


Abstract
The constitutive active receptor (CAR) in mouse primary hepatocytes undergoes okadaic acid (OA)-sensitive nuclear translocation after activation by xenobiotics such as phenobarbital (PB) and 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP). We have now mimicked this TCPOBOP-dependent and OA-sensitive translocation of mouse CAR (mCAR) in HepG2 cells and have demonstrated that protein phosphatase 2A regulates this nuclear translocation. Site-directed mutagenesis analysis of various Ser and Thr residues delineated the translocation activity to Ser-202. Mutation of Ser-202 to Asp (S202D) prevented mCAR translocation into the nucleus of TCPOBOP-treated HepG2 cells. In addition, in the livers of Car-/- mice, the YFP-tagged S202D mutant did not translocate into the nucleus after PB treatment. To examine whether Ser-202 can be phosphorylated, flag-tagged wild-type mCAR or flag-tagged S202A mutant was expressed in HepG2 cells and subjected to Western blot analysis using an antibody specific to a peptide containing phospho-Ser-202. A high molecular weight phosphorylated form of CAR was detected only with the wild-type mCAR. These results are consistent with the conclusion that the dephosphorylation of Ser-202 is a required step that regulates the xenobiotic-dependent nuclear translocation of mCAR.
Upon activation by PB and various PB-type inducers, CAR up-regulates the expression of a large set of hepatic genes that encode drug metabolizing enzymes and transporters, such as cytochrome P450 and multidrug-resistant protein (Sueyoshi and Negishi, 2001; Kast et al., 2002; Ueda et al., 2002). Thus, CAR coordinates a cellular defense mechanism elicited against xenobiotic insults by increasing hepatic capability to metabolize and excrete them. In addition to its role in drug metabolism, CAR is now implicated as a coordinating factor in regulating various types of hepatic functions, such as gluconeogenesis, fatty acid oxidation and the metabolism of steroid hormones and bilirubin (Wei et al., 2000; Sugatani et al., 2001; Ueda et al., 2002; Huang et al., 2003). For example, CAR cross-talks with the insulin response transcription factor FOXO1 to repress the genes that encode gluconeogenic enzymes, such as glucose-6-phosphatase and phosphoenolpyruvate carboxykinase 1 (Kodama et al., 2004). CAR can also be an adverse factor; Yamamoto et al. (2004) showed that CAR promotes the development of hepatocellular carcinoma in PB-treated mice. Although the diverse role of CAR in regulating various liver functions is becoming evident, the molecular mechanism of CAR activation remains elusive. CAR is unique in that it can be activated by many drugs, such as PB, without binding directly to them. The presence of a signal transduction pathway has been suggested to regulate this so-called “ligand-independent” or “indirect” activation. However, phosphorylated CAR as a direct target of the signal pathway has not yet been demonstrated.
CAR is retained in the cytoplasm of noninduced mouse hepatocytes and translocates into the nucleus after activation by drugs such as PB and 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) (Kawamoto et al., 1999). This nuclear translocation is the initial step that occurs during the process of CAR activation, providing an excellent target for investigations into whether a signaling pathway is involved in this activation. Our previous studies showed that the xenobiotic response signal, a peptide near the C terminus of the LBD, regulates PB-induced nuclear translocation of mCAR in mouse hepatocytes (Zelko et al., 2001). We have shown that a protein phosphatase inhibitor okadaic acid (OA) represses the nuclear translocation of mCAR (Kawamoto et al., 1999). Because CAR forms a complex with Hsp90 in the cytoplasm and recruits protein phosphatase 2A (PP2A) in response to PB (Yoshinari et al., 2003), it has been speculated that PB treatment may dephosphorylate the LBD to translocate CAR into the nucleus.
Unlike in mouse hepatocytes (Kawamoto et al., 1999), CAR diffuses passively into the nucleus when it is expressed in transformed cells such as HepG2. In particular, stable HepG2 cell lines, such as Ym17, express CAR predominantly in the nucleus (Swales et al., 2005). Thus, when CAR is cotransfected with a reporter gene in HepG2 cells, it fully activates the transcription of reporter gene in the absence of CAR activator such as TCPOBOP. This has discouraged us from investigating CAR nuclear translocation at the molecular level using HepG2 cells. However, we have observed that CAR first appears in the cytosol of transiently transfected HepG2 cells before the receptor spontaneously accumulates in the nucleus. We have now investigated the mechanism of CAR nuclear translocation using transient transfection conditions under which HepG2 cells mimic the TCPOBOP-dependent and OA-sensitive CAR nuclear accumulation. With this HepG2 system, PP2A regulates the nuclear translocation of CAR. We then focused on serine and threonine residues of the LBD to identify the phosphorylation site that might regulate the nuclear translocation of CAR. To this end, we took advantage of our recent discoveries that the mouse pregnane X receptor (mPXR) translocates into the nucleus of mouse hepatocytes in response to its specific activator 5-pregnen-3β-ol-20-one-16α-carbonitrile (PCN) but not to PB, the CAR activator (Squires et al., 2004). However, when the LBD of mPXR is placed after the DBD of mCAR, the chimeric receptor is found to respond to PB, translocating into the nucleus of mouse livers. If the PB-induced signal and PP2A target a residue of mCAR, that residue may be a conserved serine or threonine in the LBD of both mCAR and mPXR. Considering these findings, we employed site-directed mutagenesis to delineate the residue that regulates the nuclear translocation of mCAR in HepG2 cells as well as mouse hepatocytes. Herein, we present our experimental observations, which are consistent with the conclusion that the dephosphorylation of Ser-202 is a required step in the nuclear translocation of mCAR.
Materials and Methods
Materials. TCPOBOP was obtained from Sigma (St. Louis, MO), okadaic acid from Calbiochem (San Diego, CA), and Complete mini (protease inhibitor cocktail tablets) from Roche Diagnostics GmbH (Indianapolis, IN). Anti-V5 and Anti-V5 HRP conjugate antibodies were purchased from Invitrogen (Carlsbad, CA), anti-Flag HRP and anti-Flag M2-agarose from Sigma, and anti-hsp90 monoclonal antibody from BD Transduction Laboratories (Lexington, KY). Antibody against the peptide MEDQIpSLLKGC (corresponding to residues 197–206 of mCAR with Ser-202 phosphorylated) was produced at Affinity Bioreagents (Golden, CO). The specificity of this antibody (named pS202) to phosphorylated Ser-202 was confirmed by enzyme-linked immunosorbent assay using phosphorylated and nonphosphorylated peptides, specifically binding to the phosphorylated peptide even at the concentration 20 μg/ml. In addition, Western blot analysis was performed and showed that pS202 antibody does not bind bacterially expressed GST-mCAR.
Plasmids. mCAR in pcDNA3.1/V5-His, pEYFP-c1, or pGEX4T-3; hCAR in pEYFP-c1; and (NR1)5-tk-luciferase reporter were constructed previously (Sueyoshi et al., 1999; Zelko et al., 2001; Kobayashi et al., 2003). The full coding sequence of hCAR or hGRIP1 was amplified using appropriate primers and cloned into pcDNA3.1/V5-His-TOPO (Invitrogen). Catalytic subunit of human PP2A (hPP2Ac) was cloned into pcDNA3.1/V5-His-TOPO (Invitrogen). Flag-tag was added to the 3′-end of mCAR cDNA and cloned into pCR3 (Invitrogen). Using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and appropriately mutated primers, mutations were introduced and verified by nucleotide sequencing. A chimera, mCARDBD-Hinge-mPXRLBD, was constructed bearing the mCAR DNA binding domain (1–116 of mCAR) and mPXR ligand binding domain (141–431 of mPXR) in pEYFP-c1.
Cell Culture and Fractionation. HepG2 cells were cultured in minimal essential medium supplemented with 10% fetal bovine serum and antibiotics (100 unit/ml of penicillin and 100 μg/ml of streptomycin) on plastic dishes at 37°C for 24 h before transfection. At approximately 75% confluence, the cells were transfected with plasmid using LipofectAMINE 2000 (Invitrogen) according to a manufacturer's instruction. Cells were scraped from the plates, washed twice with phosphate-buffered saline, collected by centrifugation and homogenized in buffer A (10 mM HEPES buffer, pH 7.6, containing 10 mM KCl, 1.5 mM MgCl2, 20 mM Na2MoO4, 0.3% Nonidet P-40, and 1 mM dithiothreitol) including Complete protease inhibitor cocktail tablets (Roche). The homogenate was centrifuged at 4000g for 10 min, and the resulting supernatant was centrifuged at 17,800g for 10 min to obtain the cytosolic fraction. The 4000g pellet was washed three times with buffer A and suspended in lysis buffer (10 mM HEPES buffer, pH 7.6, containing 0.1 M KCl, 3 mM MgCl2, 0.1 mM EDTA, 1 mM Na3VO4, 10% glycerol, and Complete protease inhibitor cocktail). NaCl was added to a final concentration of 0.4 M, and suspension was incubated with constant rotation at 4°C for 1 h and centrifuged at 100,000g for 30 min to obtain a nuclear extract.
Immunoprecipitation. Anti-Flag M2-agarose (40 μl) was added to the cytosol (1–2 mg of protein) isolated in 10 mM sodium phosphate, pH 7.4, containing 0.14 M NaCl, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM NaF, 20 mM Na2MoO4, 0.3% nonidet P-40, and 1 mM dithiothreitol and incubated at 4°C for 16 h. After centrifugation (800g for 2 min), the resulting pellet was washed five times with 1 ml of 50 mM Tris-HCl, pH 7.5, containing 150 mM NaCl, 20 mM Na2MoO4, and 0.2% Nonidet P-40. The remaining agarose was boiled for 10 min in Nu-PAGE LDS sample buffer (Invitrogen) to extract the proteins.
Expression of YFP-Tagged CAR in Mouse Liver. Plasmid was injected via the tail vein of CD-1 or CAR-null mice using the TransIT in vivo gene delivery system (Mirus, Madison, WI) according to the manufacturer's instructions. By this method, generally approximately 10% of hepatocytes of the liver showed expression of YFP-CAR. The mice were injected with PB (100 mg/kg body weight) or DMSO 3 and 6 h after the injection of plasmid and sacrificed 2 h after the second drug administration. Liver sections (30 μm) were prepared and analyzed by confocal laser scanning microscopy using a Zeiss LSM510 system. A 63× oil immersion objective (1.4 numeric aperture) was used for scanning with a pixel size of 0.06 μm. YFP was excited at 514 nm and the emission was detected using a 530-nm band-pass filter. Pinhole size was set for a 1.0-μm confocal slice. At least 100 cells from four different sections were examined for each treatment to determine the pattern of intracellular localization of the receptor, either predominantly cytoplasmic, present in equal amounts in the nuclear and cytoplasmic compartments, or predominantly nuclear.
Western Blot. Proteins were separated on a 4 to 12% Nu-PAGE Novex Bis-Tris Gel (Invitrogen) or a 10% SDS-polyacrylamide gel and were transferred to Immobilon-P membrane. The membrane was blocked in 5% nonfat milk powder in 25 mM Tris-HCl, pH 7.5, containing 137 mM NaCl and 0.1% Tween 20 (TBS-Tween). When anti-pS202 was used as primary antibody, 5% bovine serum albumin in TBS-Tween was used to block membrane. The blocked membranes were incubated for 1 h at 25°C with primary antibodies; either anti-V5 (1:5000), anti-V5 HRP (1:5000), anti-Flag HRP (1:5000), or anti-pS202 antibody (0.5 μg/ml in TBS-Tween). Horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA) was used as the secondary antibody. Finally, protein bands were visualized using ECL Plus Western blotting detection reagent (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). Blots were subsequently stripped and restained for Lamin β and Hsp90 as a loading control.
Luciferase Reporter Assay. Luciferase activity was measured in cell lysates using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) as described previously (Sueyoshi et al., 1999). Promoter activity was determined in HepG2 cells from three independent transfections and calculated from firefly luciferase activity normalized against Renilla reniformis luciferase activity of an internal control pRL-SV40 plasmid.
GST Pull-Down Assay. Glutathione-S-transferase (GST)-mCAR, mCARS202A, and mCARS202D fusion proteins were expressed in Escherichia coli BL21 cells and purified using glutathione-Sepharose (Amersham Biosciences). [35S]Methionine-labeled hGRIP1 was expressed using the TnT T7 coupled reticulocyte lysate system (Promega) using pcDNA3.1/hGRIP1-V5-His. In vitro translated 35S-labeled hGRIP was mixed with GST-mCAR or the mCAR mutants in the presence of glutathione Sepharose in 50 mM HEPES buffer, pH 7.6, containing 0.1 M NaCl and 0.1% Triton X-100. The mixture was incubated for 20 min at room temperature under gentle mixing; the resin was recovered by centrifugation and washed four times with HEPES buffer, pH 7.6, containing 0.1 M NaCl and 0.1% Triton X-100. Thereafter, proteins were extracted in SDS-PAGE sample buffer and electrophoresed on a 4 to 12% gradient acrylamide Bis-Tris gel (Invitrogen). The gel was dried and subjected to autoradiography.
Gel Shift Assay. Proteins were produced using the in vitro transcription/translation system (TnT T7 quick-coupled system; Promega) and incubated with 32P-labeled NR1 double strand DNA (40,000 cpm) probe in 10 μl of binding buffer containing 10 mM HEPES, pH 7.6, containing 0.5 mM dithiothreitol, 10% glycerol, 0.05% Nonidet P-40, 50 mM NaCl, and 1.5 μg of poly(dI-dC). The proteins were separated on a 5% acrylamide gel in 7 mM Tris-acetate buffer, pH 7.5, containing 1 mM EDTA at 180 V for 1.5 h, and the gel was vacuum-dried and subjected to autoradiography at -70°C.
Results
Delineating Translocation Activity of mCAR to Ser-202. It is known that mPXR translocates into the nucleus after treatment with its activator PCN but not with PB, the mCAR activator in mouse livers (Squires et al., 2004). A strong nuclear localization signal within the DBD dictates this nuclear translocation of mPXR. Although a peptide sequence similar to the nuclear localization signal is present in the DBD of mCAR, this sequence does not regulate PB-induced nuclear translocation of mCAR (Zelko et al., 2001; Squires et al., 2004). Given these facts, we hypothesized that the LBD of mPXR might respond to PB, triggering nuclear translocation within the context of mCAR. To this end, the mPXR LBD was placed at after the DBD-hinge region of mCAR to construct the expression plasmid of YFP-tagged chimera mCARDBD-Hinge-mPXRLBD. This chimera was directly expressed in the livers of CD-1 mice by injecting the plasmid via the tail vein. Then, its intracellular localization was determined and compared with those of mCAR and mPXR. Fluorescent images of liver cells that signify the expression of a given receptor are shown in Fig. 1A. Quantitative analysis of the intracellular localization for a given receptor from at least 100 cells is summarized in Fig. 1B. As expected, all three receptors were predominantly expressed in the cytoplasm of noninduced hepatocytes. Similar to mCAR, the chimera was expressed in the cytoplasm of more than 80% of the cells analyzed, whereas more than 60% of the cells that expressed mPXR showed the specific expression in the cytoplasm. Similar to mCAR, chimeras were expressed in the nuclei of nearly 90% of the PB-treated hepatocytes. On the other hand, mPXR remained expressed predominantly in the cytoplasm even after PB treatment. Thus, the LBD of mPXR acquires the activity of PB-induced nuclear translocation when it is placed in the context of the mCAR DBD.

Intracellular localization of mCAR, mPXR and chimera in mouse livers. A, mCAR, mPXR and a chimera molecule between them (mCARDBD-Hinge-mPXRLBD) as YFP fusion proteins were expressed in the CD-1 mice livers by tail-vein injection of each expression plasmid. Mice were either untreated or treated with PB. Liver sections were prepared and examined by microscopy for YFP expression (in yellow) and Hoechest S33258 staining for nuclei (in blue). Two representative images are shown for each treatment group. B, nucleocytoplasmic distribution of the YFP fusion proteins shown in A was categorized into three groups. Mice were either untreated (-) or treated (+) with PB. The percentage of cells with the YFP fusion proteins expressed predominantly in the cytoplasm (Cyt, □), equally in the cytoplasm and the nucleus (Cyt and Nuc,  ), and predominantly in nucleus (Nuc, □) were determined using a fluorescent microscope. At least 100 cells expressing the fusion proteins were examined for each treatment.
), and predominantly in nucleus (Nuc, □) were determined using a fluorescent microscope. At least 100 cells expressing the fusion proteins were examined for each treatment.
If an OA-sensitive phosphatase dephosphorylates serine and/or threonine of mCAR to regulate the PB-induced nuclear translocation, this residue may be conserved in the LBD of mPXR. Amino acid sequence alignments of mCAR, mPXR, and human CAR identified four serine and three threonine residues that are found in all three LBDs: Thr-176, Ser-202, Thr-218, Thr-224, Ser-279, Ser-331, and Ser-358 in mCAR (Fig. 2A). Thr-176 was identified previously as one of the conserved residues in these receptors (Ueda et al., 2005). The first six residues were mutated to Asp to mimic phosphorylation and examine whether the mutation altered the intracellular localization in mouse liver. The last residue, Ser-358, which resides within the AF2 domain of mCAR, was excluded from investigations, because it is known that the AF2 is not involved in the PB-induced nuclear translocation (Zelko et al., 2001). Each YFP-tagged mCAR mutant was expressed in the livers of Car-/- mice, and intracellular localization of the expressed protein was evaluated by analyzing at least 100 hepatocytes (Fig. 2B). All mutants translocated into the nucleus after PB treatment, except S202D, as indicated by the fact that more than 80% cytoplasmic localization was reduced to 30%, whereas the nuclear localization was up from less than 10% to more than 50% after PB treatment. In contrast, the S202D mutant expressed in the cytoplasm of nearly 100% hepatocytes analyzed before PB treatment and remained unchanged even after PB treatment.
To provide further evidence that the phosphorylation of Ser-202 might be involved in regulating the nuclear translocation of the mCAR, the expression of the S202R and S202A mutants was compared with that of S202D mutant in the mouse liver (Fig. 3). Similar to the case of wild-type mCAR, the expression of S202R and S202A in the nucleus increased from 10 to 60% of cells after PB treatment, with the number of cells showing cytoplasmic expression decreasing from approximately 70% to less than 20%. Thus, placing a hydrophobic or positively charged side chain at position 202 did not change the ability of mCAR to translocate into the nucleus after PB treatment. As observed in Fig. 2, the S202D mutant was retained in the cytoplasm in 80% of the cells examined, even after PB treatment. When Asp with a negatively charged side chain substituted Ser-202, mCAR became unable to translocate into the nucleus. These results suggested that Ser-202 regulates the PB-induced nuclear translocation in the mouse livers, probably via its dephosphorylation.

Intracellular localization of various Asp mutants of mCAR in mouse livers. A, conserved Ser or Thr residues are indicated in color. B, wild-type mCAR and the mutants (mCART176D, mCARS202D, mCART218D, mCART224D, mCARS279D, and mCARS331D) were expressed as YFP fusion proteins in Car-/- mouse livers by injecting the expression plasmids via the tail vein. Mice were either untreated (-) or treated (+) with PB, and the cells expressing the YFP fusion proteins were categorized as in Fig. 2B. The percentages of cells with the YFP fusion proteins expressed predominantly in the cytoplasm (Cyt, □), equally in the cytoplasm and the nucleus (Cyt and Nuc,  ), and predominantly in nucleus (Nuc, □) were determined. At least 100 cells expressing the fusion proteins were examined for each treatment.
), and predominantly in nucleus (Nuc, □) were determined. At least 100 cells expressing the fusion proteins were examined for each treatment.
Functional Conservation of Ser-202. Ser-202 of mCAR is also conserved in hCAR and mPXR (Fig. 2A). Given the fact that Ser-202 regulated the PB-triggering nuclear translocation of mCAR, the corresponding Ser residues of hCAR and mPXR were examined as to whether these could also regulate their nuclear translocation. Ser-192 of YFP-tagged hCAR was mutated to Asp and Ala, and the mutants were directly expressed in mouse livers. Similar to wild-type hCAR, the S192A mutant translocated into the nucleus after PB treatment. On the other hand, the S192D mutant remained in the cytoplasm even after PB treatment (Fig. 4A). Ser-271 of mPXR was also mutated to Asp and Ala in the mCARDBD-Hinge-mPXRLBD chimera and expressed in mouse livers. Similar to the hCAR mutants, S271D did not translocate into the nucleus in the PB-treated mouse livers (Fig. 4B). Thus, these results suggest that the Ser residues of mCAR, hCAR, and mPXR (in the mCARDBD-Hinge-mPXRLBD chimera) are functionally conserved to regulate the nuclear translocation of the receptor. Although mPXR is known to translocate into the nucleus of PCN-treated mouse liver (Squires et al., 2004), it remains an interesting question whether Ser-271 regulates the PCN triggering nuclear translocation of mPXR.
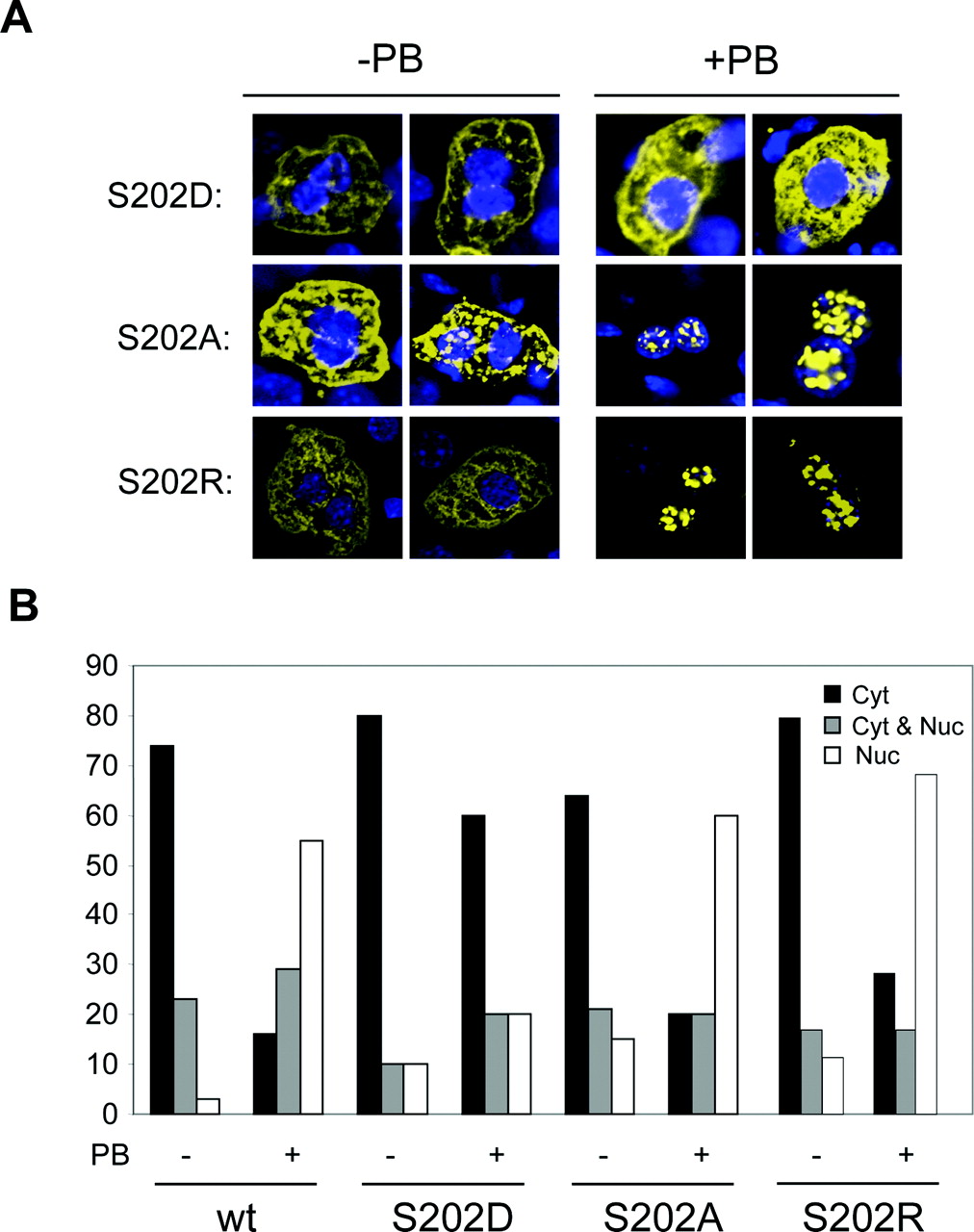
Mutational analysis of Ser-202 for mCAR nuclear translocation in mouse livers. A, mCAR, mCARS202D, mCARS202A and mCARSS202R were expressed as YFP fusion proteins in Car-/- mouse livers by injecting the expression plasmids via the tail vein. Mice were either untreated (-) or treated (+) with PB. Liver sections were prepared and examined by microscopy for YFP expression (in yellow) and Hoechst S33258 staining for nuclei (in blue). Two representative images are shown for each treatment group. B, nucleocytoplasmic distribution of the YFP fusion proteins shown in A was categorized into three groups. Mice were either untreated (-) or treated (+) with PB. The percentages of cells with the YFP fusion proteins expressed predominantly in the cytoplasm (Cyt, □), equally in the cytoplasm and the nucleus (Cyt and Nuc,  ), and predominantly in nucleus (Nuc, □) were determined. At least 100 cells expressing the fusion proteins were examined for each treatment.
), and predominantly in nucleus (Nuc, □) were determined. At least 100 cells expressing the fusion proteins were examined for each treatment.
Exogenously Expressed PP2A-Regulated Nuclear Accumulation of mCAR in HepG2 Cells. V5-tagged mCAR was transiently expressed in HepG2 cells, from which cytosolic and nuclear fractions were prepared. Thereafter, Western blotting analysis was performed to examine amount of mCAR in those fractions. PB does not bind directly to CAR and exhibited no effect on the intracellular localization of CAR in HepG2 cells; therefore, TCPOBOP was used to activate the receptor. TCPOBOP treatment decreased the amount of mCAR in the cytosolic faction and increased it in the nuclear faction at 24 or 45 h after treatment (Fig. 5A). The amount of mCAR was higher in the cytosolic faction than the nuclear fraction of DMSO-treated cells, whereas TCPOBOP treatment reversed this distribution. Thus, mCAR undergoes TCPOBOP-dependent nuclear translocation in the transfected HepG2 cells. Cotreatment with the active inhibitor OA, but not with the inactive inhibitor norokadaone, dramatically increased the level of mCAR in the cytosolic fraction (Fig. 5B). These results suggested that HepG2 cells retain a degree of capability in mimicking the regulated nuclear translocation of mCAR, as observed previously in mouse hepatocytes (Kawamoto et al., 1999). To examine whether PP2A could regulate the nuclear translocation of mCAR in HepG2 cells, V5-tagged catalytic subunit of human PP2A was coexpressed with V5-tagged mCAR (Fig. 5C). Both hPP2Ac and mCAR were detected simultaneously in Western blots of the cytosolic fraction. Similar to TCPOBOP, hPP2Ac decreased mCAR in the cytosolic fraction and increased it in the nuclear fractions. These results indicated that PP2A stimulates the nuclear translocation of mCAR in HepG2 cells.

Regulatory function of Ser-202 is conserved in hCAR and mPXR-CAR chimera. A, nucleocytoplasmic distribution of YFP-hCAR and its mutants in CAR null mouse livers in vivo. Plasmids encoding hCAR and its mutants as YFP fusion proteins were injected via the tail vein and cell counting of liver sections were carried out as described in Fig. 2B. Mice were either untreated (-) or treated (+) with PB. The percentages of cells with the YFP fusion proteins expressed predominantly in the cytoplasm (Cyt, □), equally in the cytoplasm and the nucleus (Cyt and Nuc,  ), and predominantly in nucleus (Nuc, □) were determined using a fluorescent microscope. At least 100 cells expressing the fusion proteins were examined for each treatment. B, nucleocytoplasmic distribution of YFP-mCARDBD-Hinge-mPXRLBD Ser 271 mutant in CAR-null mouse livers in vivo. Experiment was performed as in A using the mutant expression plasmid and protein localization in liver cells were categorized.
), and predominantly in nucleus (Nuc, □) were determined using a fluorescent microscope. At least 100 cells expressing the fusion proteins were examined for each treatment. B, nucleocytoplasmic distribution of YFP-mCARDBD-Hinge-mPXRLBD Ser 271 mutant in CAR-null mouse livers in vivo. Experiment was performed as in A using the mutant expression plasmid and protein localization in liver cells were categorized.
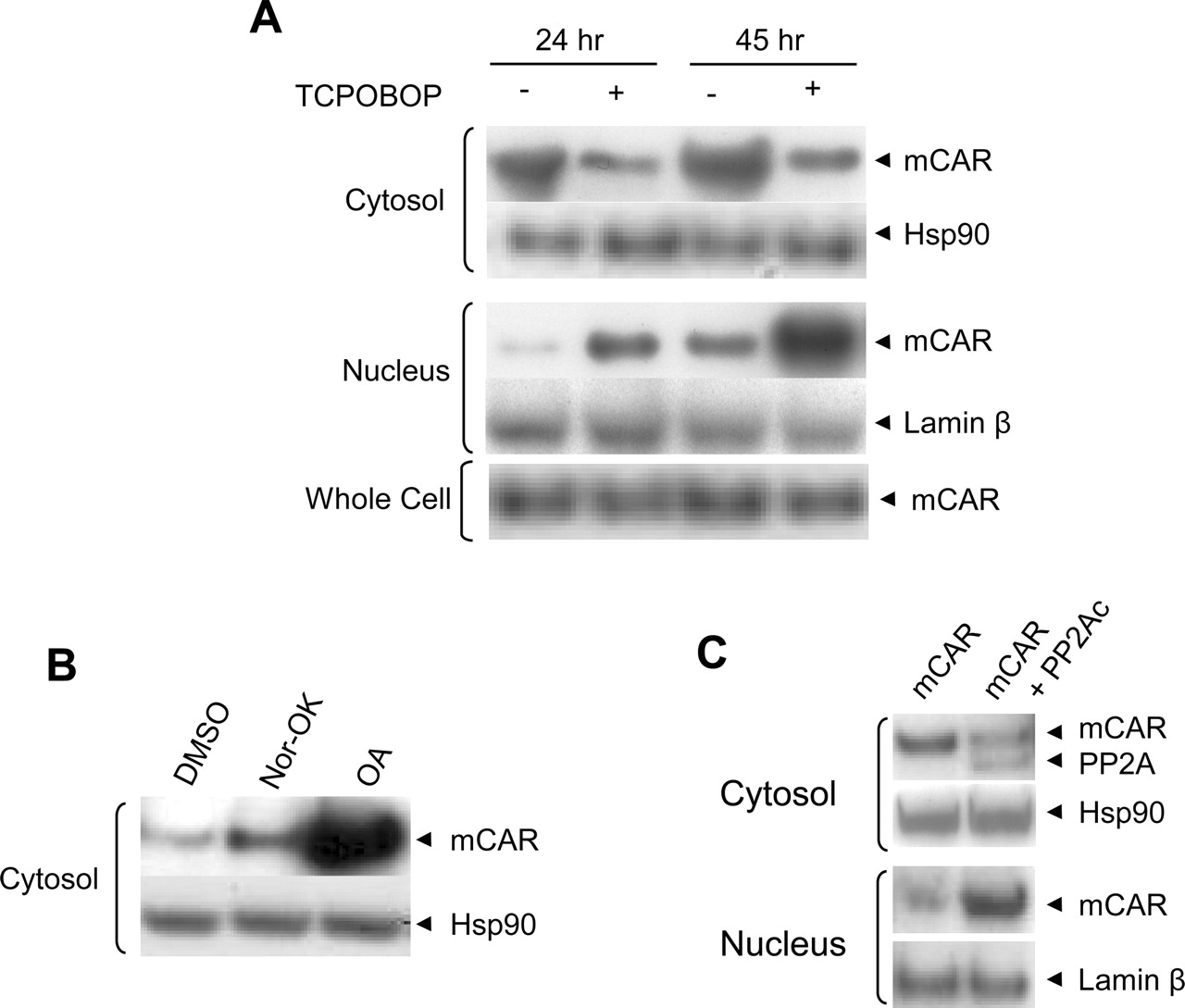
mCAR nuclear translocation in HepG2 cells. A, HepG2 cells were transfected with pcDNA3.1/V5-His-mCAR and treated with 250 nM TCPOBOP 3 and 24 h after transfection so that treatment periods are 24 and 45 h, respectively. Cells were harvested 48 h after transfection for cell fractionation. Western blotting detection was performed for cytosolic, nuclear, and whole-cell extract proteins (30, 15, and 5 μg, respectively), after electrophoresis on 10% SDS-PAGE gels using primary antibodies, anti-V5 for mCAR, anti-hsp90, and anti-laminβ. B, HepG2 cells were transfected with pcDNA3.1/V5-His-mCAR and cells were treated with 10 nM nor-okadaone (Nor-OK) or OA along with vehicle DMSO for 21 h. Cells were harvested 24 h after transfection and cytosol extracts were prepared. Western blotting detection of mCAR was performed with 30 μg of cytosolic proteins, after separation on a 10% SDS-PAGE gel using anti-V5 antibody. C, HepG2 cells were cotransfected with pcDNA3.1/V5-His-mCAR and pcDNA3.1/V5-His-hPP2A and cells were harvested after 24 h. Cytosols (40 μg) and nuclear extracts (20 μg) were subjected to Western blot analysis with anti-V5 antibody for mCAR and hPP2A, anti-hsp90, and anti-laminβ.

Nuclear translocation and trans-activation of mCAR mutants in HepG2 cells. A, HepG2 cells were transfected with pcDNA3.1/V5-His bearing mCAR, mCARS202A, or mCARS202D and 3 h after transfection cell were either treated with 250 nM TCPOBOP (+) or vehicle DMSO (-). Cytosolic (30 μg) and nuclear (10 μg) proteins were prepared 24 h after transfection and subjected to Western blotting analysis as in Fig. 1. B, pcDNA3.1/V5-His bearing mCAR, mCARS202A, or mCARS202D were cotransfected with (NR1)5-tk-luciferase plasmid and pRL-SV40 into HepG2 cells. Twenty-four hours after transfection, cells were treated with 250 nM TCPOBOP or vehicle (DMSO) alone and subjected to luciferase assay 24 h later. The luciferase activity level is indicated as an average from three wells with standard deviation. C, DNA binding activity of the mCAR Ser 202 mutants. mCAR and its mutants were prepared by in vitro translation and mixed with similarly prepared RXR for gel shift assays using 32P-labeled NR1 oligonucleotide as a probe. Mock-translated mCAR was used as the control.
Ser-202-Regulated Nuclear Translocation of mCAR in HepG2 Cells. To examine whether Ser-202 could regulate the TCPOBOP-dependent nuclear translocation, the V5-tagged S202D and S202A mutants were expressed in HepG2 cells in the presence or absence of TCPOBOP (Fig. 6A). The S202A mutant was predominantly expressed in the nuclear fraction in the absence of TCPOBOP and was unaffected after TCPOBOP treatment. On the other hand, the S202D mutant was preferentially expressed and retained in the cytosolic fraction even after treatment with TCPOBOP. The lack of nuclear translocation of the S202D mutant affected its trans-activation of the NR1 reporter construct in the HepG2 cells (Fig. 6B). Because V5 is tagged at the C terminus of mCAR in the constructs used, the tagged receptor exhibits low constitutive activity that can effectively be activated by TCPOBOP in HepG2 cells (Swales et al., 2005). Therefore, all three receptors were similarly activated by TCPOBOP by approximately 20-fold (Fig. 6B). The same activation rate suggested that Ser-202 is not directly involved in the transactivation function of mCAR in the nucleus. Every time, all three effectively formed a complex with NR1 in gel-shift assays (Fig. 6C). However, the basal as well as TCPOBOP-activated activities of the S202D mutant were approximately 50% lower than wild-type mCAR and the S202A mutant (Fig. 6B). These results indicated that phosphorylation of Ser-202 may also be a factor regulating the nuclear translocation in the HepG2 cells.

Effect of hGRIP1 on the function of mCAR in HepG2 cells. A, hGRIP1 shows no effect on localization of mCAR and its mutants in HepG2 cells. pcDNA3.1/V5-His bearing mCAR, mCARS202D, or mCARS202A were cotransfected with hGRIP1 expression vector (+) or empty pcDNA3.1 (-) in HepG2 cells. Cells were harvested, and Western blotting was performed as in Fig. 1A. B, interaction of mCAR and mCAR mutants with hGRIP1 in GST pull down assays. In vitro-translated 35S-labeled hGRIP1 was incubated with GST-mCAR, GST-mCARS202A or GST-mCARS202D fusion protein on glutathione resin in the presence of DMSO (-) or TCPOBOP (+). After washing the resin, proteins were extracted and separated on a 10% SDS-PAGE gel. Five percent of the lysate used in the binding reactions was used as input. Bound proteins were detected by autoradiography. C, hGRIP1 coactivation of mCAR and S202 mutants in HepG2 cells. hGRIP1 was coexpressed with mCAR, mCARS202A, or mCARS202D in HepG2 cells along with (NR1)5-tk-luciferase reporter. Twenty-four hours after transfection, cells were treated with 250 nM TCPOBOP or vehicle alone and subjected to luciferase assay 24 h later. The luciferase activity level is indicated as an average from three wells with standard deviation.
A coregulator GRIP1 was recently reported to mediate the nuclear translocation of CAR in both mouse livers and HepG2 cells (Xia and Kemper, 2004). Experiments were performed to investigate whether the mutation of Ser-202 affected the GRIP1-dependent nuclear translocation of mCAR. To this end, human GRIP1 was coexpressed with the V5-tagged mCAR, mCARS202A, and mCARS202D in HepG2 cells. Western blotting was carried out to investigate the expression of a given receptor in the cytosolic and nuclear fractions (Fig. 7A). As shown in Fig. 6A, S202D and S202A were predominantly accumulated in the cytoplasmic and nuclear fractions, respectively. The coexpression of hGRIP1 did not affect their subcellular translocation. GST pull-down assays revealed no difference in the binding of mutants to hGRIP1 compared with that of wild type mCAR (Fig. 7B). hGRIP1 coactivated mCAR-mediated trans-activation of the NR1 reporter in the cotransfected HepG2 cells. As observed with the mCAR, all three mutated receptors exhibited the similar rate of coactivation by hGRIP1 in both the absence and presence of TCPOBOP (Fig. 7C). These results indicated that hGRIP1 does not play a role in the Ser-202-regulated translocation of mCAR.
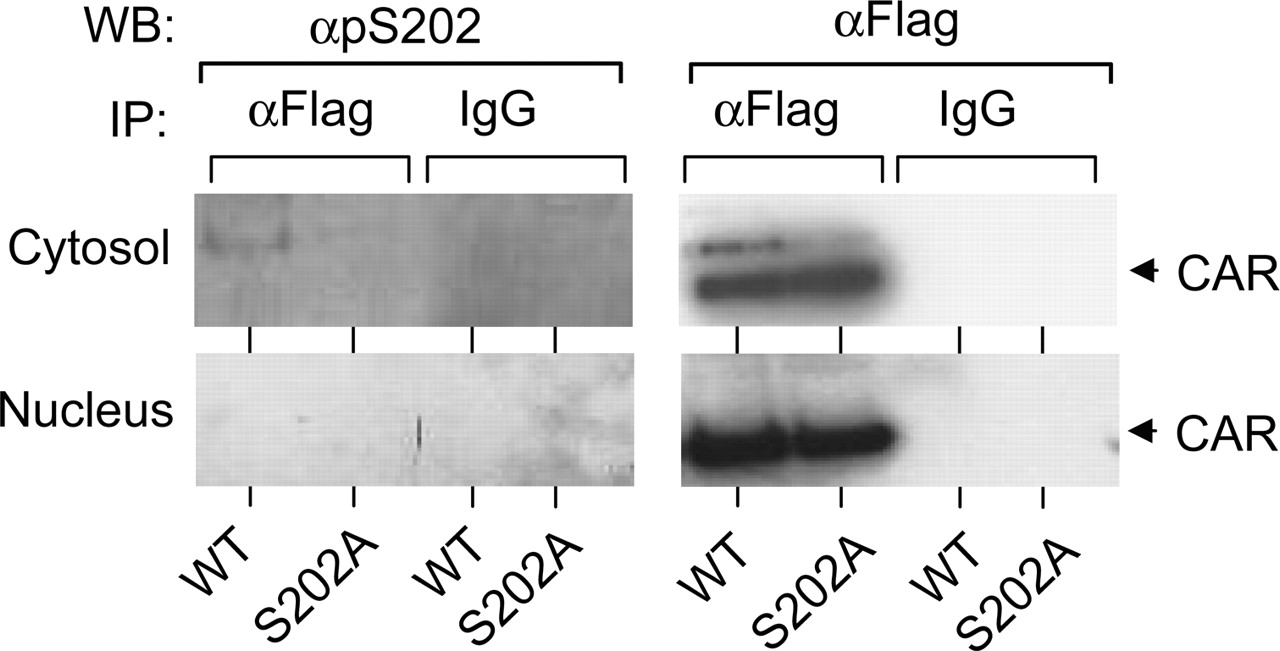
Phosphorylation of Ser-202. Flag-tagged wild-type mCAR or the flag-tagged S202A mutant was expressed in HepG2 cells, from which cytosolic and nuclear fractions were prepared. Then, the wild-type mCAR was immunoprecipitated by anti-flag antibody. Using anti-pS202 antibody, Western blot analysis was performed on immunoprecipitates to examine whether Ser-202 is phosphorylated. Anti-pS202 detected an immunoreactive band for the wild-type mCAR in the cytosolic fraction (Fig. 8). Western blotting using anti-flag antibody showed one major and minor band from this fraction, of which the major one corresponded to nonphosphorylated mCAR. The minor band appeared above the major one and comigrated with the anti-pS202-reacting band; it is thus suggested to represent the phosphorylated form of mCAR. This phosphorylated band was never detected in the nuclear faction, thus suggesting that Ser-202 can be phosphorylated only when mCAR is sequestered in the cytoplasm.
For control experiment, the Flag-tagged S202A mutant was also expressed, immunoprecipitated and subjected to Western blot analysis. As expected, no phosphorylated form of S202A mutant was detected by Western blotting with anti-pS202 antibody.
Discussion
Because PB triggering nuclear translocation of mCAR was found to be sensitive to protein phosphatase inhibitor OA, phosphorylation of mCAR has been a key subject of investigations. We have now identified Ser-202 as the phosphorylation site of mCAR. Ser-202 is phosphorylated when mCAR is sequestered in the cytoplasm; its dephosphorylation facilitates the nuclear translocation of mCAR. PP2A is a protein phosphatase that can catalyze the dephosphorylation of Ser-202. Mimicking phosphorylation, the mutation of Ser-202 to Asp makes mCAR no longer capable of PB triggering translocation into the nucleus in mouse liver. A key question that has now arisen is how PB and TCPOBOP trigger dephosphorylation of CAR. Because TCPOBOP can bind directly to mCAR, this binding may elicit signal to dephosphorylate Ser-202 of mCAR. However, the mechanism can be more complex for PB, because PB does not require direct binding to activate mCAR. Because our previous study shows that the cytoplasmic CAR-Hsp90 complex recruits PP2A upon PB treatment (Yoshinari et al., 2003), future investigations may lead us to uncover a drug-triggering signal transduction pathway that regulates PP2A and the nuclear translocation of mCAR.
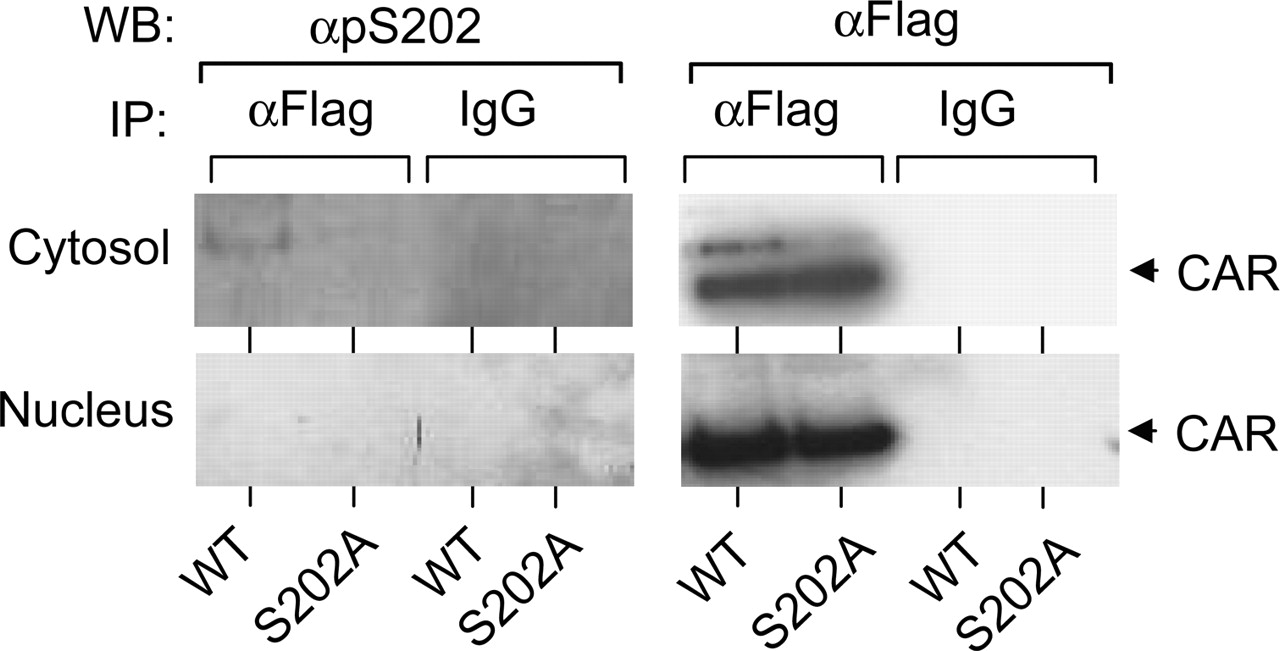
Phosphorylation of Ser 202 in HepG2 cells. HepG2 cells were transfected with Flag-mCAR/pCR3 and its S202A mutant. Immunoprecipitation was carried out by anti Flag-antibody agarose or normal mouse IgG agarose using 1.5 mg of cytosolic or nuclear extract isolated from transfected HepG2 cells. Precipitated samples were separated on a SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane. The same membrane was sequentially stained with anti pS202 antibody and then with anti-Flag HRP.
Nuclear localization of mCAR can also be augmented by multiple factors including the ability of binding to RXR, coregulators, and/or DNA elements. The mutation of Ser-202 did not alter those binding abilities, because the S202A and S202D mutants exhibited the same binding ability to RXR and NR1 as the wild-type mCAR in gel shift assays. GRIP1 has been suggested to accumulate mCAR in the nucleus through binding and stabilizing the receptor (Xia and Kemper, 2004). Western blot analysis confirmed the expression of GRIP1 in the HepG2 cells; however, the S202D mutant still accumulated predominantly in the cytosolic fraction, whereas the S202A mutant accumulated in the nuclear fraction. Although GRIP1 exhibited TCPOBOP-dependent binding to wild-type mCAR in GST pull-down assays, none of the mutations changed the binding. GRIP1 coregulated the S202D mutant-mediated activation of the NR1 reporter in the presence or absence of TCPOBOP as effectively as the wild-type mCAR and the S202A mutant. GRIP1 did not alter the inability of the S202D mutant to accumulate in the nucleus. No nuclear export signal is found in mCAR, and proactive nuclear export has not been intensively investigated. Again, a strong tendency of mCAR to diffuse passively in cells is a major difficulty facing investigations. Once the proper nuclear export assay is established in the future, the role of Ser-202 in the nuclear export of CAR should be revisited.
If the nuclear translocation of mCAR depended only on the dephosphorylation of Ser-202, the S202A mutant would be expected to accumulate in the nucleus without drug treatment. To some extent, this was observed in the HepG2 cells; the S202A mutant accumulated more effectively in the nucleus than the wild-type mCAR did in the absence of TCPOBOP. However, the S202A mutant is sequestered in the cytoplasm in nontreated mouse liver and translocates into the nucleus only after PB treatment, as does wild-type mCAR. This suggests that the dephosphorylation of Ser-202 is required but not sufficient to regulate the nuclear translocation of mCAR. mCAR is retained in the cytoplasm by forming a complex with Hsp90 and a CAR specific cochaperon CCRP (Kobayashi et al., 2003; Yoshinari et al., 2003). Receptors such as the glucocorticoid receptor and the aryl hydrocarbon receptor also form a complex with Hsp90 and contain the specific cochaperones immunophilin and X-associated protein, respectively; these cochaperones regulate their nuclear translocations (Tai et al., 1992; Pratt, 1993; Pollenz et al., 1994; Carver and Bradfield, 1997; Pratt and Toft, 1997; Meyer et al., 1998). Other factors, such as cytoplasmic CAR retention protein and xenobiotic response signal, may also regulate the nuclear translocation of mCAR, maintaining mCAR under tight control in mouse liver. Some of these regulatory factors may be lost in HepG2 cells; thus, Ser-202 becomes the dominant factor regulating nuclear translocation. Taken together, these data suggest the existence of factors that regulate the nuclear translocation in addition to Ser-202. Identifying those factors and their functions should greatly advance our understanding of PB triggering nuclear translocation of mCAR.
Two X-ray crystal structures of mCAR have now been reported. One is the ternary complex with RXR, coactivator peptide, and TCPOBOP; another is in the form of a homodimer (Shan et al., 2004; Xu et al., 2004). First, it is reasonable to have found that the mutation of Ser-202 affected neither CAR binding to RXR nor activation by GRIP1, because Ser-202 is not positioned to interact directly with either RXR or the coactivator. In both structures, Ser-202 is located on the bottom of a shallow pocket and its side chain oxygen is diagonally oriented toward the surface (loop consisting of residues 279 to 287) of the pocket. Therefore, Ser-202 is positioned not to be phosphorylated in the active state of mCAR, suggesting that Ser-202 can only be phosphorylated in its inactive form. This structural constraint may explain why it has been so difficult to demonstrate the phosphorylation; during preparation of mCAR from cells, Ser-202 is rapidly dephosphorylated as the mCAR molecule takes the most stable structure. Because of residual protein phosphatase activity under the experimental conditions used, this dephosphorylation could not have been prevented. If Ser-202 should be phosphorylated, the loop needs to move away from Ser-202 and/or the side chain oxygen of Ser-202 needs to be reoriented more toward the open surface of the pocket. When mCAR is sequestered in the cytoplasm, it forms an Hsp90-CCRP complex that is completely different from the nuclear complex with RXR and coregulators. In the cytoplasmic complex or during the complex formation, Ser-202 may be accessible for phosphorylation by exposing its side chain on the surface of the mCAR molecule. Once a three-dimensional structure of CAR-cytoplasmic CAR retention protein complex is determined in the future, we should be able to answer the question as to whether Ser-202, in fact, undergoes this conformational change.
Footnotes
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.019505.
-
ABBREVIATIONS: PB, phenobarbital; CAR, constitutive active/androstane receptor; TCPOBOP, 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene; LBD, ligand binding domain; m, mouse; h, human; OA, okadaic acid; Hsp90, 90-kDa heat shock protein; PP2A, protein phosphatase 2A; mPXR, mouse pregnane X receptor; PCN, 5-pregnen-3β-ol-20-one-16α-carbonitrile; DBD, DNA binding domain; HRP, horseradish peroxidase; hPP2Ac, human protein phosphatase 2A catalytic subunit; YFP, yellow fluorescent protein; DMSO, dimethyl sulfoxide; TBS, Tris-buffered saline; SV40, simian virus 40; GST, glutathione-S-transferase; PAGE, polyacrylamide gel electrophoresis; RXR, retinoid X receptor; hGRIP1, human glucocorticoid receptor interacting protein-1.
- Received October 4, 2005.
- Accepted December 23, 2005.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}