


In simpler times, studies of G protein-coupled receptor (GPCR) signaling focused almost exclusively on pathways characterized by a small number of sequential events, largely confined to the plasma membrane [for example, activation of adenylyl cyclase by the β2AR (Lefkowitz, 2000)]. However, in recent years, there has been a growing awareness that GPCRs stimulate much more complex pathways that ultimately connect events at the plasma membrane to nuclear events, such as gene transcription. Receptor regulation of mitogen-activated protein kinases is a case in point. The complexity, heterogeneity, and apparent redundancy of the pathways that lead from the seven membrane-spanning and tyrosine kinase receptors in the plasma membrane to ERK in the cytosol are dazzling and involve almost every known type of signaling molecule (Luttrell et al., 1999;Gutkind, 2000). Moreover, this remarkable multiplicity of pathways extends to individual receptors. Thus, a single receptor may signal to ERK by numerous pathways, the importance of which may vary dramatically between different cell types or cell lines or even among different isolates of the “same” cell line. Therefore, the very general conclusions drawn by Friedman et al. (2002) concerning the “dominant mechanism for β2AR activation of ERK” in an article in this issue of Molecular Pharmacology represent a broad and potentially misleading simplification. However, this article provides an excellent focal point for discussion of the complexity of signaling pathways that lead from GPCRs to downstream effectors such as ERK and an occasion to highlight an interesting mechanism for regulating the signaling specificity of such receptors: the “switching” of their G protein-coupling specificity by PKA-mediated receptor phosphorylation.
The most thoroughly studied actions of the Gscoupled β2AR are mediated by cAMP and PKA. The first Gs-dependent effect of β2ARs on ERK discovered was cAMP-mediatedinhibition of ERK (Cook and McCormick, 1993; Wu et al., 1993; Crespo et al., 1995). This is due to the Gαs-dependent activation of PKA, leading to phosphorylation and inhibition of c-Raf1 (Cook and McCormick, 1993; Wu et al., 1993). However, stimulation of the β2AR can also activate ERK via a Gs dependent pathway in some cell types. This has been demonstrated in human embryonic kidney (HEK) 293 cells (Schmitt and Stork, 2000) and S49 lymphoma cells (Wan and Huang, 1998). This pathway is pertussis toxin-insensitive and seems to involve Src, the small G protein Rap1, and the B-Raf isoform, rather than c-Raf1. It is independent of Ras. This pathway, originally described by Schmitt and Stork in 293 cells (Schmitt and Stork, 2000), is now recharacterized by Friedman et al. (2002).
However, a growing body of in vivo and in vitro data indicate that some actions of the β2AR are transduced via Gi. These include activation of the enzymes ERK (Daaka et al., 1997), AKT (Zhu et al., 2001), phosphoinositide-3 kinase (Zhu et al., 2001) and certain receptor tyrosine kinases (Maudsley et al., 2000), and inhibition of adenylyl cyclase (Lawler et al., 2001). Gi-mediated activation of ERK by the β2AR has been demonstrated in cultured HEK293 cells (Daaka et al., 1997), monkey kidney cells (COS-7) (Maudsley et al., 2000), Chinese hamster ovary (CHO) cells (Zamah et al., 2002), and cultured rat cardiac myocytes (Zou et al., 1999). This Gi-mediated pathway is pertussis toxin-sensitive and seems to be transduced by Gβγ subunits, Src, Ras, and c-Raf1, and may involve transactivation of the epidermal growth factor receptor (Daaka et al., 1997; Luttrell et al., 1999; Zou et al., 1999; Gutkind, 2000; Maudsley et al., 2000; Lawler et al., 2001; Zhu et al., 2001;Zamah et al., 2002).
These two pathways are schematically depicted in Fig.1. A particularly interesting aspect of the Gi-mediated stimulation of ERK via the β2AR is that it seems to require prior phosphorylation of the receptor by PKA (Daaka et al., 1997), a mechanism previously demonstrated to “desensitize” β2ARs by decreasing their coupling to Gs (Benovic et al., 1985; Pitcher et al., 1992). PKA phosphorylation simultaneously diminishes β2AR coupling to Gs and increases coupling to Gi. This mechanism has been referred to as G protein “switching” (Daaka et al., 1997). That some regulatory mechanism would be required to “switch” or alter the inherent coupling of the β2AR and thus enable its interaction with Gi was clear, in retrospect, from reconstitution experiments performed almost 20 years ago (Cerione et al., 1985). These demonstrated, in reconstituted systems consisting only of purified receptors and G proteins, that the β2AR, although coupling robustly to Gs, was virtually devoid of coupling to Gi.
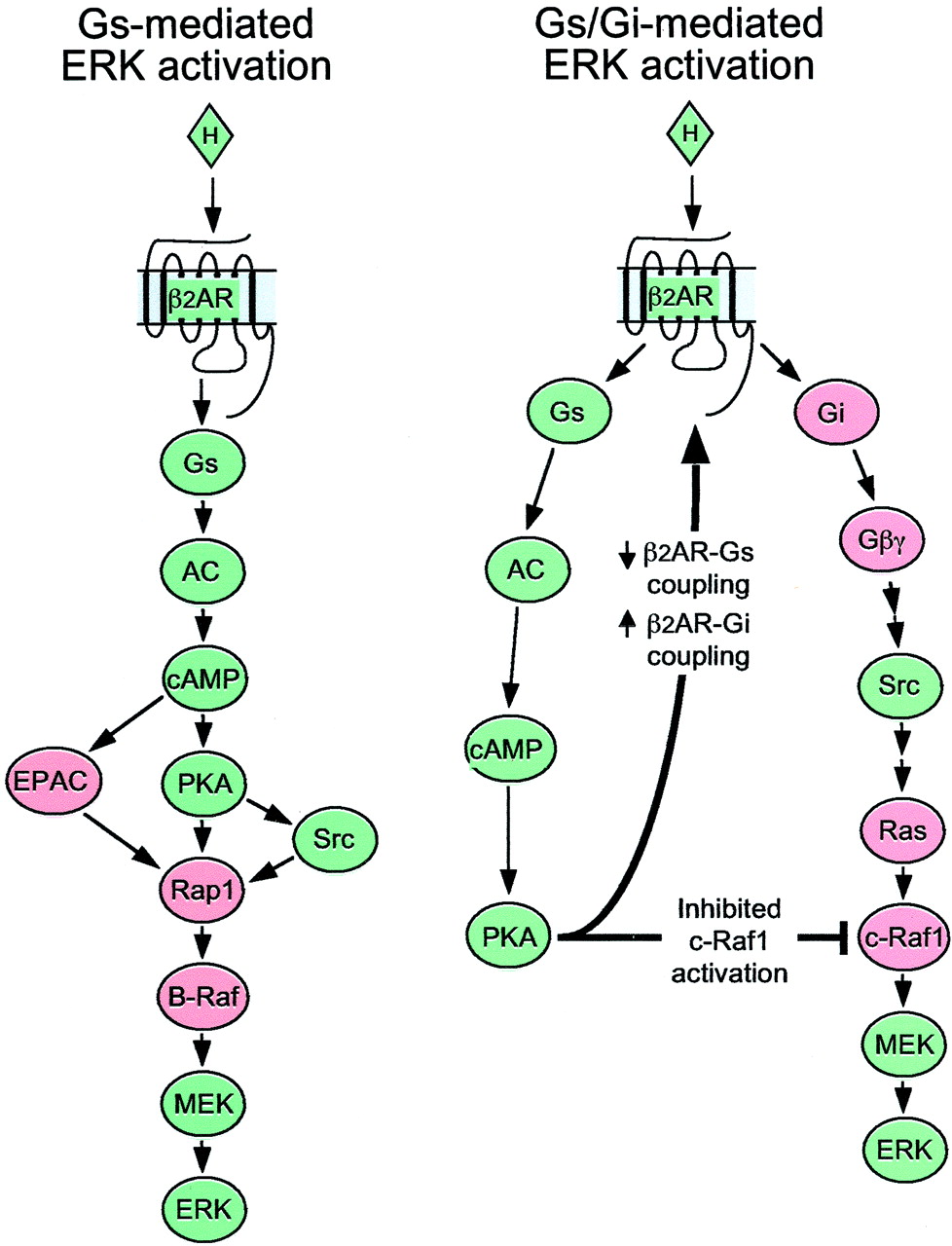
Gs- and Gi-mediated pathways for ERK activation by the β2AR. Signaling intermediates colored green are shared by both pathways, whereas those in salmon are unique to one pathway.
The first indication that PKA-mediated phosphorylation of the β2AR might do more than simply “desensitize” the receptor by inhibiting its coupling to Gs was provided by Okamoto et al. (1991). They demonstrated that a short peptide derived from the third cytoplasmic loop of the β2AR effectively activated purified Gs but only very weakly activated Gi in vitro. This peptide contains one of the two consensus PKA phosphorylation sites found in the receptor. When the peptide was phosphorylated by PKA, its ability to activate Gs was dramatically reduced, whereas its ability to activate Gi was reciprocally increased.
These results are essentially identical to findings in recent in vitro reconstitution studies with the intact recombinant human β2AR and recombinant Gsand Gi (Zamah et al., 2002). Reconstituted native β2AR mediates robust activation of Gs but not Gi. When the receptor is phosphorylated in vitro by PKA, Gscoupling is reduced, but Gi activation is markedly enhanced. The results are replicated when a purified recombinant β2AR, in which the PKA phosphorylation sites are mutated (S→D) to mimic the effects of phosphorylation, is tested in the reconstituted system. Again, Gs coupling is impaired, whereas Gi stimulation is increased (Zamah et al., 2002).
That PKA-mediated phosphorylation of the β2AR might regulate its coupling to Gs and Gi in cells was first appreciated in studies performed using a line of HEK293 cells; Daaka et al. (1997)demonstrated that activation of ERK by endogenous β2ARs was mediated largely by Gi (pertussis toxin-sensitive). However, concomitant PKA activation seemed to be required because the ERK activation was blocked by the PKA inhibitor H89, as was the ability of the receptor to stimulate [32P]GTP azidoanilido loading of Gi in cell membranes. Gi-mediated inhibition of forskolin-stimulated adenylyl cyclase by the β2AR in HEK293 cells was also blocked by pertussis toxin or H89 but not by a PKC inhibitor (Lawler et al., 2001). That the receptor was the site of the required PKA phosphorylation was suggested by the failure of a mutant β2AR (S→A), which was not a substrate for PKA, to activate ERK when transfected into these cells (Daaka et al., 1997).
Gi-mediated activation of ERK by the β2AR, which requires prior activation of PKA, has subsequently been demonstrated in murine submandibular gland cells (Luo et al., 1999), rat cardiac myocytes (Zou et al., 1999), and CHO cells (Zamah et al., 2002). Moreover in the latter two systems, as in the original studies with HEK293 cells, the β2AR itself was implicated as the locus of PKA phosphorylation, because phosphorylation site mutants of the β2AR (PKA−) failed to activate ERK or acted as dominant negatives for isoproterenol-stimulated ERK activation. Recently, evidence has been presented suggesting that this switching mechanism operates in vivo in the mouse heart (Hasseldine et al., 2002).
An entirely analogous switching mechanism has been reported for the Gs-coupled mouse prostacyclin receptor (Lawler et al., 2001). In HEK293 cells, this receptor can activate Gs (stimulate cAMP accumulation), Gi (inhibit forskolin stimulated cAMP accumulation), and Gq (activate phospholipase C leading to calcium release). However, the Gi- and Gq-mediated responses are blocked by H89, and a receptor point mutant (S357A) that cannot undergo PKA phosphorylation activates only Gs. Moreover, whereas the wild-type receptor can be immunoprecipitated with Gs, Gi, and Gq in an agonist-dependent manner, the mutant receptor interacts only with Gs. Not only do these studies confirm the Gs-to-Gi switching mechanism, they extend it to another G protein, Gq (Lawler et al., 2001). A further interesting twist is that the human prostacyclin receptor, which has a PKC site in place of the PKA site of the mouse receptor, couples to Gs and Gq but not Gi. Substitution of the PKC site by a PKA site by mutagenesis enables PKA-dependent coupling to Gi, which is lost on further mutagenesis to remove the relevant serine from this position (Miggin and Kinsella, 2002). Thus, at present, it seems that PKA, but not PKC, can mediate such switching phenomena.
The studies reviewed above (summarized in Table1), provide compelling support for the idea that PKA-mediated phosphorylation of GPCRs regulates their coupling to different G proteins, especially Gi. So why then do Friedman et al. (2002) fail to observe this switching mechanism in their 293 cells, a line previously used by others to demonstrate just such effects for both the β2AR (Daaka et al., 1997; Lawler et al., 2001) and the prostacyclin receptor (Lawler et al., 2001; Miggin and Kinsella, 2002)? The most obvious lesson is that transformed cell lines are not uniform in their properties. During prolonged culture, such lines alter chromosome number, gene expression signaling pathways used, and even morphology. Thus, it is not possible to draw from any single study of GPCR-mediated ERK activation in a cultured cell line any valid, generally applicable conclusions about the “dominant pathway of ERK activation” by that receptor.
PKA-mediated receptor phosphorylation switches G protein-coupling specificity
Lines referred to as HEK293 have a remarkable heterogeneity; for example, we recently tested the sensitivity to pertussis toxin of ERK activity stimulated by isoproterenol through endogenousβ2ARs in nine different isolates of HEK293 cells and found that this varied from 0 to 100% (Fig.2). Friedman et al. (2002) reported a minimal effect of pertussis toxin on ERK activation by the β2AR, whereas, by contrast, Daaka et al. (1997)reported that the β2AR-mediated effect on ERK was quite sensitive to pertussis toxin. Based on these findings, the divergent effects of a PKA-β2AR mutant receptor on ERK activity in the different studies are quite predictable. In the HEK 293 cells used by Friedman et al. (2002), the β2AR activates ERK almost exclusively via Gs. Because such signaling is inhibited by PKA phosphorylation of the receptor (Fig. 1), the PKA-β2AR should be normally active or superactive in these cells. Conversely, in the cells used by Daaka et al. (1997), the β2AR activates ERK primarily via Gi. Because such signaling is enabled by receptor phosphorylation (Fig. 1), the PKA-β2AR should be inactive in these cells. This is precisely what was found in the two studies (Daaka et al., 1997; Friedman et al., 2002).
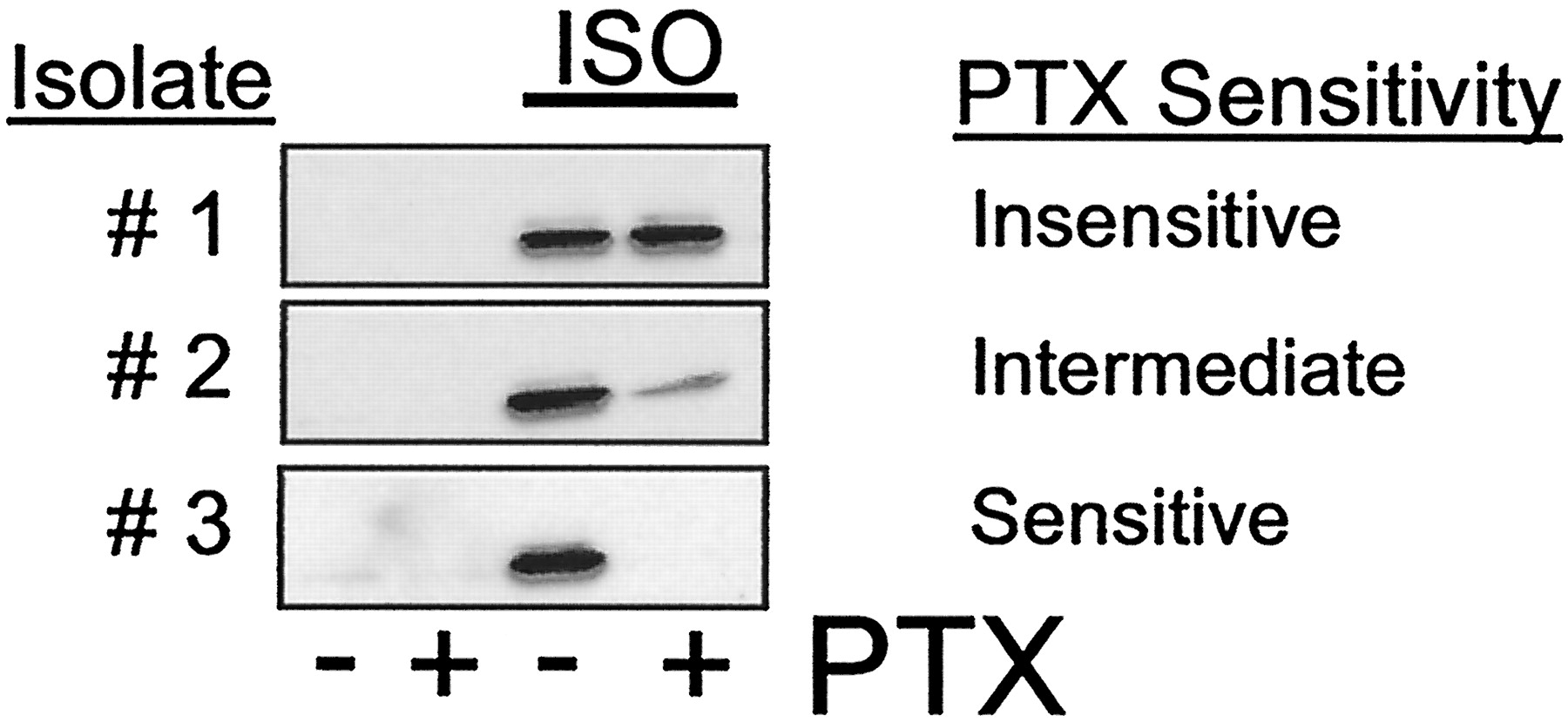
Pertussis toxin sensitivity of β2AR stimulated ERK activity in different isolates of HEK 293 cells. 293 cells plated in 12-well dishes (150,000 cells/well) were serum-starved and pertussis toxin-treated (PTX, 100 ng/ml) overnight before stimulation with 1 μM isoproterenol (ISO) for 5 min at 37°C. Cell lysates were prepared, and immunoblotting performed following standard procedures using a phospho-ERK specific antibody (Cellular Signaling, Beverly, MA). A representative experiment is shown, but experiments were repeated at least three times with each isolate. The three isolates shown are representative of nine that were studied. In two of the nine isolates, isoproterenol did not stimulate ERK phosphorylation. Isolates were obtained from various investigators at Duke University Medical Center and from the American Type Culture Collection (Manassas, VA).
Currently unclear, however, are the specific differences that account for the marked variability in pathway usage between these different isolates of 293 cells. This largely unstudied problem may hold clues to understanding important aspects of cellular regulation. Recently developed gene chip array and proteomics approaches have obvious applicability here, as do more specific, hypothesis-driven approaches focusing on expression levels of the obvious known intermediates in the various pathways. Moreover, to understand the ultimate physiological outcome, the relative contributions of each of the different pathways need to be characterized both in primary cells and in vivo.
In conclusion, the article by Friedman et al. (2002) illustrates the confusions of interpretation that result from failing to appreciate the diversity and complexity of mechanisms that can be used by a single receptor to signal to a very downstream effector in different cells. In a final ironic twist, the pathway globally assigned by Friedman et al. as “the dominant pathway for β2ARactivation of ERK” (Gs → AC → PKA → Src →→ Rap1 →→→ ERK) has recently been shown toinhibit Ras-dependent ERK activity in NIH3T3 fibroblasts (Schmitt and Stork, 2002), thereby inhibiting cell growth.
Acknowledgments
We thank Musa Zamah, Seungkirl Ahn, and Darrell Capel for help with experiments.
Footnotes
- Received August 1, 2002.
- Accepted August 6, 2002.
-
This work was supported in part by National Institutes of Health grant HL16037. R.J.L. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- GPCR
- G protein-coupled receptor
- AR
- adrenergic receptor
- ERK
- extracellular signal-regulated kinase
- HEK
- human embryonic kidney
- PKC
- protein kinase C
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}