


Abstract
Stimulation of the β2-adrenergic receptor (β2AR) in human embryonic kidney (HEK) 293 cells causes a transient activation of Extracellular Signal-Regulated Kinase (ERK) 1/2. One of the mechanisms proposed for this activation is a PKA-mediated phosphorylation of the β2AR that switches receptor coupling from Gs to Gi and triggers internalization of the receptor. To examine these phenomena, we characterized agonist activation of ERK1/2 in HEK293 cells by the endogenous β2AR and in HEK293 cells stably overexpressing either the wild-type β2AR or a substitution mutant β2AR (PKA−) that lacks the cyclic AMP-dependent protein kinase (PKA) consensus phosphorylation sites (S261A, S262A and S345A, S346A). As the baseline, we established that epinephrine stimulation of the endogenous β2AR in HEK293 cells (20–30 fmol/mg) caused a rapid and transient activation of ERK1/2 with an EC50 of 5 to 6 nM. In contrast, the potency of epinephrine stimulation of ERK1/2 in cells stably overexpressing WTβ2AR and PKA− (2–4 pmol of β2AR/mg) was increased by over 100-fold relative to HEK293 cells, the EC50 values being 20 to 60 pM. The nearly identical 100-fold shift in EC50 for ERK1/2 activation in the PKA− and WTβ2AR relative to that in the HEK293 showed that the PKA− are fully capable of activating ERK1/2. We also found maximal activation of ERK1/2 in the overexpressing cell lines at concentrations of epinephrine that cause no internalization (i.e., the EC50 for internalization was 75 nM). Pertussis toxin pretreatment caused only a weak inhibition of epinephrine activation of ERK1/2 in the HEK293 (7–16%) and no inhibition in the PKA− cells. Finally we found that the Src family kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (10 μM) caused a >90% inhibition of epinephrine or forskolin activation of ERK1/2 in both cell lines. Our results indicate that the dominant mechanism of β2AR activation of ERK1/2 does not require PKA phosphorylation of the β2AR, receptor internalization or switching from activation of Gs to Gi but clearly requires activation of a Src family member that may be downstream of PKA.
Early reports that purified β2AR or a synthetic peptide corresponding to the third intracellular loop of the β2AR could activate pure Gi in reconstituted preparations in vitro raised the possibility that the β2AR could activate Gi in vivo (Cerione et al., 1985; Rubenstein et al., 1991), although with reduced efficiency relative to Gs. It was then demonstrated that PKA phosphorylation of a IIIi loop peptide of the β2AR increased activation of Gi in reconstituted preparations and slightly reduced its activation of Gs (Okamoto et al., 1991), leading to the proposal that β2AR activation could switch from Gs to Gi after PKA phosphorylation of the IIIi loop. Recent support for this scheme was derived from studies showing that isoproterenol activation of ERK1/2 in HEK293 cells was blocked more than 85% by pretreatment with pertussis toxin, that isoproterenol activation of ERK1/2 was inhibited by H89, and that transient expression of a β2AR lacking the PKA consensus sites (S261A, S262A, S345A, S346A) inhibited isoproterenol activation of ERK1/2 by 40% (Daaka et al., 1997). Other studies are not consistent with the switching hypothesis. First, it has been shown that forskolin activates ERK1/2 consistent with a PKA-mediated pathway of activation downstream of the receptor (Daaka et al., 1997; Schmitt and Stork, 2000). Second, a recent study found that isoproterenol activation of ERK1/2 in HEK293 cells was not inhibited by pertussis toxin (Schmitt and Stork, 2000). Third, we have shown that pertussis toxin pretreatment does not alter epinephrine-induced desensitization of HEK293 cells and S49 lymphoma cells, as would be expected if a Gs to Giswitch occurred (Clark et al., 1986; Seibold et al., 2000). Fourth, isoproterenol activation of ERK1/2 does not occur in the kin− and cyc− mutants of S49 mouse lymphoma cells (Wan and Huang, 1998).
Reports on the role of internalization of the β2AR in ERK1/2 activation, studied through the use of transient expression of dominant negative dynamin (K44A) and mutant arrestins, have also been inconsistent. Several studies suggested that internalization was required for ERK1/2 activation by the β2AR in HEK293 cells (Daaka et al., 1998;Luttrell et al., 1999b) and COS-7 cells (Pierce et al., 2000), whereas others found that internalization was not required for β2AR activation of ERK1/2 in COS-1 cells (DeGraff et al., 1999).
Several lines of evidence suggest a role for Src in activation of ERK1/2 by the β2AR: i) isoproterenol activation caused the formation of a multiprotein complex of receptor, β-arrestin, and Src, and dominant negative mutants of Src reduced ERK1/2 activation (Daaka et al., 1997; Luttrell et al., 1999b; Zou et al., 1999; Miller et al., 2000); ii) Src was shown to be activated by Gsα and Giα in reconstituted in vitro preparations suggesting direct activation of Src by G proteins (Ma et al., 2000); and iii) it was suggested that Src binds to tyrosine 350 of the β2AR after phosphorylation of this residue by an unidentified tyrosine kinase (Fan et al., 2001).
Obviously there seems to be considerable complexity in β2AR activation of ERK1/2, and many issues are unresolved. In this article, we examine the role of receptor switching, Gi-internalization, and Src family kinases in β2AR activation of ERK1/2 in HEK293 cells. We had previously generated a mutant β2AR in which the two PKA consensus sites were eliminated by substitution of serines 261, 262, 345, and 346 with alanine, and this mutant receptor, termed PKA−, was stably overexpressed in the HEK293 cell line. We reported previously that the coupling efficiency of this mutant and the EC50 for activation of adenylyl cyclase were nearly identical to those of WTβ2AR overexpressed at comparable levels (Seibold et al., 1998, 2000). Stable overexpression of the PKA− and the WTβ2AR (∼100-fold relative to endogenous receptor) caused a dramatic left-shift in the EC50 for epinephrine activation of adenylyl cyclase. This property of the β2AR allows the analysis of mutant receptors overexpressed in HEK293 cells because the response of the endogenous receptor is overwhelmed if expression is high enough. Because the EC50 for epinephrine activation of adenylyl cyclase in cells expressing either the PKA− or WTβ2AR is left-shifted relative to the endogenous receptor, it follows that activation of ERK1/2 should show a similar left-shift if the mechanism involves receptor activation of Gs. The prediction of the switching model in which the PKA-phosphorylated β2AR is proposed to activate Gi is that there should be no left shift in ERK1/2 activation relative to the endogenous receptor; rather, there should be an inhibition of the activation by endogenous β2AR (Daaka et al., 1997). The potency of agonist activation of ERK1/2 in HEK293 cells by transiently expressed wild-type or PKA− mutant β2ARs was not examined in previous studies (Daaka et al., 1997; Schmitt and Stork, 2000).
In this article, we demonstrate that the stably overexpressed PKA− mutant of the β2AR shows the predicted enormous left shift in the potency of activation of ERK1/2 by epinephrine relative to that of the endogenous low level of expression in the HEK293 and that the shift is nearly identical to that found with overexpressed wild-type β2AR. This demonstrated that the PKA consensus sites were not required for full activation of ERK1/2. Our data also show only a minor role for Gi in epinephrine activation of ERK1/2, no requirement for internalization (because the EC50for epinephrine-induced internalization in the PKA− was about 1000-fold higher than that for activation of ERK1/2), and a requirement for Src family kinase activation.
Materials and Methods
Materials.
Antibodies to ERK1/2 and phospho-ERK1/2 were purchased from Cell Signaling Technology Inc. (Beverly, MA). [32P]NAD and [3H]CGP-12177 were obtained from PerkinElmer Life Sciences (Boston, MA). Pertussis toxin was from List Biological Laboratories Inc. (Campbell, CA). Enhanced chemiluminescence reagents and Hyperfilm were from Amersham Biosciences (Piscataway, NJ). Agonists and antagonists were purchased from Sigma-Aldrich. BA85 nitrocellulose was from Schleicher & Schuell (Keene, NH). Cell culture reagents were purchased from Mediatech (Herndon, VA). The Src family kinase inhibitor PP2 was from Alexis Corporation (Läufelfingen, Switzerland).
Cell Culture.
HEK293 cells purchased from the American Type Culture Collection (Manassas, VA) were grown in 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. The stably transfected HEK293 cell lines used in this study expressing either WT or mutant receptors have been described previously (January et al., 1997; Seibold et al., 1998; Seibold et al., 2000), and were as follows: double epitope-tagged wild-type receptor (HA-β2AR-His6) and the double epitope-tagged PKA− (S261A, S262A, S345A, S346A), both with the hemagglutinin (HA) tag on the N terminus and His6 tag on the C terminus; and HA-tagged wild-type (HA-β2AR). The stably transfected cell lines were cultured in medium containing 400 μg/ml G418. The levels of β2AR in these transfect lines were 2 to 4 pmol/mg membrane protein, whereas the level of endogenous β2AR was 30 to 40 fmol/mg.
Cell Treatment and Preparation of Solubilized Extract.
Cells were grown to confluence in growth medium in 12-well plates coated with poly(l-lysine). The medium was removed 18 h before treatment, cells were incubated for 30 min with serum-free DMEM, and that medium was replaced with 2 ml of serum-free DMEM with or without 100 ng/ml of pertussis toxin. Cells were treated with hormones or carrier as indicated at 37°C with continuous rocking; pretreatments with β2AR antagonists were for 2 min before agonist treatment, and PP2 was added 1 h before agonist treatment. Epinephrine was stored in 10 mM ascorbate/100 mM thiourea pH 7 (AT). Stock solutions were diluted 100-fold when additions were made to cells such that the final concentration of AT in all incubations (control and treated) was 0.1 mM ascorbate/1.0 mM thiourea. PP2 was dissolved and stored in DMSO and was diluted 100-fold for cell treatments. To terminate treatments, the medium was removed and 0.5 ml of solubilization buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 0.8% dodecyl maltoside, 1 mM EDTA, pH 7, 20 mM tetrasodium pyrophosphate, 10 mM NaF, 10 μg/ml benzamidine, 10 μg/ml trypsin inhibitor, 10 μg/ml leupeptin, and 0.1 μM okadaic acid) was added to each well. The plates were placed on ice, and the solubilized cells were pipetted into 1.5-ml microcentrifuge tubes. The tubes were rocked at 4°C for 30 min, and then centrifuged at 12,500 rpm for 15 min. The supernatants were removed and frozen at −80°.
Measurement of ERK1/2 and Phospho-ERK1/2.
To measure activation of ERK1/2 by Western blotting, 20 μl of the solubilized extracts were resolved by SDS-PAGE (10% gels) and blotted onto BA-85 nitrocellulose. The blots were blocked for 1 h in 5% nonfat dried milk in wash buffer (150 mM NaCl, 50 mM Tris pH 7.5, and 0.1% Tween 20). After two 10-min washes, the blots were incubated overnight at 4°C on a rocker with a 1:1000 dilution of anti-phospho-ERK, washed twice for 10 min each, and incubated for 1 h at room temperature with a 1:5000 dilution of secondary antibody (goat anti-rabbit HRP-conjugate, Bio-Rad, Hercules, CA) After 2 washes, the blots were exposed to ECL reagents for 1 min, dried, and exposed to Hyperfilm (Amersham Biosciences) for 15 sec to 2 min. The blots were then stripped and reprobed identically with anti-ERK. Western blots were quantitated using Scion Image software (Scion Corp., Frederick, MD). All results with the anti-phospho-ERK were normalized to the corresponding anti-ERK.
Membrane Preparation and Adenylyl Cyclase Assay.
Cells were plated into 100-mm dishes that had been precoated with poly(l-lysine). Treatments were administered at 37°C and were stopped by removal of media followed by six washes with 5 ml of ice-cold HE buffer (20 mM HEPES, pH 8.0, 1 mM EDTA, pH 7). The cells were scraped into HE containing 10 μg/ml leupeptin and 0.1 μM okadaic acid and homogenized with seven strokes in a type B Dounce homogenizer (Bellco Glass, Vineland, NJ). The homogenates were layered onto sucrose step gradients (23 and 43% prepared in HE buffer) and centrifuged at 25,000 rpm in a Beckman SW28.1 rotor for 35 min. The fraction at the 23/43% sucrose interface was removed, frozen in liquid nitrogen, and stored at −80°C. Adenylyl cyclase activity was measured as described previously (Seibold et al., 2000).
ADP Ribosylation.
Membranes from control or pertussis toxin-treated cells were incubated for 30 min at 30°C with 10 μM NAD, 0.5 mM ATP, 0.2 mM GDP, 5 mM MgCl2, 1 mM EDTA, pH 7, 20 mM Tris, pH 7.5, 5 mM dithiothreitol, 5 mM thymidine, 8 mM creatine phosphate, 8 U/ml creatine phosphokinase, and 5 μCi/tube [32P]NAD. The incubation mix was diluted in 5 ml of 20 mM Tris, pH 7.5, and centrifuged for 15 min at 30,000 rpm in a Beckman 50Ti rotor (Beckman Coulter, Fullerton, CA). The pellets were each dissolved in SDS sample buffer and resolved on 12% SDS-PAGE gels. The proteins were transferred to nitrocellulose and exposed to a Storm Phosphorimager screen (Molecular Dynamics, Sunnyvale, CA). The phosphorylated bands were quantitated using ImageQuant (Molecular Dynamics).
Internalization.
The procedure for measuring internalization of the β2AR has been described in detail previously (Seibold et al., 2000). Cells in 12-well dishes were treated with epinephrine or AT for 5 min. Cells were then washed five times with ice-cold, serum-free DMEM, placed on ice, then incubated with 5 to 10 nM [3H]CGP-12177 with or without 1 μM alprenolol in serum-free DMEM. Dishes were then incubated for 1 h on ice, washed twice with ice-cold PBS to remove [3H]CGP-12177, and cells released by trypsin were transferred to scintillation vials for counting.
Results
Characterization of β2AR Agonist Activation of ERK1/2 in HEK293 Cells.
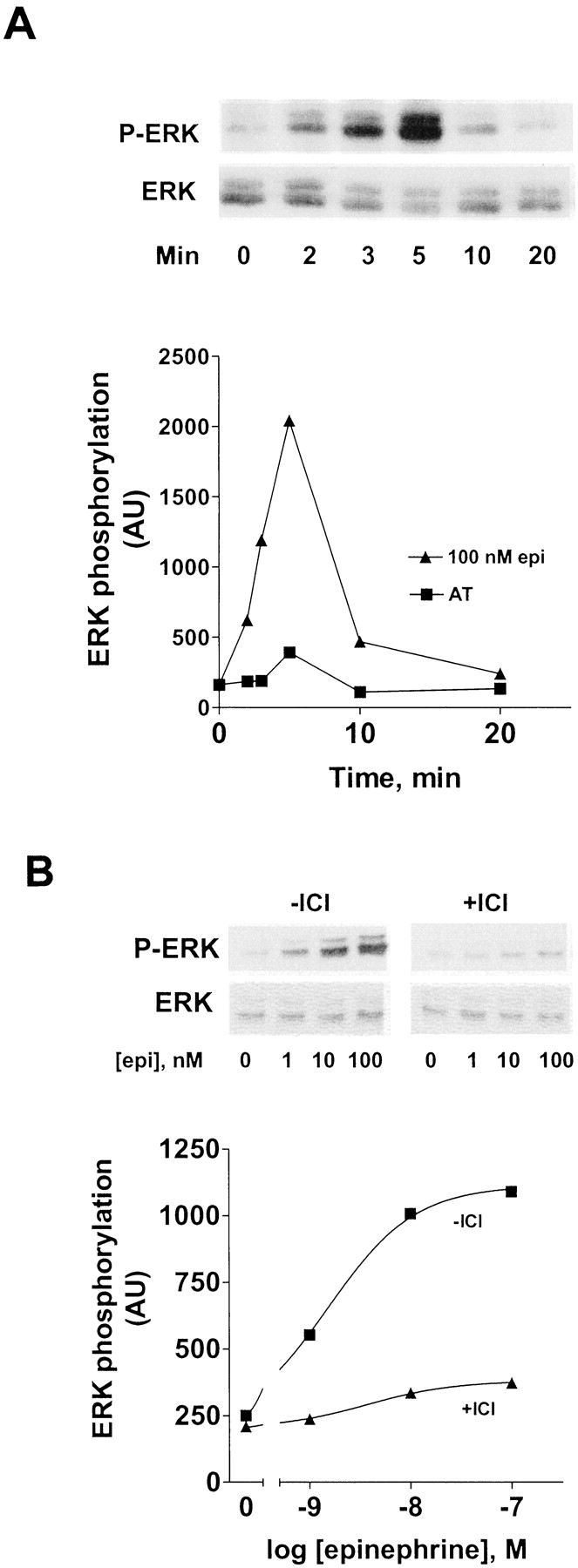
To examine the activation of ERK1/2 by the endogenous β2AR (≈20–30 fmol/mg membrane protein), HEK293 cells grown to confluence were placed in serum-free medium for 18 h before treatment with epinephrine. As shown in Fig.1A, activation of ERK1/2 by 100 nM epinephrine showed a slight lag after which levels peaked at 5 min and then sharply declined, returning to near control levels by 20 min. To determine whether the activation of ERK1/2 by epinephrine was mediated by its binding to the β2AR, several approaches were taken. We found that 5 μM ICI-118551, a selective β2AR antagonist, added 2 min before a 5-min incubation with various concentrations of epinephrine, caused a nearly complete inhibition of ERK1/2 activation (Fig. 1B), consistent with the competitive action of ICI-118551. We also made the following observations (data not shown): i) the activation of ERK1/2 by 100 nM isoproterenol was equivalent to epinephrine and was also blocked by ICI-118551; ii) 20 nM salmeterol, a partial agonist and one of the most selective β2AR agonists, showed ≈80% of the activation of ERK1/2 induced by epinephrine and was also blocked by ICI-118551; iii) 100 nM prazosin (an antagonist of both α1- and α2-adrenergic receptors) had no effect on epinephrine activation of ERK1/2. These data demonstrated that epinephrine activation of ERK1/2 was specific for the β2AR.

Characterization of epinephrine activation of ERK1/2 in HEK293 cells. A, time course of epinephrine activation of ERK1/2. HEK293 cells grown to confluence in 12-well plates were treated with 100 nM epinephrine or carrier (AT) for the indicated times. B, ICI-118551 inhibition of epinephrine-activated ERK1/2. HEK293 cells were pretreated for 2 min ± 5 μM ICI-118551, followed by 5-min treatments with the indicated concentrations of epinephrine or carrier (AT). After treatment, cells were solubilized and extracts were subjected to SDS-PAGE, transferred onto BA85 nitrocellulose membranes, and Western blotted with anti-phospho-ERK1/2 as described underMaterials and Methods. Gels were then stripped and reprobed with anti-ERK1/2. Results with anti-phospho-ERK were normalized to the corresponding anti-ERK data and shown as arbitrary units (AU) on the y-axis.
To determine the EC50 for epinephrine activation of ERK1/2, HEK293 cells were incubated for 5 min with a range of epinephrine concentrations. Immunoblots from one representative experiment are shown in Fig. 2, as well as a data summary from five identical experiments. The EC50 for epinephrine activation of ERK1/2 was 5.7 nM. It should be noted that this EC50 is about 100- to 200-fold lower than that for epinephrine activation of adenylyl cyclase in membranes prepared from these cells (600–800 nM, data not shown).

Dose response of epinephrine activation of ERK1/2 in HEK293 cells. HEK293 cells were treated for 5 min with the indicated concentrations of epinephrine. The cells were solubilized and ERK1/2 phosphorylation measured and quantitated as described underMaterials and Methods. The data on the graph are the mean values ± S.E.M. from five experiments, each performed in duplicate. The data from each experiment were converted to percentage of the maximum stimulation before determination of the means. The Western blot is from one representative experiment.
Epinephrine Stimulation of ERK1/2 in HEK293 Cells Overexpressing Either PKA−, HA-β2AR-His6, or HA-β2AR.
As discussed above, overexpression of β2AR in a variety of cell types causes a progressive decrease in the EC50 (increased potency) for β2AR activation of adenylyl cyclase (Whaley et al., 1994;Seibold et al., 1998; Clark et al., 1999). We have shown previously that the EC50 values for epinephrine activation of adenylyl cyclase in cells overexpressing either the WTβ2AR or the PKA− were left-shifted 50- to 100-fold relative to the endogenous receptor in HEK293 cells. It follows that activation of ERK1/2 should show a similar left shift if the mechanism involved for both processes was similar at the level of receptor coupling to Gs/adenylyl cyclase.
To determine whether the EC50 for epinephrine activation of ERK1/2 in the PKA− overexpression line was left-shifted relative to the endogenous receptor and whether the shift in the potency of the PKA− was similar to that of two lines overexpressing the WTβ2AR to similar levels, the experiments shown in Fig.3 were performed. HEK293 cells stably overexpressing either the PKA−, HA-β2AR-His6, or HA-β2AR (2–4 pmol/mg) were incubated with a range of epinephrine concentrations for 5 min and the EC50 values for ERK1/2 activation determined. The EC50 for epinephrine activation of ERK1/2 in the PKA− cell line was ≈ 36 pM, more than 100-fold left-shifted relative to epinephrine activation of ERK1/2 by the endogenous receptor in HEK293 cells. Concentrations as low as 10 pM gave a significant increase in ERK1/2 phosphorylation. The EC50 values for epinephrine activation of ERK1/2 in the HA-β2AR-His6 and HA-β2AR were in the range of 20 to 40 pM. These data demonstrated that the PKA−β2AR was unimpaired in its activation of ERK1/2 because the potency of epinephrine activation was similar in the two lines overexpressing the wild-type receptors. Furthermore, as with the endogenous receptor, the left shift in the EC50for ERK1/2 activation by the three overexpressing clones relative to their EC50 for adenylyl cyclase activation (January et al., 1998; Seibold et al., 2000) was more than 100-fold.

Dose response of epinephrine activation of ERK1/2 in HEK293 cells stably expressing either the PKA−, HA-β2AR-His6, or HA-β2AR. Cells expressing the various receptors were treated for 5 min with the indicated concentrations of epinephrine. The cells were solubilized and ERK1/2 phosphorylation was measured and quantitated as described underMaterials and Methods. A, Western blots are from one representative experiment for each cell line. B, the data are the mean ± S.E.M. of seven (PKA−) or four (HA-β2AR-His6 and HA-β2AR) experiments, each performed in duplicate. The data from each experiment were converted to percentage of maximum epinephrine stimulation of ERK1/2 phosphorylation before calculating the means.
Because there was the possibility that the time course of ERK1/2 activation by epinephrine was altered at these very low concentrations, we followed the time course of ERK1/2 activation in the PKA− by 1.0 and 10.0 nM epinephrine as shown in Fig. 4. The data show that the time course of activation by 1.0 and 10.0 nM epinephrine in the PKA− cells was similar to that of the endogenous receptor in HEK293 cells. With lower concentrations of epinephrine, there is some indication that the lag is extended; however, the peak level of ERK1/2 remained at 5 min. A similar time course of ERK1/2 activation was found in the cells expressing the two wild-type receptors.

Time course of epinephrine activation of ERK1/2 in PKA− cells. PKA− cells were treated with either 1.0 nM epinephrine (▪) or 10.0 nM epinephrine (▴) for the times indicated on the graph. The value at zero time is the mean of controls determined at each of the time points. The cells were solubilized and ERK1/2 phosphorylation measured and quantitated as described under Materials and Methods. The data are from one representative experiment.
Effect of Pertussis Toxin Pretreatment of HEK293 Cells on the β2-Adrenergic Activation of ERK1/2.
To determine the role of Gi in epinephrine activation of ERK1/2, we examined the effect of pertussis toxin pretreatment. HEK293 cells expressing only endogenous levels of receptor and the PKA− cell line were pretreated with or without 100 ng/ml of pertussis toxin for 18 h in serum-free medium. Cells were then incubated with various concentrations of epinephrine for 5 min. As shown in Fig.5A (summary of seven independent experiments with a typical Western blot above), pertussis toxin pretreatment of HEK293 cells expressing the endogenous receptor caused only a 7 to 16% inhibition of ERK1/2 activation by the various concentrations of epinephrine. We also found only marginal pertussis toxin inhibition of either isoproterenol or salmeterol activation of ERK1/2 in the HEK293 cell line (data not shown). No significant pertussis toxin inhibition of epinephrine activation of ERK1/2 in the PKA−-expressing cells was observed (Fig. 5B).

Effect of pertussis toxin on epinephrine activation of ERK1/2 in HEK293 cells. HEK293 cells expressing either the endogenous β2AR only (A) or the PKA−β2AR (B) growing in 12-well plates were placed in serum-free medium and then treated with or without 100 ng/ml pertussis toxin (PTOX) for 18 h as described under Materials and Methods. Cells were incubated with increasing concentrations of epinephrine for 5 min as indicated. Phosphorylation of ERK1/2 was measured as described under Materials and Methods. The data are the means ± S.E.M. of seven (HEK293) or three (PKA−) experiments. One representative Western blot is shown for each cell line.
Because pertussis toxin treatment caused only a weak inhibition of epinephrine activation of ERK1/2 by the endogenous receptor, and no inhibition of ERK1/2 activation by the PKA−, we performed several controls to determine the extent of toxin activity. First, cells were pretreated overnight with pertussis toxin, and membranes derived from control and pertussis toxin-treated HEK293 or PKA− cells were ADP-ribosylated in the presence of [32P]NAD. Overnight treatment with pertussis toxin resulted in a >90% inhibition of ADP-ribosylation of Gi/Go in membrane preparations (Fig. 6A). As a second control, we examined pertussis toxin's effect on epinephrine stimulation of adenylyl cyclase. We found that pertussis toxin treatment of HEK293 cells caused an approximate doubling of epinephrine-stimulated adenylyl cyclase activity with no change in the EC50 (Fig. 6B). This result is consistent with our prior study of this toxin's effects on epinephrine stimulation of adenylyl cyclase in the HEK293 cells overexpressing the wild-type and mutant β2AR (Seibold et al., 2000) (i.e., a doubling of the V max and no change in the EC50).

Evidence for the efficacy of pertussis toxin treatments. A, ADP ribosylation of Gi/Go in membranes from HEK293 and PKA− cells pretreated for 18 h ± pertussis toxin. HEK293 or PKA− cells plated into 100-mm dishes were treated with or without pertussis toxin (100 ng/ml) and membranes were harvested and frozen as described underMaterials and Methods. Thawed membranes were then incubated for 30 min in the presence or absence of pertussis toxin in the ADP-ribosylation mix containing [32P]NAD. Membranes were washed and aliquots resolved on 12% SDS-PAGE. B, epinephrine-stimulated adenylyl cyclase activity in membranes from HEK293 cells treated with pertussis toxin or not. HEK293 cells in 100-mm dishes were treated overnight with or without 100 ng/ml of pertussis toxin, membranes were prepared, and adenylyl cyclase stimulation by the indicated concentrations of epinephrine was measured in the membranes. The data are the means ± S.E.M. of triplicate determinations from one representative experiment.
Internalization of the β2AR in PKA−.
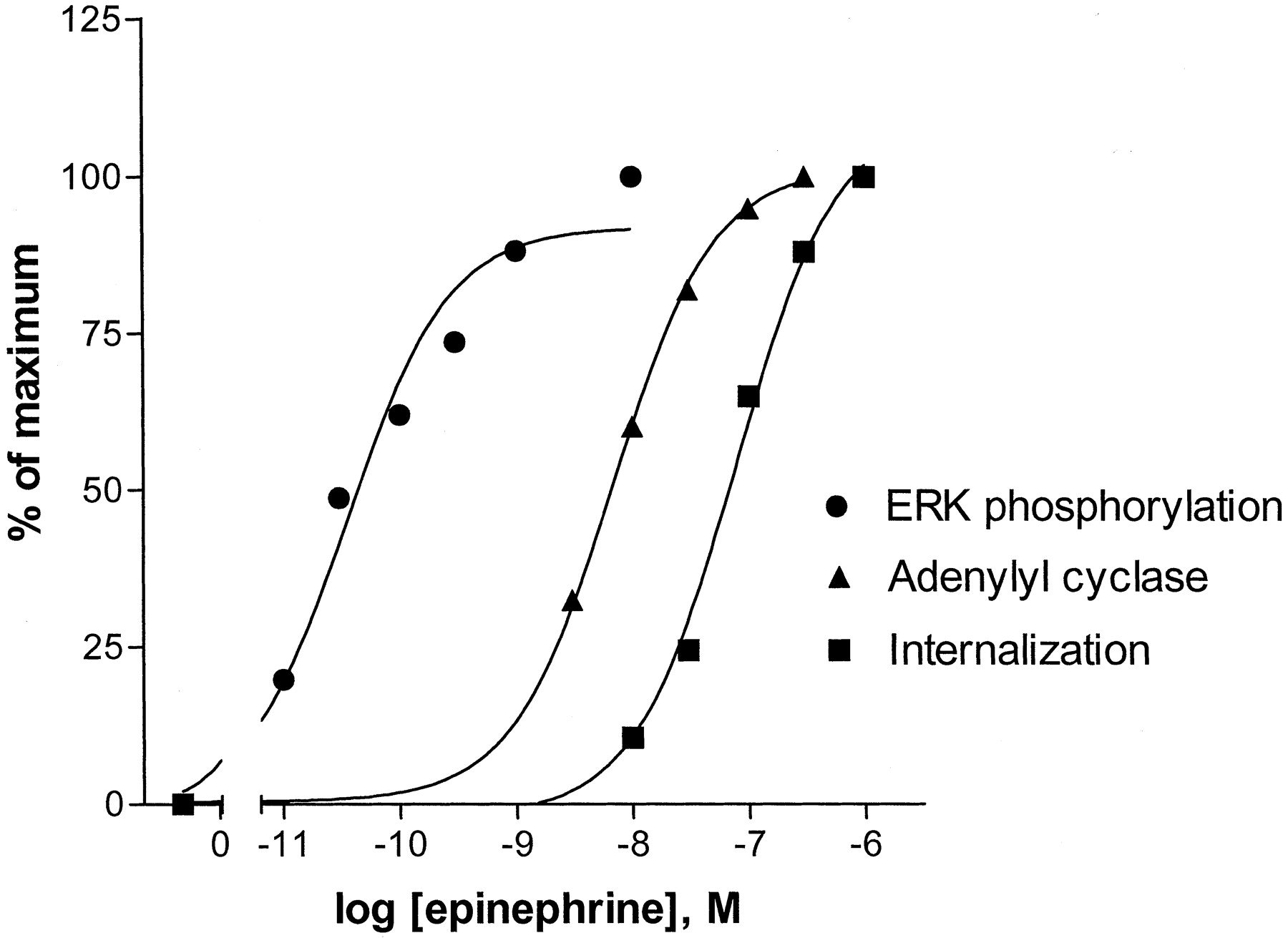
As discussed, there have been some inconsistencies concerning the role of β2AR internalization in activation of ERK1/2. It occurred to us that we could determine how closely these two processes were related by comparing their EC50 values. To measure the EC50 for epinephrine-induced internalization of the PKA− β2AR, cells were treated for 5 min with or without various epinephrine concentrations as indicated in Fig. 7(▪). Internalization was measured using [3H]CGP-12177 as described underMaterials and Methods. Epinephrine (10 nM) produced a barely detectable level of internalization, and the EC50for epinephrine-induced internalization was 75 nM. To compare the EC50 for epinephrine-induced internalization with that for activation of adenylyl cyclase and ERK1/2, the PKA− data from Fig. 3 and the results from a typical adenylyl cyclase assay are also plotted in Fig. 7. It can be seen that the EC50 for epinephrine activation of internalization is approximately 1000-fold higher than that for ERK1/2 and 10-fold higher relative to activation of adenylyl cyclase.

Dose response of epinephrine-induced internalization in the PKA− and comparison with that for activation of ERK1/2 and adenylyl cyclase. Cells in 12-well dishes were treated with various concentrations of epinephrine for 5 min, and internalization of the β2AR was measured by surface binding of [3H]CGP-12177 as described under Materials and Methods. The data shown are the average values of two identical experiments, each performed in triplicate at the various concentrations of epinephrine (variation in the values was less than 5% at each time point). Results are expressed as the percentage of the total surface binding that was internalized. The data for epinephrine activation of ERK1/2 for the PKA− expressing cells were taken from Fig.3. The dose response for epinephrine stimulation of adenylyl cyclase in PKA− membrane preparations is from one representative assay.
Inhibition of ERK1/2 Activation by the Src Family Kinase Inhibitor PP2.
To assess the role of the Src family in ERK1/2 activation, PKA− cells were pretreated with the Src family inhibitor PP2 for 1 h before stimulation with either epinephrine, forskolin, or EGF (Fig. 8). PP2 blocked 90 to 100% of epinephrine and forskolin activation of ERK1/2, but produced only a 33% inhibition of EGF stimulation. Similar results were obtained with the HEK293 cells expressing only endogenous β2AR (data not shown). These data strongly suggest that Src family kinases play an essential role in β2AR activation of ERK1/2. Furthermore, the inhibition of forskolin activation of ERK1/2 shows that it is unlikely that a direct receptor or G protein interaction with Src is required.

Inhibition of epinephrine-, EGF-, and forskolin-activated ERK1/2 by PP2. To assess a possible role for Src in ERK1/2 activation, PKA− cells were pretreated for 1 h at 37°C with 1% DMSO or 10 μM PP2. The cells were then incubated for 5 min with 100 pM or 10 nM epinephrine, 100 ng/ml EGF, or 20 μM forskolin. Phosphorylation of ERK1/2 was measured as described underMaterials and Methods. The data from each experiment were normalized as percentage of maximum ERK1/2 stimulation. The data are the means ± S.E.M. from four experiments.
Discussion
In this article, we focused on the proposal that PKA phosphorylation of the β2AR switches receptor activation from Gs to Giand that this switch of receptor activation of G proteins was required for ERK1/2 activation, as was internalization of the receptor. Two approaches were used to assess the capacity of the PKA− receptor to activate ERK1/2, both of which were based on our prior studies demonstrating that β2AR overexpression predictably left-shifts the EC50 for agonist activation of adenylyl cyclase relative to that for the endogenous β2AR. First, we found that the potencies (EC50 values) for epinephrine activation of ERK1/2 in the PKA−and the clones overexpressing WTβ2AR were similar (20–60 pM) and about 100- to 200-fold lower than the EC50 for epinephrine activation of endogenous receptor in HEK293 cells (5–6 nM). Second, we determined whether the amplification of epinephrine activation of ERK1/2 relative to its activation of adenylyl cyclase for the overexpressed β2ARs was similar to that for the endogenous receptor. For the endogenous receptor, the EC50for epinephrine activation of adenylyl cyclase in cell-free membrane preparations (≅600–800 nM) was about 100-fold greater than the EC50 for ERK1/2 activation (5–6 nM), whereas the comparable ratio for the PKA− was about 200- to 300-fold (15 nM for adenylyl cyclase activation, and 30–60 pM for ERK1/2 activation). The shift in sensitivity of the PKA− for ERK1/2 activation relative to adenylyl cyclase was actually somewhat greater than that for the HEK293 cell line expressing only endogenous β2AR.
Our work also addressed the role of Gi in β2AR activation of ERK1/2. Previous studies had found either a major role for Gi for agonist activation of ERK1/2 after stimulation of the endogenous β2AR in HEK293 cells (Daaka et al., 1997) or none (Schmitt and Stork, 2000). In the latter study, activation of ERK1/2 in HEK293 cells by isoproterenol stimulation of the endogenous β2AR involved Gs/PKA activation of Rap1 and B raf with no role for Gi. In agreement with this study, we found no pertussis toxin inhibition of epinephrine activation of ERK1/2 in HEK293 cells expressing the PKA− and only a slight inhibition of activation in HEK293 cells expressing only the endogenous β2AR, suggesting that Giplayed little if any role. In further support of our conclusion concerning the effects of pertussis toxin, we previously reported that conditions (low concentrations of epinephrine) that provoke a PKA-mediated desensitization of the β2AR result in only a modest 2- to 3-fold increase in the EC50 for epinephrine stimulation of adenylyl cyclase in L cells (Yuan et al., 1994). If the β2AR switched to activation of Gi, a considerably larger loss of epinephrine-stimulated adenylyl cyclase activity would be expected. Also, we have not found that pertussis toxin treatment alters the extent of epinephrine-induced desensitization of HEK293 cells (Clark et al., 1996; Seibold et al., 2000) as would be predicted if PKA-mediated switching occurred.
There are a number of reasons that might account for the differences between our work and that of others (Daaka et al., 1997) that were also performed on clones of the HEK293 cells. Clonal cell lines are adapted for fast growth and it is possible that there are important differences in the expression of factors involved in the complex activation of ERK1/2 that shunt activation to alternate pathways in different clones, as has been suggested previously (Liebmann, 2001). That is, it is entirely possible that for unknown but potentially very interesting reasons, the clonal lines of HEK293 cells used by other groups (Daaka et al., 1998; Luttrell et al., 1999a; Maudsley et al., 2000) express such factors as reduced levels of Gi-specific RGS proteins and different levels of receptor binding proteins that alter localization to microdomains in the plasma membrane thus diminishing the coupling efficiency of β2AR activation of Gi (Hall et al., 1998; Ostrom et al., 2001). For reasons such as these (and one could imagine many other scenarios) the role of Gi may vary significantly from cell line to cell line. Important experimental differences in the present work were that we used only stable transfection of the β2ARs and determined EC50values for epinephrine activation of ERK1/2 with overexpression of the PKA− and wild-type β2ARs. EC50 values were not determined in the previous study of the PKA−mutant (Daaka et al., 1997). It is possible that the use of transient overexpression paradigms in previous studies actually resulted in much higher levels of receptor expression in a fraction of the cells and/or differential localization, and these factors could affect specificity and/or coupling efficiency of β2AR activation of Gs versus Gi. Our studies with transiently expressed β2ARs in HEK293 indicate that they couple poorly to Gs/adenylyl cyclase (i.e., they show almost no left shift in EC50 for epinephrine activation of adenylyl cyclase with high expression). Finally, it is important not to overinterpret results after an 18-h pretreatment with pertussis toxin (Piiper et al., 2000), because cAMP levels are elevated for prolonged periods.
With regard to a role of internalization in ERK1/2 activation, previous studies based on the use of transient expression of dominant negative blockers of internalization have given somewhat ambiguous results. Taking a different approach that circumvents the need for expression of dominant-negative dynamin or arrestins, which may have nonspecific effects, we found that activation of ERK1/2 in the PKA− overexpressing cells occurred at approximately 1000-fold lower concentrations (EC50 = 30–60 pM) of epinephrine than those required for internalization (EC50 = 75 nM), demonstrating a huge amplification of ERK1/2 activation relative to internalization. Our result contrasts with the prior study showing a correlation of the two processes (Daaka et al., 1998). Because we found full ERK1/2 activation with extremely low levels of β2AR occupancy that do not cause internalization, there should be no GRK phosphorylation of the β2AR and arrestin binding, because it is generally accepted that they are correlated with and required for internalization. The very small amplification of internalization (EC50 = 75 nM) we observed relative to occupation of the β2AR (K d for epinephrine is ∼500–600 nM) is entirely consistent with previous work that GRK-mediated phosphorylation and binding of β-arrestin occurs at much higher concentrations of epinephrine than those required for PKA activation.
A number of findings suggest that Src activation plays a role in β2AR stimulation of ERK1/2 (Daaka et al., 1997;Ma et al., 2000; Maudsley et al., 2000; Fan et al., 2001), and it has been suggested that Src is activated by either formation of a β2AR/β-arrestin/Src complex or by direct Gs or Gi activation of Src. Consistent with a role for Src, we found that the Src family kinase inhibitor PP2 (10 μM) caused a >90% inhibition of epinephrine and forskolin activation of ERK1/2 in the HEK293 cells. However, that we see full activation of ERK1/2 by very low concentrations of both epinephrine (which are most unlikely to cause formation of a β2AR/β-arrestin/Src complex) and forskolin (which bypasses the need for receptor or G proteins by direct binding to adenylyl cyclase), raises the question of the mechanism of Src activation. In this regard Schmitt and Stork (2002) recently reported that PKA activates Src by phosphorylation of serine 17 in NIH3T3 cells, as well as in HEK293 cells (P. Stork, personal communication). Whereas this phosphorylation site on Src has been known for some time (Collett et al., 1979), the physiological relevance has not been understood. These studies provide evidence that PKA phosphorylates and activates Src directly and, combined with our studies showing full activation of ERK1/2 by extremely low receptor occupancy in the PKA− cells, leads us to propose that Src is activated by the β2AR by the classic Gs/adenylyl cyclase(AC)/PKA pathway and that the dominant pathway for β2AR activation of ERK1/2 in HEK293 cells is as follows: β2AR → Gs → AC → PKA → Src →→ ERK. The similar amplification in the potency of ERK1/2 activation relative to adenylyl cyclase activation by the endogenous and overexpressed β2ARs is further indication that Gs and PKA are probably major upstream players in ERK1/2 activation.
Acknowledgments
We are grateful for the technical contributions of Bruce Williams and to Dr. Anita Seibold for providing the stably transfected cell lines.
Footnotes
- Received February 26, 2002.
- Accepted July 5, 2002.
-
This work was supported by National Institutes of Health grant GM31208 (to R.B.C.).
Abbreviations
- β2AR
- β2-adrenergic receptor
- HEK
- human embryonic kidney
- ERK
- extracellular signal-regulated kinase
- PKA
- cyclic AMP-dependent protein kinase
- WT
- wild-type
- CGP-12177
- 4-[3-[(1,1-dimethylethyl)amino]2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one
- PP2
- 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- DMEM
- Dulbecco's modified Eagle's medium
- HA
- hemagglutinin
- AT
- ascorbate/thiourea
- HE
- HEPES/EDTA
- PAGE
- polyacrylamide gel electrophoresis
- ICI-118551
- (±)-1-[2,3-(dihydro-5,7-methyl-1H-inden-4-yl)oxy]-3-[(methylethyl)amino]-2-butanol
- EGF
- epidermal growth factor
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}