


Abstract
The group III metabotropic glutamate receptors (mGluRs) represent a family of presynaptically expressed G-protein-coupled receptors (GPCRs) with enormous therapeutic potential; however, robust cellular assays to study their function have been difficult to develop. We present here a new assay, compatible with traditional high-throughput screening platforms, to detect activity of pharmacological ligands interacting with Gi/o-coupled GPCRs, including the group III mGluRs 4, 7, and 8. The assay takes advantage of the ability of the Gβγ subunits of Gi and Go heterotrimers to interact with G-protein regulated inwardly rectifying potassium channels (GIRKs), and we show here that we are able to detect the activity of multiple types of pharmacophores including agonists, antagonists, and allosteric modulators of several distinct GPCRs. Using GIRK-mediated thallium flux, we perform a side-by-side comparison of the activity of a number of commercially available compounds, some of which have not been extensively evaluated because of the previous lack of robust assays at each of the three major group III mGluRs. It is noteworthy that several compounds previously considered to be general group III mGluR antagonists have very weak activity using this assay, suggesting the possibility that these compounds may not effectively inhibit these receptors in native systems. We anticipate that the GIRK-mediated thallium flux strategy will provide a novel tool to advance the study of Gi/o-coupled GPCR biology and promote ligand discovery and characterization.
The metabotropic glutamate receptors (mGluRs) are among the most abundantly expressed receptors in the mammalian central nervous system and are thought to play critical roles in regulating activity in a broad range of CNS circuits (Conn and Pin, 1997). Despite an increasing appreciation of the involvement of mGluRs in CNS function, however, progress in understanding the signaling pathways and functional properties of some members of the mGluR family has been relatively slow. This is especially true for the group III mGluRs, which include mGluRs 4, 6, 7, and 8. These G-protein-coupled receptors (GPCRs) are heavily expressed in presynaptic terminals, where they are likely to regulate voltage-dependent ion channels and play important roles in regulating synaptic transmission (Conn and Pin, 1997). Although these receptors can couple to Gi/o G proteins and inhibit adenylyl cyclase, it has been difficult to measure robust and consistent responses to their activation. Studies of coupling of these receptors to promiscuous GTP-binding proteins have also been more challenging than has been the case for many other Gi/o-coupled receptors. For example, Corti et al. (1998) examined the signaling profiles of splice variants of mGluRs 7 and 8 using chimeric and promiscuous G proteins to couple these receptors to the phosphoinositide hydrolysis pathway. In these experiments, the best G protein/mGluR7 combination induced only a 2-fold increase in signal over the basal response. Because of difficulties in measuring activity of group III mGluRs, our understanding of the pharmacological properties of this important receptor subfamily remains rudimentary, and effects of many compounds commonly used as group III mGluR ligands have not been closely examined using in vitro assays with individual receptors. These results have suggested that alternate methods to more easily examine group III mGluR pharmacology should be explored to enable a better understanding of their pharmacology and potential therapeutic utility.
In general, most techniques used to assess Gi/o-coupled GPCR activity rely upon the transduction of signal from the receptor through the G protein α subunit. Heterotrimeric G proteins are composed of Gα, β, and γ subunits; the complex dissociates to generate a free Gα subunit and a βγ complex after agonist stimulation. It is becoming increasingly appreciated that βγ proteins, particularly those liberated from Gi/o heterotrimers, play key signaling roles in the modulation of targets such as ion channels (for reviewed, see Dascal, 2001). The group III mGluRs have been shown to regulate the function of a number of ion channels (Saugstad et al., 1996, 1997; Guo and Ikeda, 2005; Bertaso et al., 2006; Pelkey et al., 2006), and patch-clamp recordings suggest that group III mGluRs can induce robust responses in cell lines or oocytes coexpressing these receptors and the G-protein regulated inwardly rectifying K+ channel (GIRK) (Saugstad et al., 1996, 1997). These findings suggested that group III mGluR coupling to GIRK channels might represent an ideal way to explore ligand pharmacology at these receptors. Electrophysiological approaches, however, are not readily amenable to the high-throughput assays that are needed for detailed pharmacological analysis and new ligand discovery.
We have now developed a novel assay for rapid parallel analysis of GPCR function that monitors the signaling of Gβγ subunits and permits easy assessment of Gi/o-coupled GPCR activity, including the group III mGluRs. The assay takes advantage of the ability of the Gβγ subunits of Gi and Go heterotrimers to interact with GIRK channels, an event that leads to rearrangements within the channel pore region and alterations in the kinetics of channel opening (for review, see Sadja et al., 2003). Exploiting the ability of potassium channels to conduct the ion thallium (Weaver et al., 2004), we examined the regulation of heteromeric GIRK 1/2 channels by endogenous and transfected Gi/o-coupled GPCRs. We show here that thallium flux through GIRK channels provides a sensitive and HTS-compatible method to examine the βγ-mediated signaling properties of agonists, antagonists, and allosteric modulators of various Gi/o-linked GPCRs. This method has now allowed us to study functional responses and pharmacological properties of the group III mGluRs. Note that a number of compounds that are commercially sold as group III antagonists that have not been well characterized simultaneously at all three receptors show very weak activity in this assay, questioning their effectiveness in antagonism of these receptors in vivo. Development of a novel and HTS-compatible assay that takes advantage of βγ-mediated signaling downstream of Gi/o-coupled receptors that have been challenging for in vitro examination, such as the group III mGluRs, represents a major breakthrough for this important class of receptors.
Materials and Methods
Cell Culture, Plating, and Dye Loading. HEK/GIRK cells stably expressing the M4 muscarinic receptor were grown in 45% Dulbecco's modified Eagle's medium (DMEM), 45% Ham's F12, 10% fetal bovine serum (FBS), 100 units/ml penicillin/streptomycin, 20 mM HEPES, pH 7.3, 1 mM sodium pyruvate, 2 mM glutamine, and 700 μg/ml G418. Rat mGluR4a, 7a, and 8a cell lines were prepared by PCR amplification of the entire coding sequence of each receptor and cloning into pIRES puro 3 (Invitrogen). For mGluR4 and 7, cloning sites were BamHI/NotI and for mGluR8, cloning sites were EcoRI/NotI. HEK/GIRK/M4 cells were transfected with 24 μg of DNA and stable transfectants were selected with puromycin. Monoclonal cell lines were established for the mGluR4/GIRK and mGluR7/GIRK lines; the mGluR8/GIRK line is polyclonal. Cells were grown in 45% DMEM, 45% Ham's F12, 10% FBS, 100 units/ml penicillin/streptomycin, 20 mM HEPES, pH 7.3, 1 mM sodium pyruvate, and 2 mM glutamine (growth media). mGluR/GIRK lines were supplemented with 600 ng/ml puromycin dihydrochloride (Sigma-Aldrich, St. Louis, MO) and 700 μg/ml G418 (Mediatech, Inc., Herndon, VA). Cells for experiments were generally maintained for approximately 15 to 20 passages; this was particularly important for experiments examining the endogenous α2C receptor.
Assays were performed within Vanderbilt University's High-Throughput Screening Center. Cells were plated into 384-well, black-walled, clear-bottomed poly-d-lysine-coated plates (Greiner Bio-One, Longwood, FL) at a density of 15,000 cells/20 μl/well in DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 100 units/ml penicillin/streptomycin (assay media). Plated cells were incubated overnight at 37°C in the presence of 5% CO2. The following day, the medium was removed from the cells, and 20 μl/well of 1.7 μM BTC-AM, an indicator dye (Invitrogen; prepared as a stock in DMSO and mixed in a 1:1 ratio with Pluronic acid F-127) in assay buffer [Hanks' balanced salt solution (Invitrogen) containing 20 mM HEPES, pH 7.3] was added to the plated cells. Cells were incubated for 1 h at room temperature, and the dye was replaced with the appropriate volume of assay buffer to generate a final volume of 50 μl after agonist addition (i.e., 40 μl for agonist experiments and 20 μl for experiments including antagonists/potentiators).
Test compound Preparation. Agonists were diluted in one of four recipes of thallium buffer [gluconate buffer: 125 mM sodium gluconate, 1 mM magnesium sulfate, 1.8 mM calcium gluconate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES, pH 7.3; sulfonate buffer: 62.5 mM methanesulfonic acid, 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES, pH 7.3; bicarbonate buffer: 125 mM sodium bicarbonate (added fresh the morning of the experiment), 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES, pH 7.3; chloride buffer: Hanks' balanced salt solution, 10 mM HEPES, pH 7.3, and 4.9 mM thallium sulfate] at 5 times the final concentration to be assayed. For antagonist/potentiator experiments, compounds were diluted to 2.5 times their final desired concentration in assay buffer. For pertussis toxin experiments, cells were incubated with 80 ng/ml pertussis toxin in assay media for approximately 18 h before the experiment. Thallium sulfate requires special handling and disposal precautions, and investigators are cautioned to contact their Environmental Health and Safety Department to ensure that proper procedures are followed.

M4 muscarinic receptors mediate thallium flux in HEK/GIRK/M4 receptor cells. A, HEK/GIRK/M4 cells were loaded with the indicator dye BTC-AM as described under Materials and Methods, and cells were placed into a kinetic imaging plate reader (Hamamatsu FDSS 6000). The assay was initiated by reading baseline fluorescence for 10 s followed by the addition of increasing concentrations of carbachol or vehicle in the presence of 2.4 mM Tl+. Vehicle traces were subtracted from traces generated in the presence of carbachol. B, the slope of the curves in A was calculated between the time window of 10 to 20 seconds after agonist addition and plotted versus carbachol concentration and fitted using GraphPad Prism software as described under Materials and Methods. Data are mean ± S.E.M. of 10 individual experiments; an individual experiment represents the average of quadruplicate determinations for each data point.
Kinetic Imaging, Data Analysis, and Statistics. Cell plates and compound plates were loaded onto a kinetic imaging plate reader (FDSS 6000; Hamamatsu Corporation, Bridgewater, NJ). Appropriate baseline readings were taken (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm) and test compounds were added. When included in an experiment, antagonists/potentiators were added in a 20-μl volume and incubated for 5 min before the addition of 10 μl of thallium buffer with or without agonist. After the addition of agonist, data were collected for an additional 2 min.
Data were analyzed using Excel (Microsoft Corp, Redmond, WA). Raw data were opened in Excel and each data point in a given trace was divided by the first data point from that trace (static ratio). For experiments in which antagonists/potentiators were added, data were again normalized by dividing each point by the fluorescence value immediately before the agonist addition to correct for any subtle differences in the baseline traces after the compound incubation period. The slope of the fluorescence increase beginning 5 s after thallium/agonist addition and ending 15 s after thallium/agonist addition was calculated. The data were then plotted in Prism software (GraphPad Software, San Diego, CA) to generate concentration-response curves after correcting for the slope values determined for baseline waveforms generated in the presence of vehicle controls. Potencies were calculated from fits using a four-point parameter logistic equation. All statistical data were calculated using either paired or unpaired Student's t tests (specific test outlined in appropriate figure legends).
Compounds. Carbamylcholine chloride (carbachol), atropine, epinephrine, bupivacaine HCl, and pertussis toxin were purchased from Sigma-Aldrich. Scopolamine hydrobromide, UK 14,304, tropicamide, AF-DX 116, BRL 44408 maleate, imiloxan hydrochloride, rauwolscine hydrochloride, N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC), 2-methyl-6-(phenylethynyl)pyridine hydrochloride, (R,S)-α-methyl-4-phosphonophenylglycine (MPPG), (R,S)-α-methyl-4-sulfonophenylglycine (MSPG), (R,S)-α-methylserine-O-phosphate (MSOP), (R,S)-α-cyclopropyl-4-phosphonophenylglycine (CPPG), UBP1112, (S)-MAP4, LY341495, and pertussis toxin were purchased from Tocris Bioscience (Ellisville, MO).
Results
The Muscarinic Agonist Carbachol Stimulated Thallium Flux in HEK/GIRK Cells. As an initial step in developing a new assay system capable of detecting and characterizing compound activity at the group III mGluRs, we examined the ability of GIRK 1/2 channels to mediate thallium flux in response to direct agonist activation of other Gi/o-coupled GPCRs for which previous assay development has been more straightforward. HEK/GIRK cells used for these experiments stably express the Gi/o-coupled human M4 muscarinic receptor (Chuang et al., 1998). Application of the general muscarinic agonist carbachol to HEK/GIRK/M4 cells preloaded with the fluorescent indicator dye BTC-AM resulted in a concentration-dependent increase in thallium conductance in HEK/GIRK/M4 cells (Fig. 1A) but not in untransfected HEK 293 cells (not shown). The potency of carbachol was almost identical to that observed by Bräuner-Osborne and Brann (1996) using a reporter assay [270 ± 0.07 nM versus 275 ± 27 nM in the current study (mean ± S.E.M.)] and the data generated via thallium flux measurement were very consistent.
Final concentrations of 2.4 mM thallium sulfate and 1.7 μM BTC-AM dye were used for these studies to balance the solubility limitations of thallium (less than 5 mM in chloride-containing buffers such as the Hanks' balanced salt solution used for dye loading and equilibration) with the generated signal and to delay apparent saturation of the dye with incoming thallium. Examination of the fluorescence traces from these experiments revealed that the rate of curve rise, measured from the slope calculated within a 10-second time window beginning 5 s after agonist addition, produced a good, concentration-dependent fit using a four parameter logistic nonlinear regression algorithm (Fig. 1B, EC50, 275 ± 27 nM, mean ± S.E.M., n = 10 independent experiments performed in quadruplicate). These data were generated over a series of several months and, as can be seen from Fig. 1B, the variability in both the potency and maximal response was quite low. As observed in Fig. 1A, within approximately 30 to 35 s, the trace begins to reach an asymptote. This suggests that there is some saturation in the system, presumably saturation of the dye with incoming thallium. Monitoring the initial slope allows analysis of concentration-dependent relationships within a portion of the trace that should be less affected by this observed plateau of the signal.
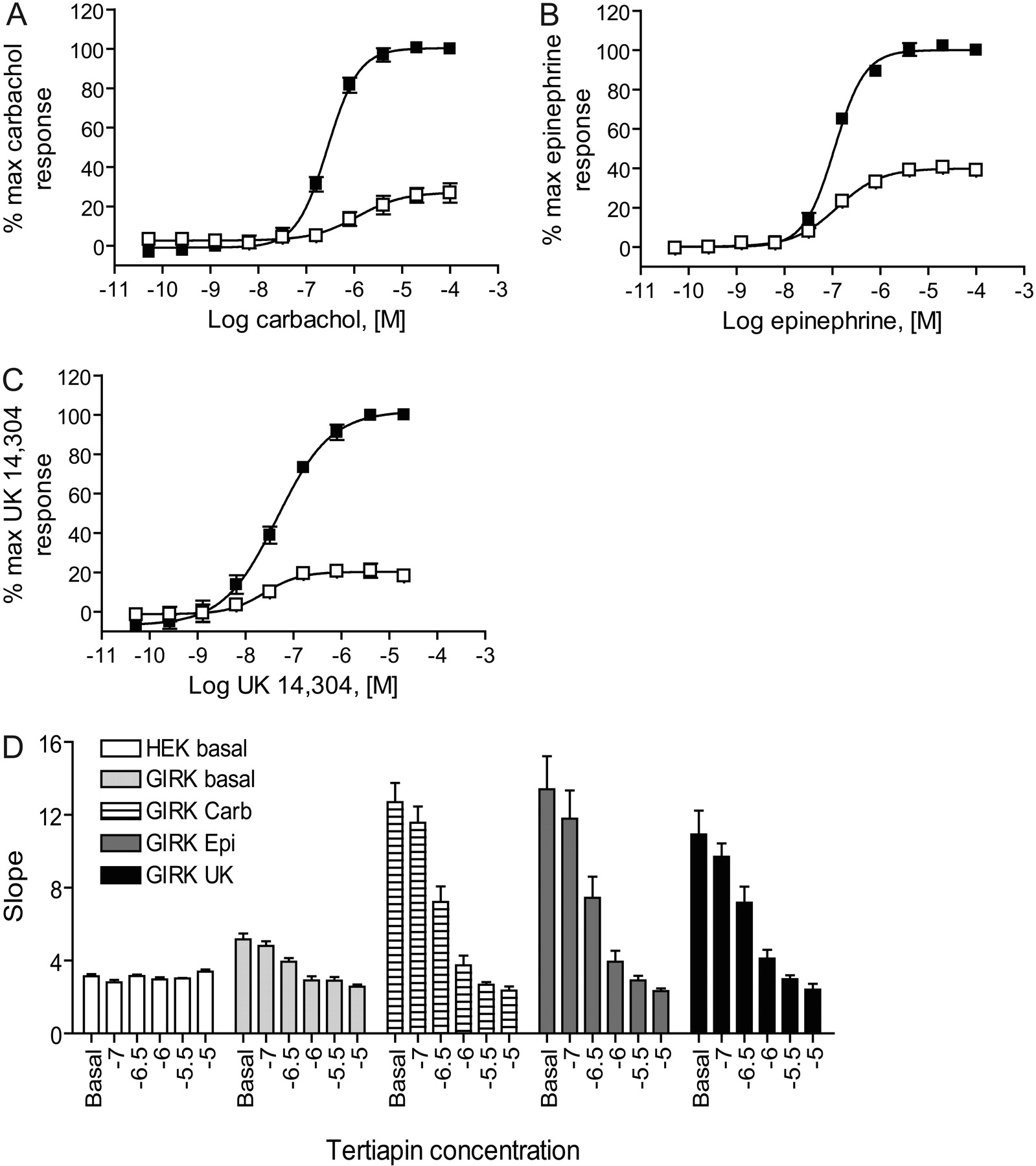
Muscarinic and Adrenergic Agonist-Induced GIRK Stimulation Was Primarily Mediated by Gi/o-G Proteins and Blocked in a Concentration-Dependent Manner by a Compound That Directly Inhibits GIRK Channels. Pretreatment of M4/HEK/GIRK cells overnight with 80 ng/ml pertussis toxin (PTX), a drug that ADP-ribosylates and inactivates Gi/o proteins, significantly blocked carbachol-mediated thallium influx (Fig. 2A), indicating that this response was dependent upon the presence of Gi/o G proteins. It has been reported that HEK cells express mRNA for the α2C adrenergic receptor (http://www.mbi.ufl.edu/~shaw/293.html; Shaw et al., 2002), and application of either epinephrine or the α2-selective agonist UK 14,304 also generated concentration-response curves for agonist-dependent thallium flux in the HEK/GIRK cell line used for these studies (EC50 for epinephrine, 110 ± 10 nM, n = 8; EC50 for UK 14,304, 45 ± 4 nM, n = 5; Fig. 2, B and C); the responses of these agonists were also significantly blocked by PTX. PTX treatment also significantly increased the basal influx of thallium through GIRK channels by 14.2 ± 2.5% (p = 0.0012, paired t test), a finding that is consistent with reports that the basal inhibitory activity of Gi/o α subunits on the GIRK channel is relieved when these subunits are inactivated (Schreibmayer et al., 1996). Basal thallium flux and the responses induced by concentrations of carbachol, epinephrine, or UK 14,304 required to induce an 80% maximal response (EC80) were blocked in HEK/GIRK cells in a concentration-dependent manner by pretreatment with tertiapin-Q, an antagonist of inwardly rectifying potassium channels (Fig. 2D) (Jin et al., 1999). Control experiments revealed that HEK 293 cells untransfected with GIRK channels did flux a small amount of thallium and that this response was not blocked by tertiapin-Q (Fig. 2D). Components of this response in HEK 293 cells can be blocked by ouabain, an antagonist of the Na+/K+ ATPase, and bumetanide, an antagonist of cation-coupled chloride transporters (C.D.W., unpublished observations), suggesting that these alternate mechanisms contribute to the thallium flux remaining in HEK/GIRK cells after treatment with tertiapin-Q.

Thallium-flux induced by muscarinic and adrenergic agonists in HEK/GIRK/M4 cells is mediated primarily by pertussis-toxin sensitive G proteins and is blocked in a concentration-dependent fashion by an antagonist of inwardly rectifying potassium channels. Increasing concentrations of carbachol (A), epinephrine (B), or UK 14,304 (C) were added to HEK/GIRK/M4 cells in the presence (□) and absence (▪) of an overnight treatment with 80 ng/ml pertussis toxin and thallium flux was measured (data shown are mean ± S.E.M., n = 3-4 independent experiments performed in quadruplicate). Data have been normalized to maximal response in the absence of PTX to show percentage activity remaining in the presence of PTX; untransformed slope values are as follows: mean maximal slope of fluorescence ratio ± S.E.M., minus PTX versus plus PTX, respectively: A, carbachol, 22.0 ± 1.7 versus 5.5 ± 1.2, p = 0.0012, paired t test, n = 4; B, epinephrine, 18.0 ± 2.9 versus 7.2 ± 1.5, p = 0.0178, paired t test, n = 3; C, UK 14,304, 13.7 ± 2.0 versus 2.9 ± 0.7, p = 0.0147, n = 3). D, basal and agonist-induced responses are blocked by tertiapin-Q in HEK/GIRK cells but not in untransfected HEK 293 cells. Increasing concentrations of tertiapin-Q were were applied before either vehicle or EC80 concentrations of the indicated agonists and thallium flux was measured. Basal HEK 293 response (white boxes), basal HEK/GIRK response (light gray boxes), HEK/GIRK response in the presence of an EC80 concentration of carbachol (striped boxes), epinephrine (dark gray boxes), or UK 14304 (black boxes). Data are the mean ± S.E.M., n = 3 independent experiments performed in quadruplicate.
The GIRK-Thallium Flux Assay Detected the Activity of GPCR Antagonists and Positive Allosteric Modulators. The results above show that the GIRK-mediated thallium flux assay was capable of detecting agonist activity at Gi/o-coupled receptors. To determine the capacity of the assay to detect other important ligands for these GPCRs, we assessed the ability of the technique to detect the activity of receptor antagonists. For the muscarinic response, we observed inhibition of the carbachol-induced GIRK current in the presence of the M4-preferring antagonist tropicamide (IC50, 8.0 ± 0.5 nM, n = 4) in addition to blockade with the nonselective muscarinic antagonists atropine and scopolamine (IC50, 86 ± 5 and 117 ± 9 pM, respectively, n = 4; Fig. 3A). For the adrenergic response elicited by UK 14,304 and epinephrine (not shown), we observed blockade with rauwolscine, a drug with a preference for binding to α2C receptors (IC50, 655 ± 30 pM, n = 3; Fig. 3B). In contrast, we observed antagonism with α2A receptor (BRL-44408 maleate; IC50, 2.6 ± 1.3 μM, n = 5) and α2B receptor (imiloxan; IC50, 6.8 ± 1.7 μM, n = 3) preferring agents only at high concentrations of drug (Fig. 3B). These results are consistent with previous reports indicating the presence of α2C adrenergic receptor mRNA in HEK 293 cells (http://www.mbi.ufl.edu/~shaw/293.html) (Shaw et al., 2002).

The GIRK-mediated thallium flux assay sensitively detects the activity of antagonists and positive allosteric modulators of GPCRs. A, increasing concentrations of the muscarinic antagonists atropine (▪), scopolamine (□), tropicamide (•), and AFDX-116 (○) were applied 5 min before the addition of an EC80 concentration of carbachol, and thallium flux was measured (n = 3-4 independent experiments performed in quadruplicate). B, increasing concentrations of the adrenergic antagonists BRL 44408 (•), imiloxan (▪), and rauwolscine (▴) were applied 5 min before the addition of an EC80 concentration of UK 14,304 and thallium flux was measured (n = 3-4 independent experiments performed in quadruplicate). C, 3 μM compound 12 from Shirey et al. (2008) potentiated M4-mediated thallium flux as shown by a 9-fold shift to the left of the concentration-response curve. Data are mean ± S.E.M., n = 3 independent experiments performed in quadruplicate.
Allosteric modulation is a mechanism that is emerging as an attractive strategy for both pharmacological tool development and drug discovery for GPCRs. By binding to a site other than the orthosteric ligand binding site on a given receptor, these compounds can often discriminate between highly related receptor subtypes and may exhibit better blood-brain barrier penetration or pharmacokinetics compared with orthosteric ligands. It is now possible to develop highly selectivity compounds that distinguish between the mGluRs using allosteric modulation strategies (for review, see Kew, 2004); therefore, it was of interest to us to validate the thallium flux assay for its capability to detect compounds that could modulate receptors in an allosteric fashion. To determine whether the GIRK-thallium flux assay could sensitively detect compounds that “potentiate” the response to endogenous ligands, we analyzed the ability of a compound derived from a family of newly described positive allosteric modulator (PAM) of the M4 receptor (Shirey et al., 2008), to shift the carbachol concentration-response curve to the left. These studies showed that preapplication of a 3 μM concentration of this M4 PAM (compound 12 from Shirey et al., 2008) shifted the carbachol concentration-response significantly to the left (Fig. 3C; EC50 for carbachol alone, 501 ± 20 nM; EC50 for carbachol plus positive allosteric modulator, 56 ± 10 nM, p = 0.0004). This indicates that the thallium flux method is useful for the detection of ligands with different modes of efficacy, ranging from agonist to antagonist to positive allosteric modulator, at Gi/o GPCRs.
The GIRK-Thallium Flux Assay Can Be Translated to High-Throughput Format and Generates Valid Screening Statistics for the Detection of Multiple Types of Ligands. In addition to its utility in the day-to-day study of receptor function, we sought to determine whether the assay was compatible with high-throughput formats and could potentially be used for compound library screening. To examine large compound collections over a series of many days, it is important to establish the sensitivity and reliability of the assay from plate to plate and from day to day. In terms of screening for novel agonists, for example, we would need to establish that we have adequate separation and low variability between vehicle and ECmax concentrations of agonist. The reproducibility and sensitivity of the assay can be assessed using a parameter termed Z′ (Zhang et al., 1999), which is a representation of the difference in signal between two populations (for example, the signal induced by vehicle versus agonist), as well as the consistency of the signal, as a function of standard deviation, within each population. Generally, Z′ values greater than 0.5 are desirable for a high-throughput assay. Measurement of Z′ screening statistics for three different types of high-throughput screens: agonist (ECmax versus vehicle), antagonist (EC80 versus vehicle), and positive allosteric modulator (EC20 versus ECmax) for the M4 muscarinic response resulted in Z′ values of 0.63 ± 0.07 (mean ± S.D.) comparing vehicle versus ECmax concentrations of carbachol, 0.53 ± 0.03 comparing EC80 versus vehicle, and 0.65 ± 0.03 for EC20 versus ECmax (Fig. 4A, vehicle versus ECmax shown). These data highlight the consistency of the assay from between days and indicate that the GIRK-mediated thallium flux assay should be useful for high throughput screening for several different types of ligands.

Demonstration that the thallium flux assay should be useful for high-throughput screening and optimization of the assay for the group III mGluRs. A, cells were plated in 384-well format and stimulated in a “checkerboard” pattern with alternating vehicle or ECmax (100 uM) concentrations of carbachol. Slope values were plotted versus well number. B, concentration-response curves of carbachol were performed in the presence of increasing concentrations of DMSO. The slope in the presence of vehicle plus the corresponding concentration of DMSO was subtracted from each point, n = 3-4 experiments performed in quadruplicate. C, rat mGluR4a was stably expressed in HEK/GIRK cells and thallium flux was measured using different anionic buffer components as described under Materials and Methods. Potencies are listed in Table 1.
In addition to reliability across plates and adequate separation of control and test populations, another potential variable for an HTS-compatible assay is tolerance to the vehicle chosen for compound library storage. The majority of chemical libraries in existence today are dissolved and stored in DMSO. Therefore, it is necessary to determine the effects of DMSO on thallium flux through GIRK channels or by non-selective mechanisms. We observed increased basal thallium flux in HEK/GIRK cells in the presence of increasing concentrations of DMSO (basal slope in absence of DMSO, 6.6 ± 0.6; in the presence of 0.1% DMSO, 7.7 ± 0.3; 0.5% DMSO, 10.0 ± 0.5, and 1% DMSO, 16.3 ± 0.8). Despite these effects on basal thallium flux, when we performed concentration-response curves for carbachol in the presence of increasing concentrations of DMSO and found that, whereas the baseline response increases, we were still able to generate a concentration-response curve of similar efficacy and potency up to 1% DMSO (Fig. 4B), suggesting that, if all vehicle concentrations are matched, the assay is quite tolerant to DMSO effects.
Optimization of the GIRK-Mediated Thallium Flux Assay for the Detection of Ligand Activity at the Group III mGluRs. We reasoned that the activity of the group III mGluRs might be easily monitored via GIRK-mediated thallium flux because of their their demonstrated ability to couple to ion channels, including GIRK, in electrophysiology studies. For example, mGluR7 has been shown to inhibit P/Q-type voltage-gated calcium channels (Pelkey et al., 2006), and mGluR7-liberated Gβγ subunits have been shown to be important for regulation of voltage-gated calcium channels (Bertaso et al., 2006). mGluR8 couples to calcium channels in rat sympathetic neurons (Guo and Ikeda, 2005) and in mouse photoreceptors (Koulen et al., 2005); in this latter study, the response was blocked in the presence of a Gβγ-inhibiting peptide. It has been shown that both mGluR7 and mGluR8 can couple to GIRK channels when the receptors and channels are coexpressed in cell lines or oocytes (Saugstad et al., 1996, 1997; Sorensen et al., 2002), suggesting that these receptors should induce thallium flux in HEK/GIRK cells and that this method might be an ideal way to monitor Gβγ signaling downstream of these receptors.
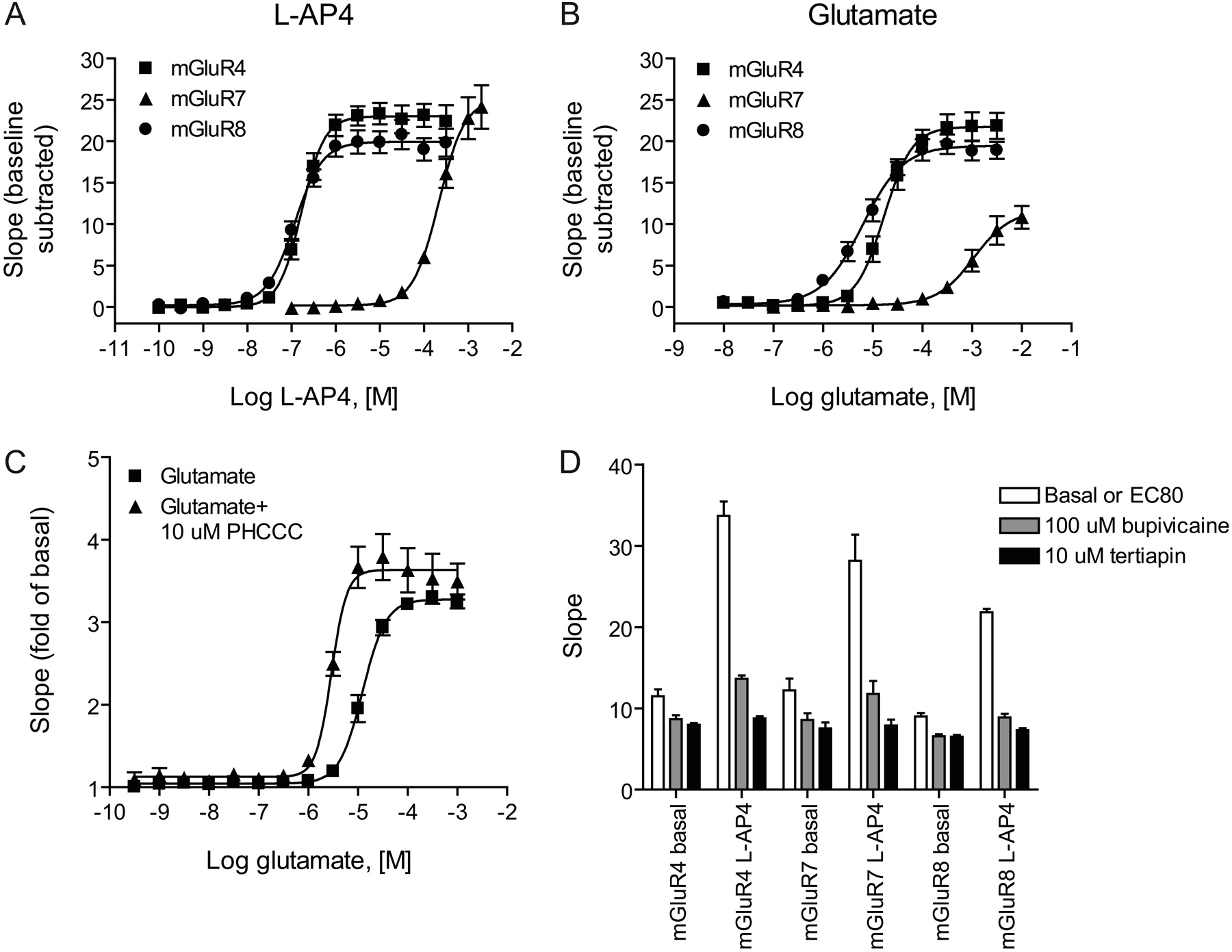
Glutamate did not induce a response in HEK/GIRK cells untransfected with mGluRs (data not shown), suggesting that signals induced after transfection of these receptors would not be compromised by responses induced by endogenous proteins responsive to glutamate. We stably expressed the three major group III mGluRs in the HEK/GIRK cell background and measured responses to agonist application using the assay buffer described above for the muscarinic and adrenergic receptors. In contrast to results seen with these family A GPCRs, however, we were unable to obtain good curve fits for glutamate, and the potencies of both glutamate and the group III agonist l-AP4 were substantially lower than those observed using other assays that rely upon Gα signaling (for example, Wu et al., 1998). Because of the use of Gβγ as a readout of receptor function, it was possible that these agonists were simply less potent in transducing a signal via Gβγ or that the Gβγ subunits used by the group III receptors were different from those used by the M4 and α2C receptors and were less efficient in activating GIRK. In contrast, because glutamate and other orthosteric glutamate receptor analogs are anions, it was possible that the buffer components interfered with the ability of these ligands to interact with the receptors. We compared three different thallium flux buffers using the anions gluconate, sulfonate, and bicarbonate and assessed the activity of ligands at the M4, mGluR4, and mGluR8 receptors. We also analyzed a buffer containing chloride as the anion, but, because of the low solubility of thallium in chloride-containing solutions, we were required to use 2.5 times less thallium, and the resulting signal was much lower than buffers that tolerated higher thallium concentrations (Fig. 4C). As can be seen in Fig. 4C and Table 1, both the glutamate and l-AP4 concentration-response curves were substantially shifted to the left by substituting the anion bicarbonate for gluconate, and the resulting potencies were much similar to those reported in the literature using Gα readouts to measure receptor activity. For example, Wu et al. (1998) reported potencies of 320 ± 30 and 61 ± 10 nM for l-AP4 on human mGluR4 and -8, respectively, using cAMP inhibition, and we obtained potencies of 331 ± 40 and 89 ± 14 nM (Table 1) for these receptors using the thallium flux assay performed in the presence of bicarbonate buffer
Agonists of mGluR4 and mGluR8 are more potent in bicarbonate-based buffer compared with other anionic buffer systems
As can be seen in Fig. 5, A and B, and Table 2, use of the bicarbonate buffer system revealed that each of the group III mGluRs coupled well to GIRK activity, and the signals were very robust and consistent between individual experiments (results shown are the mean ± S.E.M. of three to nine individual experiments). mGluR4 activity was potentiated by the known positive allosteric modulator PHCCC (Fig. 5C; EC50 glutamate, vehicle control, 12.6 ± 1.7 μM; PHCCC, 2.9 ± 0.22 μM; mean ± S.E.M., n = 3) and the extent of potentiation was consistent with values previously reported in the literature using other measurement techniques (Maj et al., 2003; Marino et al., 2003). Again, responses to the receptors were blocked in a concentration-dependent manner by preapplication of tertiapin-Q (Fig. 5D, 10 μM tertiapin shown). For these studies, we also performed concentration-response experiments using the local anesthetic bupivacaine, which has been shown to block the GIRK channel (Zhou et al., 2001) and found that preapplication of bupivacaine also almost completely blocked the response of the mGluRs (Fig. 5D, 100 μM bupivacaine shown). These studies indicate that these receptors are mediating thallium flux via GIRK activation rather than another mechanism within the cells.
Potencies of various agonists at the group III mGluRs as assessed using the GIRK thallium flux assay
Potencies of compounds are shown in micromolar concentrations for EC50 values.
Comparison of the Ability of Orthosteric Antagonists to Block Group III mGluR-Mediated GIRK Activation. Establishment of cell lines coexpressing GIRK and each of the group III mGluRs that is expressed in the CNS provides a rapid and robust way to compare responses among these receptors using the same cell culture system and signaling pathway. Compounds have been identified that have been described to inhibit l-AP4-induced responses in brain tissue, various neuronal preparations, and cell lines and have been classified as antagonists of the group III mGluRs (for review, see Yang, 2005). In some of these cases, however, the activity of a compound at each of the cloned group III receptors has not been directly compared using the same assay system. In fact, for the compound MSPG, there is no information available for the activity of this ligand at any of the cloned group III mGluRs, and the ability of the compound to act as a group III mGluR antagonist has been inferred from its ability to block l-AP4 responses in various neuronal preparations (Jane et al., 1995; Bedingfield et al., 1996). Generation of the cell lines described above allowed us to rapidly assess the antagonist activity of a number of commercially available ligands described as group III mGluR antagonists. These compounds include MPPG, MSPG, MSOP, CPPG, UBP1112, (S)-MAP4, and LY341495. Although each of these compounds is commonly considered a general group III mGluR antagonist and activity of some of these compounds has been determined at some of the group III receptors (Kingston et al., 1998; Wu et al., 1998; Schoepp et al., 1999; De Colle et al., 2000; Naples and Hampson, 2001), affinity and potency values for most of these compounds have not been simultaneously established in the same assay across the cloned rat group III mGluR subtypes.
Pharmacological assessment of antagonist activity of the above compounds at rat mGluRs 4a, 7a and 8a is shown in Fig. 6 and Table 3. In these experiments, antagonists were evaluated for their ability to inhibit an EC80 concentration of agonist appropriate for each receptor. For mGluRs 4 and 8, these comparisons were made using both l-AP4 and glutamate as agonists (Tables 2 and 3); data for antagonism of l-AP4 are shown in Fig. 6 to compare results with those obtained for mGluR7, a receptor with an extremely low affinity for glutamate. LY341495, a group II mGluR-preferring antagonist with demonstrated ability to either bind to or functionally inhibit human (Kingston et al., 1998) and rat (Wright et al., 2000) group III receptors was used as a reference ligand to determine whether potencies in an assay that monitors Gβγ signaling downstream of these receptors might be very different from assays that examine signaling downstream of Gα. Using thallium flux measurements, LY341495 was the most potent at mGluR8 (209 ± 10 nM), followed by mGluR7 (3.2 ± 0.3 μM) and then mGluR4 (27 ± 12 μM) (Fig. 6A, Table 3). Although performed using a distinct assay and independent cell lines, we note that these potencies are in good agreement with values reported by Kingston et al. (1998) for LY341495 activity at the human group III receptors using a cAMP inhibition assay (22 ± 1.3 μM, human mGluR4; 0.99 ± 0.07 μM, human mGluR7; and 0.17 ± 0.02 μM, human mGluR8). These results also correlated well with our own data for mGluR4 obtained using a chimeric G protein approach (EC50 for LY341495 using l-AP4 as agonist, 22.6 ± 7.4 μM, n = 3 experiments performed in triplicate). These results suggest that, at least for LY341495, potencies obtained with this Gβγ-dependent assay were very similar to those obtained using assays relying on Gα signaling. For the remaining antagonists, in every case potency was highest at mGluR8 (most potent compounds shown in Fig. 6B). Despite use of 1 mM concentrations of antagonists as the highest concentration, there were a number of compounds for which reliable curve fits could not be obtained, particularly at mGluR4 (Table 3). IC50 values for compounds are shown in Table 3; Fig. 6C shows the maximal level of inhibition of the l-AP4 response induced by a 1 mM concentration of each antagonist. Development of a robust and reliable cell based assay for these receptors has now allowed us to expand the known pharmacology of the group III mGluRs by comparing the rat receptors simultaneously in a similar assay system. Surprisingly, these results reveal that many of the available described group III antagonists have very weak activity at these receptors, particularly at concentrations below 1 mM.
Potencies of various antagonists at the group III mGluRs as assessed using the GIRK thallium flux assay
Potencies of compounds are shown in micromolar concentrations for IC50 values.

The GIRK-mediated thallium flux assay is useful for the examination of group III mGluR activity. l-AP4 (A) and glutamate (B) concentration-response curves were performed using HEK/GIRK cell lines expressing rat mGluR4a, rat mGluR7a, and rat mGluR8a. Data are the mean ± S.E.M. of three to nine independent experiments; each experiment was performed in quadruplicate. Potencies of agonists are listed in Tables 2 and 3. C, potentiation of the glutamate concentration-response at mGluR4 in the presence of the positive allosteric modulator PHCCC. D, blockade of the responses of the group III mGluRs by tertiapin and bupivacaine. White boxes, basal or EC80 agonist response; gray boxes, 100 μM bupivacaine; black boxes, 10 μM tertiapin-Q. Data for C and D are the mean ± S.E.M. of three independent experiments performed in quadruplicate.
Discussion
GPCRs are the most successful class of drug targets to date (Fang et al., 2003). Of the GPCRs, it has been perhaps most difficult to develop robust assays to detect activity downstream of the Gi/o-coupled receptors. One such group of receptors that has suffered from a lack of robust in vitro pharmacological assays is the group III mGluRs. Building upon the observation that these receptors can couple to various calcium and potassium channels, we surmised that an assay designed to easily assess Gβγ subunit signaling might offer a new approach to assess the pharmacology of these receptors. In addition, this assay could then serve as a generally applicable method to assess ligand activity at other Gi/o-coupled receptors, expanding our ability to explore signaling mediated by Gβγ subunits.

Commercially available group III mGluR antagonists are most active at inhibiting thallium flux via mGluR8, and several compounds have only weak activity at the group III mGluRs. A, increasing concentrations of LY 341495 were applied to cells expressing mGluR4 (squares), mGluR7 (triangles), or mGluR8 (circles) in the presence of an EC80 concentration of l-AP4 and thallium flux was measured. B, antagonists which completely inhibited mGluR8 responses in the concentration range examined are shown: LY 341495 (▪), CPPG (▴), MPPG (□), and UBP1112 (•). C, maximal inhibition induced by a 1 mM concentration of each antagonist is compared for mGluRs 4 (black bars), 7 (gray bars), and 8 (white bars). Data are the mean ± S.E.M. of three to four independent experiments performed in triplicate or quadruplicate.
We first validated the assay using several Gi/o-coupled GPCRs (Figs. 1, 2 and 3) for which assay development has been more straightforward. We also were able to show that the assay was amenable to high-throughput screening platforms (Fig. 4), which should enhance ligand discovery for Gi/o GPCRs. Establishment of this assay then provided a robust and sensitive way to rigorously assess the pharmacological properties of the group III mGluRs (mGluRs 4, 7, and 8), perhaps the most poorly understood of the three major subgroups of this important receptor family. Using LY341495 as a reference ligand, we observed similar potencies in comparison with previous literature values. This provided us with a mechanism to easily assess the activity of compounds that are commercially sold as group III antagonists; a categorization often based on limited information in complex systems such as neuronal preparations. The antagonist studies presented here show that several compounds commonly viewed as group III mGluR antagonists do inhibit responses to activation of these receptors, but they also revealed several critical findings. For instance, most of the antagonists tested were relatively inactive at mGluRs 4 and 7 and induced an inhibition of only approximately 50 to 60% (or less) at concentrations up to 1 mM. Every antagonist tested was observed to be more potent at mGluR8. CPPG, MPPG, and UBP1112, compounds considered to be general group III antagonists, inhibited agonist-induced GIRK activation in mGluR8-expressing cells with potencies in the mid-micromolar range, although their potencies on mGluR4 and 7 were much lower. One of the most surprising findings of these studies was that three compounds that are viewed as group III mGluR antagonists, (S)-MAP4, MSPG, and MSOP, were not very potent at any of the group III mGluR subtypes in this assay, and we were unable to generate reliable curve fits for any of the receptors at the concentrations examined. Note that the compound MSPG has been described as a nonselective antagonist of group II/III receptors and was shown to inhibit l-AP4 mediated responses in rat cortical slices with a potency of 170 ± 76 nM (Bedingfield et al., 1996). The present findings would suggest that this might not be due to blockade of any of the known group III mGluR subtypes expressed in brain. The relatively low potencies of the antagonists in this assay suggests that these compounds should be used with caution when employed, for example, in electrophysiology experiments designed to determine the role of the group III mGluRs in synaptic transmission.
Using the group III mGluRs as an example, development of this assay provides a major breakthrough in our ability to measure activity of previously intractable GPCRs as well as expand our ability to easily detect and measure the coupling of GPCRs to ion channels in HTS format. The assay is capable of measuring the activity of multiple ligands for GPCRs including agonists, antagonists, and PAMs. Like other HTS assays, the thallium flux method is not immune to false positives. Calcium mobilization assays, for example, can detect ligands that interact with receptors other than the GPCR of interest, ligands that induce calcium influx through channels, or compounds that modulate intracellular calcium handling. Likewise, compounds identified as modulating thallium flux might do so via a variety of mechanisms such as via other endogenous GPCRs that might modulate GIRK or by regulation of other components in these cells, such as the Na/K ATPase, that mediate thallium flux. The hit rate determined in an HTS campaign is also certainly dependent on the individual target, the protocol used for screening (i.e., time of compound addition versus thallium addition, etc.), the compound collection used for screening, compound handling and management, data analysis and hit selection criteria. For the thallium flux assay described here, appropriate follow-up steps, such as screening with cells untransfected with the GPCR of interest, using pertussis toxin to determine whether the effect of a compound is at the level of a Gi/o-coupled GPCR, and exploiting the presence of the endogenous α2C receptor are all encouraged as mechanisms to validate hit specificity.
In contrast to the technique described here, many of the assays commonly used to assess Gi/o-coupled GPCR activity employ more indirect approaches; although useful, they can suffer from a number of deficiencies. For example, decreases in cAMP levels must be measured indirectly by first stimulating cells with a compound, such as forskolin, which increases intracellular cAMP. Compared with kinetic-based assays such as calcium mobilization, cAMP measurements also require significant amounts of time between receptor manipulation and response measurement, which may decrease the sensitivity of the assay for certain types of ligands, such as those that affect receptor desensitization. Several of the assays commonly used for Gi/o-coupled GPCRs, most notably cAMP enzyme-linked immunosorbent assays and guanosine 5′-O-(3-thio)triphosphate binding/proximity assays, can also be quite expensive, particularly if one wishes to perform high-throughput screening of large random chemical libraries to search for new tools or drug leads.
Another assay often used for the study of ligands interacting with GPCRs involves coexpression of promiscuous or chimeric Gα proteins along with the receptor of interest to couple the receptor to an easily measured signal, such as calcium mobilization. It has been observed, however, that some receptors do not couple well to these altered G protein subunits, resulting in very low or nonexistent signals; these altered/chimeric G protein subunits can potentially affect receptor/ligand pharmacology (Kostenis et al., 2005). For example, it has been shown for the Gi/o-coupled serotonin 1A receptor that coupling to a chimeric G protein results in a loss of activity of ipsapirone, a known 5-HT1A agonist with good activity in cAMP inhibition assays (Kowal et al., 2002). This suggests that there is a need to confirm the activity of receptor ligands using signaling pathways that may more accurately reflect the activity of a receptor in vivo. For the group III mGluRs in particular, there is substantial evidence that these receptors regulate ion channel function (Millán et al., 2002; Guo and Ikeda, 2005; Bertaso et al., 2006; Pelkey et al., 2006), and it is widely accepted that Gi/o coupled receptors mediate the regulation of these channels via βγ subunits (for review, see Dascal, 2001). This suggests that the search for new, relevant tools for these receptors may best be found, or at least confirmed for activity before in vivo studies, using an assay that can detect βγ activation. In addition, there is substantial evidence that compounds can induce different conformational states of a GPCR and that distinct ligands interacting with the same receptor can induce completely different signaling pathways or different degrees of signaling through several pathways. This phenomenon has been termed “agonist directed trafficking” or “functional selectivity” (for review, see Urban et al., 2007). Development of an easy assay that allows monitoring of Gβγ subunit signaling should promote further examination of the ability of ligands to potentially induce differential signaling via Gβγ versus Gα subunits of the G protein complex.
In conclusion, we have developed a new strategy to examine activity of Gi/o-coupled receptors that complements current methods by providing a mechanism to examine Gβγ-mediated signaling. This assay can sensitively detect the activity of a number of important types of GPCR ligands and is useful for monitoring ligand activity at both family A (i.e., M4 and α2C) as well as several family C (mGluRs) receptors for which HTS-compatible assay development has been particularly difficult (i.e., mGluRs 7 and 8). The method is compatible with HTS formats and generates robust screening statistics, indicating that it can be used for high-throughput screening of diverse chemical libraries. The method has provided a way to quickly and simultaneously assess ligand activity at the group III mGluRs, a group of receptors that may heavily rely upon Gβγ-mediated signaling in vivo. These studies have revealed that several compounds described as antagonists of these receptors may have very weak activity at these targets, suggesting caution when using these compounds for in vivo studies or in slice preparations. We anticipate that the GIRK-mediated thallium flux strategy will add a critical tool to the battery of assays that can be used for pharmacological tool development, daily pharmacological assays, and drug discovery for Gi/o-coupled GPCRs.
Acknowledgments
HEK 293 cells stably expressing GIRK 1, GIRK 2, and the human M4 muscarinic receptor (HEK/GIRK cells) were generously provided by Drs. Huai Hu Chang and Lily Jan (University of California San Francisco, San Francisco, CA).
Footnotes
-
This work was supported by National Institutes of Health grants NS053536 and NS051342 and the Michael J. Fox Foundation. Vanderbilt University Medical Center is the Molecular Libraries Screening Center Network (MLSCN) site for GPCRs, Ion Channels and Transporters.
-
ABBREVIATIONS: mGluR, metabotropic glutamate receptor; CNS, central nervous system; GPCR, G-protein-coupled receptor; GIRK, G-protein regulated inwardly rectifying K+ channel; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; HEK, human embryonic kidney; DMSO, dimethyl sulfoxide; UK 14,304, 5-bromo-6-(2-imidazolin-2-ylamino)quinoxaline tartrate; AF-DX 116, 11-[[2-[(diethylamino)methyl]-1-piperidinyl]-acetyl]-5,11-dihydro-6H-pyrid[2,3-b][1,4]benzodiazepin-6-one; BRL 44408 maleate, 2-[(4,5-dihydro-1H-imidazol-2-yl)methyl]-2,3-dihydro-1-methyl-1H-isoindole maleate; PHCCC, N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide; MPPG, (R,S)-α-methyl-4-phosphonophenylglycine; MSPG, (R,S)-α-methyl-4-sulfonophenylglycine; MSOP, (R,S)-α-methylserine-O-phosphate; CPPG, (R,S)-α-cyclopropyl-4-phosphonophenylglycine; UBP1112, α-methyl-3-methyl-4-phosphonophenylglycine; (S)-MAP4, (S)-2-amino-2-methyl-4-phosphonobutanoic acid; LY341495, (2S)-2-amino-2-[(1S, 2S)-2-carboxyclycloprop-1-yl]-3-(xanth-9-yl) propanoic acid; BTC-AM, benzothiazole coumarin-acetoxymethyl ester; PTX, pertussis toxin; PAM, positive allosteric modulator; HTS, high-throughput screening; l-AP4, l(+)-2-amino-4-phosphonobutyric acid.
- Received August 20, 2007.
- Accepted December 31, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}