm, respectively.
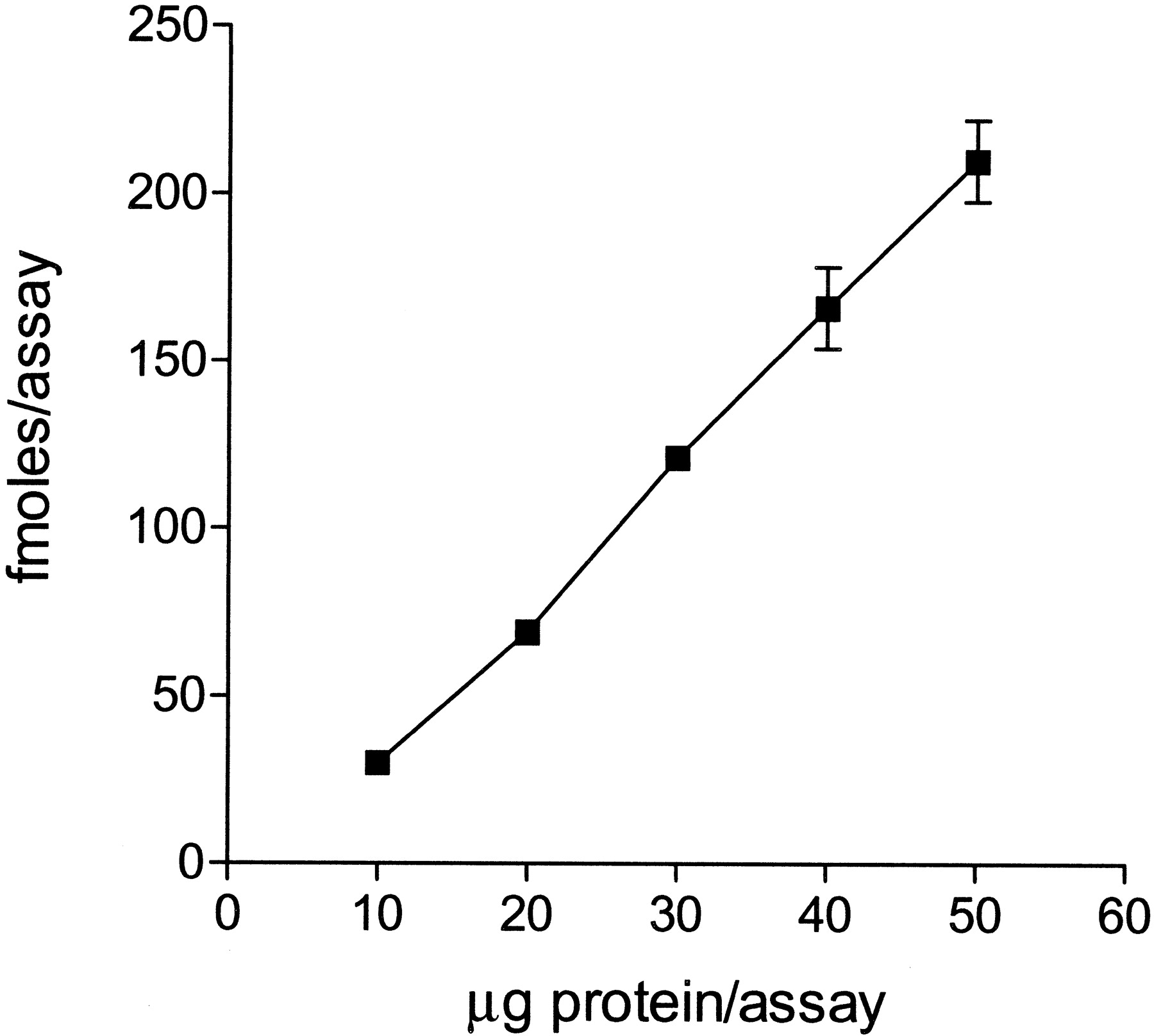
Specific [3H]R214127 binding is linear with amount of membrane protein. Rat mGlu1a receptor CHO-dhfr− membranes (10–50 μg) were incubated for 30 min on ice with 2.5 nM [3H]R214127. Data are expressed as mean ± S.D. of triplicate determinations and are from a representative experiment.
Addition of 1.2 mM MgCl2 and 2 mM CaCl2 caused a slight increase in specific binding (data not shown). Further addition of NaCl (10, 100, or 300 mM) had no effect. Although specific binding decreased by 22% at pH 6, increasing the pH up to 10 had no effect (data not shown).
Association kinetics were measured as described under Materials and Methods. Decreasing the incubation temperature to 4°C dramatically enhanced specific binding, whereas a low binding was found at 37°C (Fig. 4). The association of [3H]R214127 to membranes was extremely fast. At 4°C, 2-min incubation resulted in a specific binding corresponding to about 70% of the quantity bound at equilibrium. Maximal binding was reached within 5 min of incubation for each incubation temperature. Analysis of the association curves resulted in observed association rate constants (kob) of 0.6285, 2.571, and 1.523 min−1 at 4, 25, and 37°C, respectively. The kinetics of dissociation were also rapid (Fig. 5). At 25°C, the radioligand dissociated within as little as 2 min after 1 μM R193845 was added to the reaction tubes. The rapid dissociation kinetics at 25°C did not allow us to calculate an accurate dissociation rate constant (koff). Dissociation occurred more gradually when incubated at 4°C. [3H]R214127 was displaced completely within approximately 45 min after the addition of excess R193845. Analysis of the dissociation curve at 4°C resulted in an koff of 0.1249 min−1. kon(kob −koff/radioligand concentration) at 4°C was 0.1007 nM−1min−1.
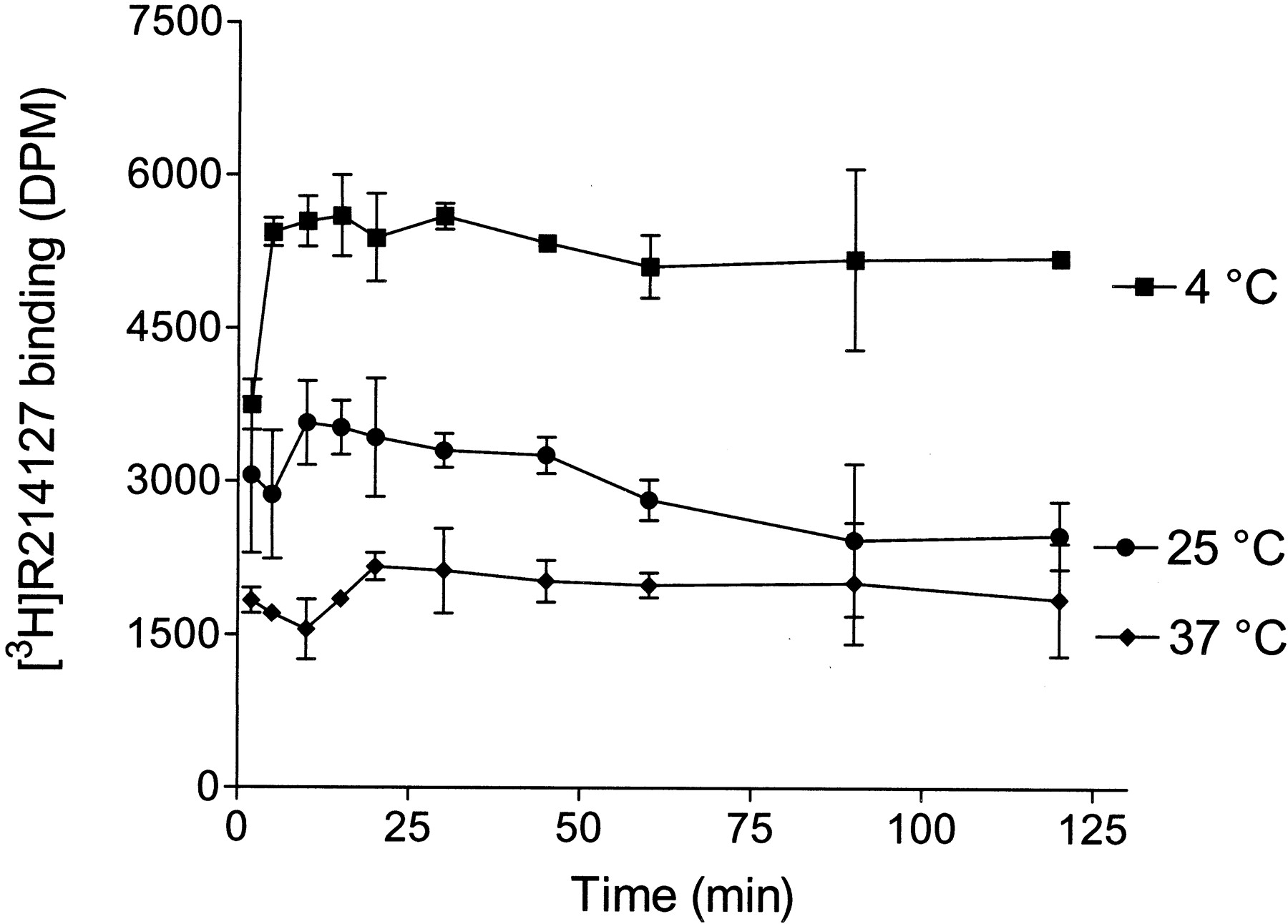
Association time course curve for [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes. Association kinetics were measured by adding 2.5 nM [3H]R214127 at different times before filtration and was determined at three different temperatures. Data are mean ± S.D. of three independent experiments performed in duplicate.

Time course for dissociation of [3H]R214127 to rat mGlu1a receptor CHO-dhfr−membranes at 4 and 25°C. Samples were incubated for 30 min at 4 or 25°C; then, an excess of R193845 was added, followed by rapid filtration at the time indicated for each data point. Values are mean ± S.D. of two independent experiments performed in duplicate.
Ligand saturation experiments were performed at apparent binding equilibrium (30-min incubation) and with 10 concentrations of radioligand. Figure 6 shows the saturation curve and Scatchard plot of [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes. Scatchard plots were linear, indicating the presence of a single, saturable, high-affinity binding site. Nonlinear regression analysis of the rectangular hyperbola revealed a Bmax of 6512 ± 1501 fmol/mg of protein and a KD of 0.90 ± 0.14 nM (n = 3).

Representative saturation binding curve and Scatchard plot of [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes. Specific binding (SB) was obtained by calculating the difference between total binding (TB) and nonspecific binding (BL), measured in the presence of 1 μM R193845. For each experiment, data points were determined in duplicate. The experiment was repeated three times.
A series of mGlu1 receptor agonists and antagonists was tested for inhibition of [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes. Inhibition curves for some antagonists are shown in Fig. 7, and the Ki values of all compounds tested are listed in Table 1. Remarkably, all ligands that bind to the glutamate binding site [i.e., glutamate, quisqualate, (1S,3R)-ACPD, (S)-3,5-DHPG, LY367385, (S)-4C3HPG, (S)-4CPG, MCPG, and AIDA] did not inhibit [3H]R214127 binding. In contrast, the noncompetitive mGlu1 receptor antagonists CPCCOEt, BAY 36-7620, NPS 2390, and R214127 inhibited [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes with potencies generally consistent with their potencies to inhibit mGlu1a receptor function. R214127 and NPS 2390 showed the highest affinity, with Ki values of 1.35 ± 0.99 and 1.36 ± 0.50 nM, respectively. BAY 36-7620 inhibited the binding also at nanomolar concentrations, whereas CPCCOEt displaced at micromolar concentrations.

Inhibition of 2.5 nM [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes by various mGlu receptor antagonists. Data points represent mean percentage of total binding ± S.D. (three to four individual experiments).
Potencies of various mGlu1 receptor agonists and antagonists in inhibition of [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes
We also investigated the specificity of [3H]R214127 binding toward the mGlu1 versus mGlu2, -3, -4, -5, and -6 receptors. Using [3H]LY341495, 95, 98, and 40% specific binding was found when using membranes prepared from CHO-dhfr− cells expressing the mGlu2, mGlu3, or mGlu6 receptor, respectively. [3H]MPEP was used as a positive control for the mGlu5 receptor and produced 95% specific binding to rat mGlu5 receptor-containing membranes. Total binding of 20 nM [3H]R214127 to membranes prepared from CHO-dhfr− cells expressing the rat mGlu2, -3, -4, -5, or -6 receptor was not higher than the binding to membranes from wild-type CHO-dhfr− cells or the nonspecific binding to rat mGlu1a receptor CHO-dhfr− membranes. Furthermore, specific binding of [3H]R214127 to these membranes was investigated using various blanks: 1 μM R193845, which is expected to bind to the same site as R214127, glutamate and l-SOP, which bind to the glutamate binding pocket and MPEP that binds to an allosteric site on the mGlu5 receptor (see Table2). None of these blanks displaced [3H]R214127. Together, these data demonstrate the specificity of [3H]R214127 for the mGlu1 receptor relative to mGlu2, -3, -4, -5, and -6 receptor subtypes.
[3H]R214127 is specific for the mGlu1 receptor relative to the mGlu2, -3, -4, -5 or -6 receptor
Comparison with [3H]Quisqualate Binding.
Saturation binding experiments were performed using 30 μg of protein per incubate and 10 concentrations (1, 2, 5, 10, 20, 40, 60, 90, 120, and 150 nM) of the mGlu1 receptor agonist [3H]quisqualate (Fig.8). Fitting of the curves revealed a single binding site with KD andBmax values of 22.0 ± 10 nM and 3912 ± 436 fmol/mg of protein, respectively (n = 3). Clearly, [3H]R214127 bound to mGlu1a with a much higher affinity than [3H]quisqualate does. The number of binding sites labeled with [3H]quisqualate was ∼60% of the number of binding sites labeled by [3H]R214127.

Representative saturation binding curve and Scatchard plot of [3H]quisqualate binding to rat mGlu1a receptor CHO-dhfr− membranes. For each experiment, data points were determined in duplicate. The experiment was repeated two times.
The same compounds were evaluated for their inhibitory action on [3H]quisqualate binding to rat mGlu1a receptor CHO-dhfr− membranes. Inhibitory potencies of the tested agonists and antagonists as well as Hill coefficients are summarized in Table 3. In this case, the compounds known to exert a competitive interaction with glutamate inhibited [3H]quisqualate binding, whereas CPCCOEt, BAY 36-7620, and NPS 2390 did not affect [3H]quisqualate binding. In addition, R214127 did not displace binding of [3H]quisqualate to the rat mGlu1a receptor. The competitive mGlu1 receptor ligands displaced [3H]quisqualate binding with the following rank order of potency: quisqualate > glutamate > LY367385 > (S)-3,5-DHPG > (S)-4C3HPG > (1S,3R)-ACPD > (S)-4CPG > AIDA > MCPG.
Potencies of various mGlu1 receptor agonists and antagonists in inhibition of [3H]quisqualate binding to rat mGlu1a receptor CHO-dhfr- membranes.
Nature of Competition between CPCCOEt, BAY 36-7620, NPS 2390, and [3H]R214127 Binding.
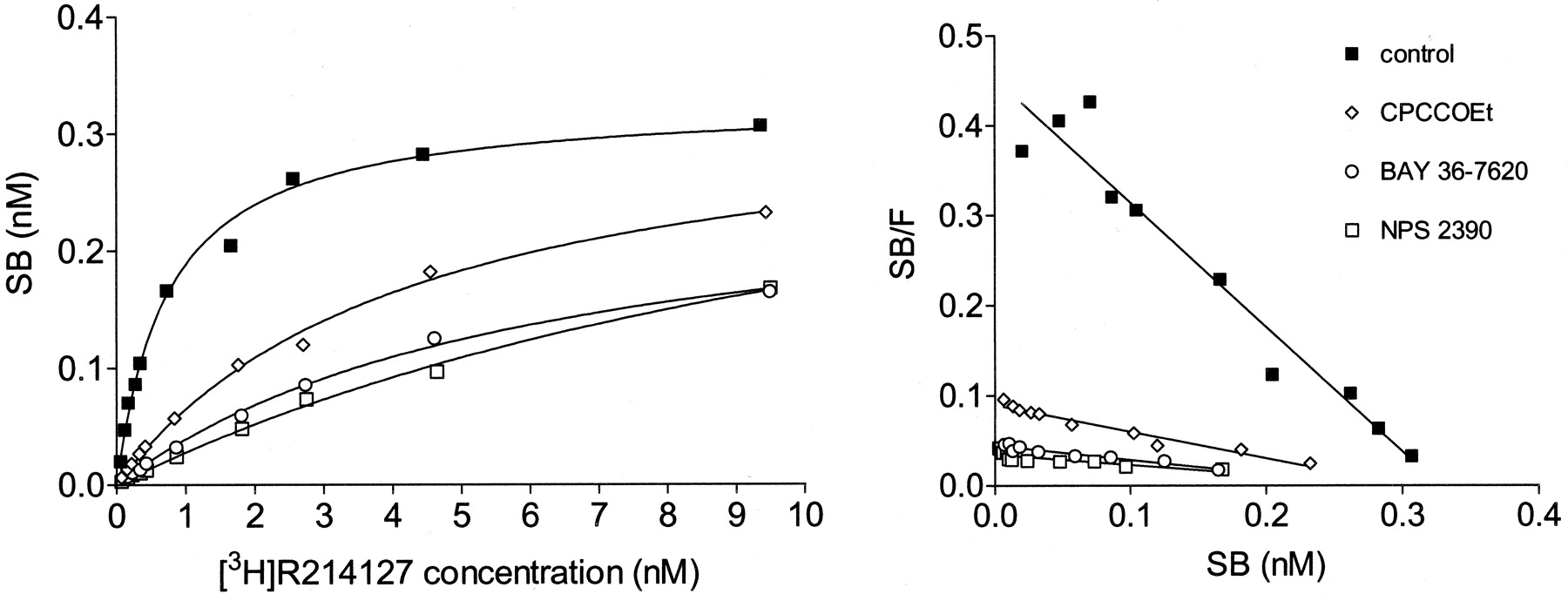
The fact that the noncompetitive compounds all displaced [3H]R214127 binding without affecting the binding of [3H]quisqualate suggested that these antagonists bound a site other than the glutamate binding site. To assess whether the reference compounds CPCCOEt, BAY 36-7620, NPS 2390, and the newly identified mGlu1 receptor antagonist R214127 compete for the same site or mutually exclusive sites, saturation experiments with [3H]R214127 concentrations from 0.1 to 10 nM in the absence and the presence of CPCCOEt (30 μM), BAY 36-7620 (100 nM), and NPS 2390 (10 nM) were performed. The presence of these competitors did not affect the Bmaxvalues but caused a significant increase in theKD value of [3H]R214127 (Table4). This is shown in Fig.9, where the data are plotted using linear regression. In Scatchard plots, the obtained linear lines indeed merge to the same intercept on the x-axis (i.e., theBmax value).
KD and Bmax values obtained from analyses of [3H] R214127 saturation binding curves obtained in the absence and presence of the mGlu1 receptor antagonists CPCCOEt (30 μM), BAY 36–7620 (100 nM), and NPS 2390 (10 nM)

Saturation binding curves and Scatchard plots of [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes in the absence and presence of CPCCOEt (30 μM), BAY 36-7620 (100 nM), and NPS 2390 (10 nM). The graph shown is a representative of three independent experiments. Data are expressed as nanomolar specifically bound. For each experiment, data points were determined in duplicate.
[3H]R214127 Binding in Rat Brain Membranes and Sections.
We used the specific mGlu1 receptor radioligand [3H]R214127 to examine receptor binding in different regions of the rat brain. Membranes from rat cortex, striatum, cerebellum, and hippocampus were prepared, and [3H]R214127 binding was measured. Nonspecific binding compared with total binding was 10% in cerebellum, 30% in hippocampus, and 25% in cortex and striatum.KD andBmax values were determined for each brain region (Table 5).KD values were about 1 nM for all structures. The Bmax values were significantly different among the various areas. [3H]R214127 labeled a remarkably high number of mGlu1 receptors in the cerebellum. Compared with the number of sites labeled in the cerebellum, only ∼16% of the striatum and hippocampus were labeled, and only 11% was bound in the rat cortex. Importantly, incubation with 10 μM of the structurally unrelated compound BAY 36-7620 maximally inhibited [3H]R214127 binding (Fig. 10). The mGlu5 receptor selective compound MPEP (tested up to 30 μM) did not affect [3H]R214127 binding to rat cerebellar membranes, again showing the mGlu1 receptor selectivity of R214127.
Equilibrium binding constants of [3H]R214127 binding to membranes from rat cortex, hippocampus, striatum, and cerebellum

Inhibition of 2.5 nM [3H]R214127 binding to rat cerebellar membranes by BAY 36-7620. Data points represent percentage of total binding and are mean ± S.D. of two individual experiments.
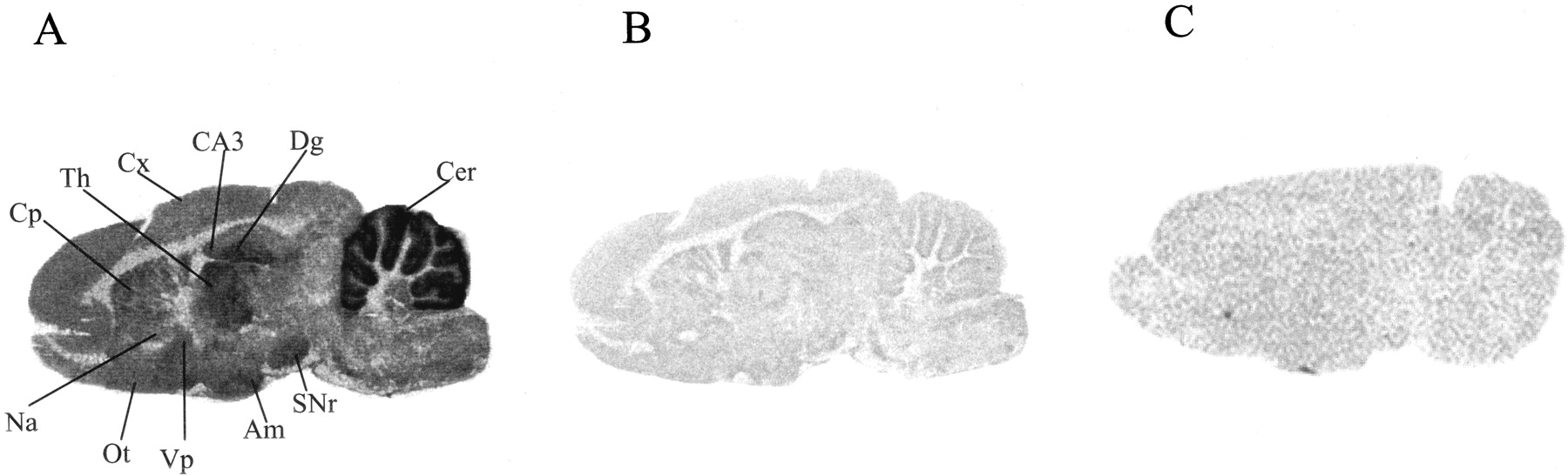
Using radioligand autoradiography, we examined [3H]R214127 binding distributions in rat brain sections in further detail (Fig. 11). [3H]R214127 autoradiography was investigated in sagittal rat brain sections; nonspecific binding was determined using R193845 (Fig. 11A). Very high specific binding was observed in the molecular layer of the cerebellum. A moderate signal was observed in the CA3 field and dentate gyrus of the hippocampal formation, thalamus, olfactory tubercle, amygdala, and substantia nigra reticulata. The cerebral cortex, caudate putamen, ventral pallidum, and nucleus accumbens showed lower labeling. In addition, incubation with BAY 36-7620 completely inhibited [3H]R214127 binding to rat brain sections (Fig. 11C).

[3H]R214127 binding to sagittal rat brain sections using autoradiography. A, representative section showing total binding with 1.5 nM [3H]R214127. B, representative and adjacent section showing nonspecific binding with 1.5 nM [3H]R214127 in the presence of 1 μM R193845. C, representative section showing nonspecific binding with 1.5 nM [3H]R214127 in the presence of 10 μM BAY 36-7620. Sections from A and B were exposed to [3H]Hyperfilm, whereas the section from C was exposed to a Fuji imaging plate. Th, thalamus; SNr, substantia nigra reticulata; CA3, CA3 region of the hippocampus; Dg, dentate gyrus of the hippocampus; Cer, cerebellum; Cp, caudate putamen; Cx, cerebral cortex; Ot, olfactory tubercle; Am, amygdala; Vp, ventral pallidum; Na, nucleus accumbens.
Discussion
Up to now, only a few mGlu1 receptor subtype-selective antagonists have been found. The mGlu1 receptor has been shown to be selectively blocked by CPCCOEt (Litschig et al., 1999) and BAY 36-7620 with potencies that vary from micromolar for CPCCOEt (6.6 μM) to high nanomolar for BAY 36-7620 (160 nM). In the present study, R214127 is identified as a novel mGlu1 receptor antagonist with low nanomolar functional antagonistic potency on the rat mGlu1a receptor (21.6 nM) and the human mGlu1a receptor (10.4 nM). The antagonist action of R214127 was found to be noncompetitive, because the maximal glutamate-induced mGlu1 receptor activation was decreased in the presence of R214127. The observed increase in glutamate EC50 in the presence of R214127 can be explained by the presence of spare receptors. In the presence of low concentrations of a noncompetitive antagonist, the concentration response curve will be shifted to the right because more agonist is needed to compensate for the ‘nonspare’ receptors that are blocked by the antagonist. These antagonist concentrations will not at first affect the maximal agonist response, but higher antagonist concentrations will eventually suppress the maximum response (Zhu, 1993). This phenomenon has also been reported for BAY 36-7620 (Carroll et al., 2001) and CPCCOEt (Hermans et al., 1998). Our data further show that R214127 may act as an inverse agonist toward the mGlu1a receptor and that R214127 acts selectively on the mGlu1 receptor with regard to other mGlu receptor subtypes and ionotropic glutamate receptors. Signal transduction data showed that R214127 does not display agonist, antagonist, or positive allosteric action on the mGlu2, -3, -4, -5, and -6 receptors, and radioligand binding studies revealed that [3H]R214127 does not bind to the mGlu2, -3, -4, -5, and -6 receptors, furthermore excluding the possibility that R214127 acts as a neutral ligand at any of these receptor types. The lack of selective mGlu1 receptor radioligands and the interesting pharmacological properties of R214127 were compelling reasons to label R214127 for the investigation of mGlu1 receptors in binding studies.
[3H]R214127 binding met all the requirements for a ligand very well suited to study binding properties, pharmacology, and distribution of mGlu1 receptors. First, [3H]R214127 binding studies were performed in rat mGlu1a receptor CHO-dhfr− membranes. Specific binding was very high and increased linearly with protein concentration (Fig. 3). Specific binding showed a modest increase in the presence of MgCl2 and CaCl2, whereas binding decreased by 22% at pH 6 and was unaffected by an increase in pH. Concerning the effects of pH on binding, it is worth noting the calculated physicochemical properties of R214127: calculated pKaand clogP are 6.2 and 4.5, respectively. At pH 7.4, the degree of ionization of R214127 is thus very low (only 5.9%). The percentage of ionization decreases further at higher pH (1.6% at pH 8, 0.2% at pH 9, and no protonation at pH 10). The clogD value remains 4.5 from pH 7.4 to 10. At pH 6, however, 61.3% of R214127 is in the protonated form. Accordingly, the clogD decreases to 4.1. The lower binding of the ligand in ionized form suggests that the nonionized ligand has the highest binding affinity. This is remarkable and is in contrast with findings for ligands for monoamine G protein-coupled receptors (e.g., the dopamine receptor), which are often strong bases and bind in a cationic form. For such compounds, the driving force for the binding to the receptor is electrostatic in nature (Van de Waterbeemd et al., 1986). Our data may indicate that ionic interactions are not a driving force in the binding to the receptor and that there is no contribution of ionic surface effects. Additionally, although R214127 is a strong lipophilic compound, the very low nonspecific [3H]R214127 binding might occur because no electrostatic interaction can take place between the nonionized form of R214127 and the negatively charged cell membrane. Binding was temperature dependent and increased substantially at 4°C (Fig. 4). By virtue of its fast association and dissociation kinetics, binding equilibrium was rapidly reached. [3H]R214127 labeled apparently a single population of sites with a very high affinity (KD = 0.90 ± 0.14 nM). In contrast, [3H]quisqualate, the mGlu1 receptor radioligand of choice up to now, exhibited a much higherKD value of 22.0 ± 10 nM, which correlated well with the value of 37 nM obtained by Mutel et al. (2000). Besides the considerable higher affinity, [3H]R214127 labeled significantly more (∼40%) binding sites than [3H]quisqualate.Bmax values of 6512 ± 1501 and 3912 ± 436 fmol/mg of protein were found for [3H]R214127 and [3H]quisqualate, respectively. This discrepancy can be explained based on the G protein coupling of the receptor. Agonists facilitate the coupling of the receptor to the G protein, which results in a receptor conformation with high affinity for agonists. According to this theory, a full agonist such as quisqualate would predominantly label the high-affinity or G protein-coupled receptor state. An antagonist would have equal affinity for coupled and uncoupled receptors, and thus for both the high- and low-affinity states of the receptor. Our finding that theBmax for [3H]R214127 is considerably higher than for [3H]quisqualate is in line with this theory.
A striking finding in this study was that the natural agonist glutamate and also quisqualate were unable to inhibit [3H]R214127 binding to rat mGlu1a receptor CHO-dhfr− membranes, whereas CPCCOEt, BAY 36-7620, NPS 2390, and R214127, known as noncompetitive antagonists, all inhibited [3H]R214127 binding to the same maximal level (Fig. 7). Inhibition of [3H]R214127 binding by the latter compounds followed sigmoidal curves with Hill coefficients of about 1.0 (Table1), which gave no indication for binding to multiple sites. It is important to mention that although a structurally related analog was used to define nonspecific binding, a similar low nonspecific binding was obtained with structurally unrelated compounds such as BAY 36-7620 when used at ≥1 μM in rat mGlu1a receptor CHO-dhfr− membranes (Fig. 7). For [3H]quisqualate, all the amino acid-like structures, known as competitive ligands, could displace [3H]quisqualate from its binding site. Inhibitory potencies of quisqualate, glutamate, LY367385, (S)-3,5-DHPG, (S)-4C3HPG, (1S,3R)-ACPD, (S)-4CPG, AIDA, and MCPG (Table 3) were in good agreement with the values reported by Mutel et al. (2000). In contrast, the above noncompetitive compounds did not affect its binding. For CPCCOEt, it has been reported that it does not affect [3H]glutamate binding to membranes prepared from rat mGlu1a receptor-expressing cells (Litschig et al., 1999). Furthermore, it has been suggested that rather than binding to the glutamate binding site, CPCCOEt interacts with Thr815 and Ala818 in transmembrane domain VII. CPCCOEt is proposed to interfere with receptor signaling by disrupting an intramolecular interaction between the glutamate-bound extracellular domain and the transmembrane domain VII. Carroll et al. (2001) demonstrated that BAY 36-7620 did not displace [3H]quisqualate from the glutamate binding pocket. Transmembrane helices 4 to 7 were shown to play a crucial role for binding of BAY 36-7620. Our inhibition experiments performed with [3H]R214127 and [3H]quisqualate suggest that CPCCOEt, BAY 36-7620, and NPS 2390 bind to the same site as R214127. Saturation experiments using [3H]R214127 in the absence and the presence of 30 μM CPCCOEt, 100 nM BAY 36-7620, and 10 nM NPS 2390 further support the idea that these compounds bind to the same or mutually exclusive sites. KD values significantly increased, whereas theBmax value was unaltered (Table 4). These results indicate that although the affinity of [3H]R214127 decreases, high concentrations of [3H]R214127 are still able to displace binding of the other compound from its binding site, which is a typical property of a competitive interaction. In conclusion, our data support the notion that CPCCOEt, BAY 36-7620, NPS 2390, and R214127 act on a site different from the glutamate binding pocket; presumably, they compete for the same transmembrane segment VII.
Previous group I mGlu receptor binding studies in brain were performed using [3H]glutamate or [3H]quisqualate (Schoepp and True, 1992; Wright et al., 1994; Mutel et al., 2000). These radioligands have the disadvantage of labeling more than one type of glutamate receptor. Therefore, selective inhibitors had to be added to the incubation buffer to prevent labeling to other metabotropic or ionotropic glutamate receptor subtypes. To date, there is no radioligand available to specifically study the binding and distribution of the mGlu1 receptor. The specific mGlu1 receptor labeling of [3H]R214127 makes it particularly useful for the investigation of native mGlu1 receptors in rat or human brain. Experiments using rat cortex, hippocampus, striatum, and cerebellum membranes revealed that [3H]R214127 specific binding, defined in the presence of 1 μM R193845, was high, especially in the cerebellum (only 10% nonspecific binding). Saturation experiments showed that [3H]R214127 again apparently labeled a single binding site with very high affinity.KD values of about 1 nM were found for all the different brain areas (Table 5). A striking difference inBmax values was found: a large population of binding sites was labeled in the cerebellum, whereas in hippocampus, striatum, and cortex, moderate to low levels of receptor expression were detected.
Because of its specificity, [3H]R214127 proved to be particularly suitable for investigation of mGlu1 receptor distribution in brain sections using radioligand autoradiography. mGlu1 receptor autoradiography revealed that the highest level of mGlu1 specific binding was present in the molecular layer of the cerebellum. The granule cell layer was very weakly labeled. These results were also found by Mutel et al. (2000), who investigated group I mGlu receptor distribution using [3H]quisqualate. In the hippocampal formation, the CA3 dendritic field and the molecular layer of the dentate gyrus showed abundant labeling. The CA1 area showed very weak [3H]R214127 binding, corresponding well with immunohistochemistry data from Lujan et al. (1996) andShigemoto et al. (1997), who showed that in CA1 dendritic fields, an antibody specific for the mGlu5 receptor, but not a specific mGlu1 receptor antibody, yielded intense immunolabeling. Autoradiography experiments using [3H]quisqualate indeed revealed staining in both the CA1 and CA3 regions of the hippocampus, indicating binding to both the mGlu1 and mGlu5 receptor, respectively (Mutel et al., 2000). [3H]R214127 binding was also quite high in the thalamus, olfactory tubercle, amygdala, and substantia nigra reticulata and was somewhat lower in the cerebral cortex, caudate putamen, nucleus accumbens, and ventral pallidum. The same structures were labeled using [3H]quisqualate (Mutel et al., 2000). In addition, immunocytochemical findings on the cellular localization of the mGlu1a receptor, using an antibody selective for the mGlu1a receptor, were generally consistent with our data (Martin et al., 1992). Because [3H]R214127 is expected to label all mGlu1 receptor splice variants known to date, the distribution of one splice variant may, however, differ from that of our radiolabel. For example, in the CA3 region and the caudate putamen, which are labeled by the radioligand, mGlu1b receptor but little or no mGlu1a receptor immunoreactivity was found (Martin et al., 1992; Shigemoto et al., 1997; Ferraguti et al., 1998). An important point in the demonstration of the identity of the [3H]R214127-labeled sites was the finding that the structurally different compound BAY 36-7620 also fully displaced [3H]R214127 binding to rat brain membranes (Fig. 10) and to brain sections (Fig. 11), providing a good guarantee that the inhibited binding is purely receptor specific and not related to a structural moiety of the radioligand.
In this article, we have shown that [3H]R214127 is an excellent radioligand to study mGlu1 receptors in an heterologous expression system, rat brain homogenates, and brain sections. We can conclude that because of its minimal nonspecific binding, high binding affinity, and marked selectivity, [3H]R214127 is the ligand of choice for further exploration of the mGlu1 receptor. [3H]R214127 opens perspectives for a detailed investigation of subcellular and cellular localization of the mGlu1 receptor and for the study of the functional role and regulation of the receptor in various areas.
Acknowledgments
We thank Liesbeth Lenaerts, Ilse Biesmans, and Luc Peeters for helpful advice during the experiments. We also thank David Ashton for consistent support and Luc Bijnens for help in statistical analysis.
Footnotes
- Abbreviations:
- mGlu
- metabotropic glutamate
- CHO-dhfr− cells
- dihydrofolate reductase-deficient Chinese hamster ovary cells
- R214127
- 1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone
- R193845
- 2-amino-3-ethyl-6-quinolinyl)(4-methoxycyclohexyl)methanone
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- MK-801
- (5R,10S)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine
- CGP39653
- dl-(E)-2-amino-4-propyl-5-phosphono-3-pentenoic acid
- LY341495
- (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)-glycine
- MPEP
- 2-methyl-6-(phenylethynyl)pyridine
- (S)-4C3HPG
- (S)-4-carboxy-3-hydroxyphenylglycine
- (1S,3R)-ACPD
- (1S,3R)-1-aminocyclopentane-trans-1,3-dicarboxylic acid
- (S)-3,5-DHPG
- (S)-3,5-dihydroxyphenylglycine
- (S)-4CPG
- (S)-4-carboxyphenylglycine
- AIDA
- (R,S)-1-aminoindan-1,5 dicarboxylic acid
- MCPG
- (S)-α-methyl-4-carboxyphenylglycine
- CPCCOEt
- 7-(hydroxyimino)cyclo-propa[b]chromen-1a-carboxylate ethyl ester
- BAY 36-7620
- (3aS,6aS)-6a-naphtalan-2-ylmethyl-5-methyliden-hexahydro-cyclopenta[c]furan-1-on
- NPS 2390
- 2-quinoxaline-carboxamide-N-adamantan-1-yl
- IP
- inositol phosphate
- Ro 48-8587
- 9-imidazol-1-yl-8-nitro-2,3,5,6-tetrahydro[1,2,4]triazolo[1,5-c]quinazoline-2,5-dione
- L689560
- trans-2-carboxy-5,7-dichloro-4-phenylaminocarbonyl amino-1,2,3,4-tetrahydroquinoline
- LY367385
- (+)-2-methyl-4-carboxyphenylglycine
- Ka
- acid dissociation constant
- P
- partition coefficient
- D
- partition coefficient at particular pH
- Received October 21, 2002.
- Accepted February 4, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}