


Abstract
The WNK [with no lysine (K)] kinases and their downstream effector kinases, oxidative stress responsive 1 (OSR1) and SPS/STE20-related proline-alanine–rich kinase (SPAK), have well established functions in the maintenance of cell volume and ion homeostasis. Mutations in these kinases have been linked to an inherited form of hypertension, neurologic defects, and other pathologies. A rapidly expanding body of evidence points to the involvement of WNKs in regulating multiple diverse cellular processes as well as the progression of some forms of cancer. How OSR1 and SPAK contribute to these processes is well understood in some cases but completely unknown in others. OSR1 and SPAK are targeted to both WNKs and substrates via their conserved C-terminal (CCT) protein interaction domains. Considerable effort has been put forth to understand the structure, function, and interaction specificity of the CCT domains in relation to WNK signaling, and multiple inhibitors of WNK signaling target these domains. The domains bind RFxV and RxFxV protein sequence motifs with the consensus sequence R-F-x-V/I or R-x-F-x-V/I, but residues outside the core motif also contribute to specificity. CCT interactions are required for OSR1 and SPAK activation and deactivation as well as cation-chloride cotransporter substrate phosphorylation. All four WNKs also contain CCT-like domains that have similar structures and conserved binding residues when compared with CCT domains, but their functions and interaction specificities are mostly unknown. A better understanding of the varied actions of these domains and their interactions will better define the known signaling mechanisms of the WNK pathway as well as uncover new ones.
SIGNIFICANCE STATEMENT WNK [with no lysine (K)] kinases and their downstream effector kinases, oxidative stress responsive 1 (OSR1) and SPS/STE20-related proline-alanine–rich kinase (SPAK), have been shown to be involved in an array of diverse cellular processes. Here we review the function of modular protein interaction domains found in OSR1 and SPAK as well as related domains found in WNKs.
Introduction
The WNK [with no lysine (K)] kinase cascade consists of the four upstream WNK kinases and their downstream effector kinases, oxidative stress responsive 1 (OSR1) and SPS/STE20-related proline-alanine–rich kinase (SPAK). The catalytic lysine important for ATP binding is located in a unique position in the four WNKs compared with all other members of the eukaryotic protein kinase superfamily (Verissimo and Jordan, 2001; Xu et al., 2000). Physiologic functions of WNKs were first discovered through positional cloning by Lifton’s group revealing that mutations in WNK1 and WNK4 caused an inherited form of hypertension, pseudohypoaldosteronism type II (PHAII) or Gordon’s syndrome, thereby connecting overexpression of these WNKs to blood pressure regulation (Wilson et al., 2001). Subsequent works revealed that WNKs bind to and activate OSR1 and SPAK by phosphorylation, which then phosphorylate a variety of substrates, many of which are linked to the control of cell volume and ion transport (Anselmo et al., 2006; de Los Heros et al., 2014; Gagnon et al., 2007a; Moriguchi et al., 2005; Pacheco-Alvarez et al., 2012; Piechotta et al., 2003; Piechotta et al., 2002; Ponce-Coria et al., 2008; Richardson et al., 2008; Richardson et al., 2011; Taylor et al., 2018; Vitari et al., 2005; Vitari et al., 2006). A common finding in many of these studies was that an interaction between the conserved C-terminal (CCT) domains of either OSR1 or SPAK, and interaction motifs present in upstream WNKs and downstream substrates with the sequence R-F-x-V/I or R-x-F-x-V/I, was required for signaling. Beyond ion homeostasis, WNK kinases have been linked to a variety of cellular processes (Gallolu Kankanamalage et al., 2018; Gallolu Kankanamalage et al., 2016; McCormick and Ellison, 2011; Siew and O’Shaughnessy, 2013; Tu et al., 2011). In some cases, what is not well understood is whether these processes are mediated all or in part via OSR1 or SPAK. Interactions between the CCT domains of these two kinases and signaling partners are an attractive target of study to discover new information related to the roles of WNK signaling. This review will first provide an overview of WNKs and OSR1/SPAK and then focus on what is known about CCT domains, including their structures, specificity, and other functions in signaling, as well as on the much less studied CCT-like domains that are found in all four WNKs.
WNK Kinases
Structure and Activation
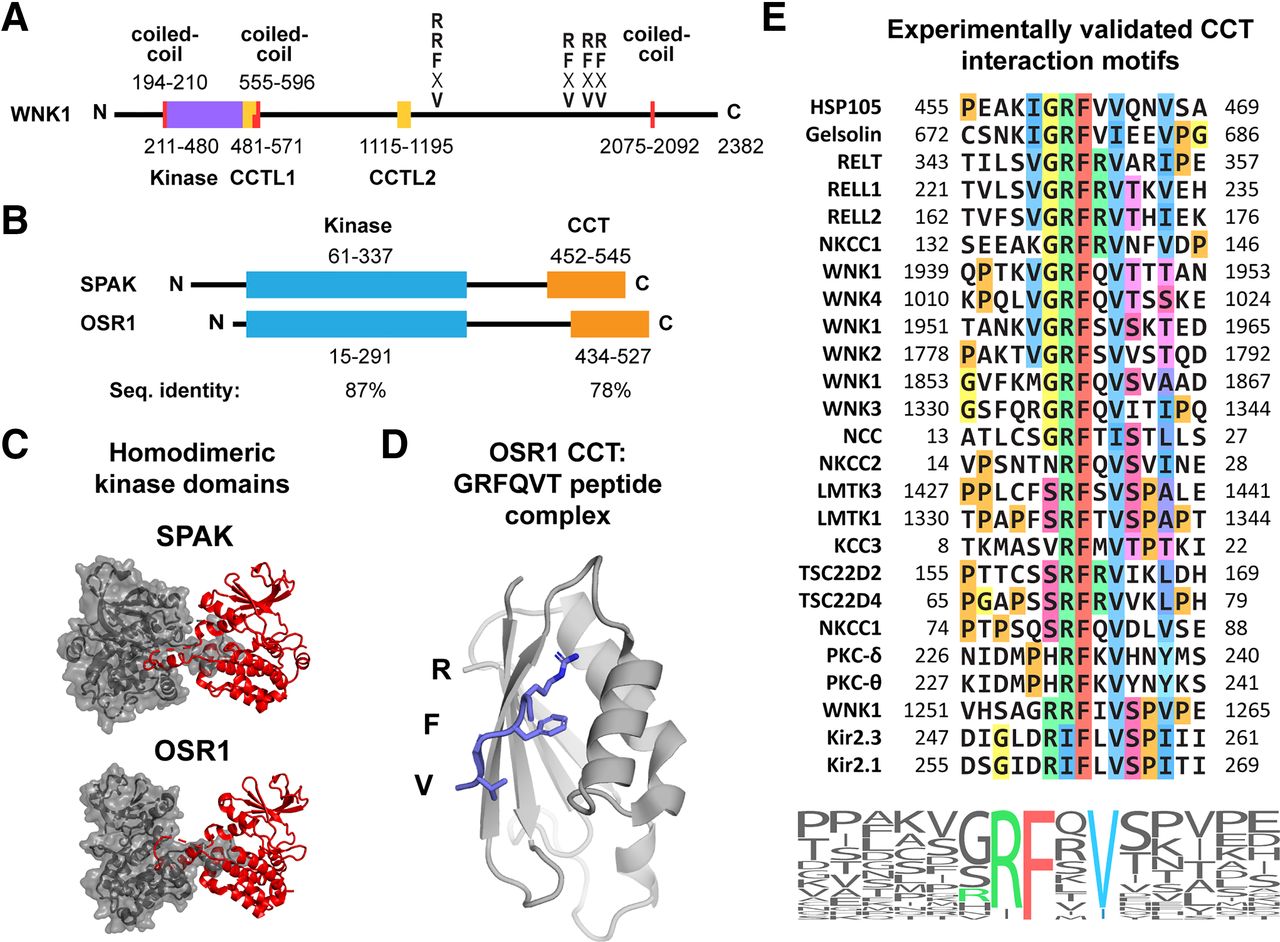
The full-length isoforms of all four WNK family members are large proteins with kinase domains near their N termini (Fig. 1A), and a wide variety of splice variants exist, some with distinct signaling mechanisms. Depending on the specific WNK and splice isoform, various RFxV motifs are interspersed throughout the C-terminal regions of the proteins to facilitate interactions with downstream kinases OSR1 and SPAK (Sengupta et al., 2012).

WNK1, OSR1, and SPAK domain organization and functions. (A) Schematic of WNK1 kinase indicating structural domains and RFxV motifs that bind OSR1 and SPAK CCT domains. Numbering for coiled-coils (red), kinase domain (purple), and CCT-like domains (CCTL1/2, yellow) is indicated. The majority of WNK1 is predicted by PSIPRED to be unstructured (Buchan and Jones, 2019). Coiled-coils predicted by MARCOIL version 1.0 (Delorenzi and Speed, 2002). (B) Schematic of SPAK and OSR1 kinases indicating structural domains. Numbering for kinase domains (blue) and CCT domains (orange) is indicated. The regions between kinase and CCT, as well as the N terminus of SPAK, are predicted to be unstructured. CCT domains interact with motifs with the consensus sequence R-F-x-V/I or R-x-F-x-V/I (Piechotta et al., 2002; Taylor et al., 2018). Sequence identity between OSR1 and SPAK of the kinase and CCT domains is indicated. (C) Crystal structures of the homodimeric SPAK (PDB: 5D9H) and OSR1 (PDB: 3DAK) kinase domains. One monomer is depicted as a gray surface and the other as a red cartoon. The activation loops from each monomer mediate the dimerization via domain-swapping. (D) Crystal structure (PDB: 2V3S) of the OSR1 CCT domain (gray) bound to a GRFQVT peptide (blue). The peptide side chains (R, F, and V) that mediate the interaction with the CCT primary binding pocket are shown. (E) Alignment of all CCT interaction motifs for which experimental evidence exists for the interaction with SPAK or OSR1. The alignment is also presented as a sequence logo, where the size of the letter depicts the frequency in the above alignment. Core motif residues are colored. Alignment generated with ClustalW2 (Thompson et al., 1994). Sequence logo generated with WebLogo (Crooks et al., 2004).
Sequence alignments of WNK1 with other kinases showed that the usual catalytic lysine was replaced with cysteine and a lysine in an alternative location served in catalysis (Xu et al., 2000). From this alternative lysine placement came the name WNK [with no lysine (K)]. We also found that WNK1 immunoprecipitated from HEK293 cells treated with NaCl exhibited greatly increased autophosphorylation, suggesting that WNK1 was a salt-sensitive kinase. Subsequently, we showed that WNK1 activation involves autophosphorylation at S382 in the kinase activation loop (Xu et al., 2002). It is now established that all four WNK kinases are activated by autophosphorylation induced by both low intracellular chloride or cellular hyperosmotic stress as a means to regulate ion homeostasis and cell volume (Kahle et al., 2005; Lenertz et al., 2005; Richardson and Alessi, 2008; Xu et al., 2000; Zagorska et al., 2007).
Dual mechanisms exist for WNK kinase activation and the molecular mechanisms underlying these modes of activation have only recently been uncovered, primarily through work of the Goldsmith laboratory. In the case of low chloride, a chloride ion binds directly to the kinase domain and inhibits autophosphorylation (Min et al., 2004; Piala et al., 2014). Specifically, chloride caps the N terminus of a 3/10 helix that initiates right after the regulatory DLG motif (DFG in most kinases) at the N terminus of the activation loop. The crystal structure suggests that stabilization of this short helix inhibits kinase activity and thus autophosphorylation. Interestingly, the canonical placement of the catalytic lysine would sterically clash with the chloride binding site, which provides a rationale for its unusual placement in WNK kinase domains. Within the last two years the molecular mechanism of WNK activation by hyperosmotic stress has begun to be elucidated. The model put forward is that osmotic pressure shifts the equilibrium from an activation incompetent dimer to an activatable monomer, and this is due to the relatively large number of ordered waters bound to the surface of WNKs (Akella et al., 2020; Akella et al., 2021). Compared with other proteins, WNKs have a higher abundance of charged amino acids on average, which might contribute to the osmosensing mechanism.
WNK Family Members
As introduced above, the four WNK family members (WNK1, WNK2, WNK3, WNK4) all have a similar domain organization (Fig. 1A), including an N-terminal kinase domain immediately followed by a CCT-like (CCTL) domain (CCTL1, discussed below), and after several hundred residues another CCT-like domain (CCTL2, discussed below). Interspersed throughout the C-terminal regions of WNKs are one to several OSR1/SPAK binding motifs. WNKs are relatively large proteins of several thousands of amino acids, and isoform sizes vary considerably depending on splicing events and alternative transcription start sites (de los Heros et al., 2018).
WNK1
WNK1 is the most studied WNK family member, and it was the first to be identified (Xu et al., 2000). A wide variety of WNK1 isoforms exist. Long WNK1 is derived from a transcript with 28 exons and encodes a more ubiquitously expressed protein of 2382 amino acids (Xu et al., 2000). Kidney-specific WNK1 (KS-WNK1) is primarily expressed in the kidney and is derived from a transcript with an alternative transcription start site in exon 4a (O’Reilly et al., 2003; Vidal-Petiot et al., 2012). KS-WNK1 therefore lacks the N-terminal kinase domain but still retains the CCTL domains and OSR1/SPAK binding motifs. Exon 4a of KS-WNK1 has been implicated in the formation of WNK bodies in the kidney distal convoluted tubule, which contain multiple WNK pathway components (Boyd-Shiwarski et al., 2018). It has been proposed that these foci facilitate WNK signaling by increasing the local concentration of signaling molecules (Boyd-Shiwarski et al., 2018; Thomson et al., 2020). A nervous system–specific exon of WNK1, termed hereditary sensory neuropathy type II, generates two different nervous system–specific isoforms of WNK1 (Shekarabi et al., 2008). Mutations within this exon lead to HSANII (hereditary sensory and autonomic neuropathy type II), a disease characterized by loss of peripheral sensory nerves leading to lack of perception to touch, heat, and pain (Ota et al., 1973). The WNK1-hereditary sensory neuropathy type II isoforms still retain kinase domains, CCTL domains, and OSR1/SPAK interaction motifs (Shekarabi et al., 2008). A variety of other isoforms exist, and many have yet to be characterized. Additionally, although they do not affect the protein amino acid sequence, mutations that lead to large intronic deletions in WNK1, causing increased WNK1 expression, are the cause of a form of familial hypertension, PHAII (Wilson et al., 2001).
The study of WNK1 function has primarily focused on its role in ion and cell volume regulation and homeostasis. Perhaps the best characterized function of WNK1 is its activation in response to both low intracellular chloride and hyperosmotic stress. WNK1 autophosphorylates its kinase domain activation loop and then activates OSR1 or SPAK by phosphorylation of residues within and immediately after the kinase domain (Fig. 1, A and B). Via phosphorylation (discussed in depth below), SPAK and OSR1 then activate Na+-K+-2Cl− cotransporter (NKCC) 1 and 2 and Na+-Cl− cotransporter (NCC) and deactivate K+-Cl− cotransporter (KCC) 1–4 (Richardson and Alessi, 2008). Interactions between the OSR1 or SPAK CCT domain and binding motifs found in WNKs and the cotransporters (Fig. 1, B and E) are required for this signaling cascade to function properly (de Los Heros et al., 2014; Gagnon et al., 2007a; Pacheco-Alvarez et al., 2012; Ponce-Coria et al., 2008; Richardson et al., 2008; Richardson et al., 2011; Sengupta et al., 2012; Vitari et al., 2006). WNK1 is also involved in the regulation of the activity of ion channels, renal outer-medullary K+ channel [ROMK, also known as inward-rectifying K+ channel (Kir) 1.1], Kir2.1, and epithelial Na+ channel (ENaC), by modulating cell surface expression (Cope et al., 2006; Huang and Kuo, 2007; Lazrak et al., 2006; Taylor et al., 2018; Xu et al., 2005). In the case of Kir2.3, and likely Kir2.1, localization of the channel is dependent on the interaction between the OSR1 CCT domain and the more recently identified RxFxV motif variant, which is found in half of the sixteen Kir K+ channel family members but not ROMK/Kir1.1 (Taylor et al., 2018).
WNK1 has been implicated in various processes not directly related to membrane ion transport. Disruption of the WNK1 gene in mice causes embryonic lethality, not observed for deletions of other WNK genes. WNK1 null mice fail at embryonic day ∼12 with a poorly developed cardiovascular system indicative of impaired angiogenesis (Zambrowicz et al., 2003). Additional work by the Huang laboratory showed that WNK1 deletion exclusively from endothelial cells caused the lethal phenotype (Xie et al., 2009). WNK1 has also been implicated in endothelial and cancer cell migration, immune responses, autophagy, and mitosis, for example, expanding on the diverse repertoire of WNK1 actions revealed from human genetic diseases and mouse knockout phenotypes (Dbouk et al., 2014; Gallolu Kankanamalage et al., 2016; Hoorn et al., 2011; Mendes et al., 2010; Pichlmair et al., 2012; Tu et al., 2011).
WNK2
WNK2 is one of the least studied of the four WNK family members. The canonical isoform is 2297 residues, but multiple isoforms exist. Perhaps one of the best studied aspects of WNK2 function, particularly within the last few years, is its function as a tumor suppressor. Multiple studies from a variety of cancer types, such as glioma, meningioma, breast cancer, lung cancer, hepatocellular carcinoma, and pancreatic ductal adenocarcinoma, have indicated that WNK2 expression is downregulated (Costa et al., 2015; Dutruel et al., 2014; Jia et al., 2020; Moniz et al., 2013; Zhou et al., 2019). The antiproliferative property of WNK2 is likely, in part, due to its ability to downregulate extracellular signal-regulated kinase–mitogen-activated protein kinase (ERK MAPK), which does require WNK2 kinase activity (Moniz et al., 2007).
WNK2, unlike the other WNKs, is little expressed in the kidneys but is instead enriched in various neuronal cell types (Rinehart et al., 2011). In the central nervous system, WNK2 regulates both cell volume and GABAergic signaling via reciprocal activation of the ubiquitous NKCC1 and inactivation of KCC2 to regulate intracellular chloride concentration in a WNK2 kinase activity–dependent manner (Rinehart et al., 2011).
WNK3
WNK3, like WNK1 and WNK4, is expressed in diverse secretory epithelial tissues but is enriched in the brain and kidney; several isoforms exist, with the canonical isoform being 1800 amino acids (Holden et al., 2004). WNK3 unlike WNK1 and WNK4 is found throughout the nephron, localized to intracellular junctions, and regulates the activity and localization of cotransporters (Rinehart et al., 2006). WNK3 also regulates the plasma membrane localization of ROMK, and in a kinase activity–dependent manner, WNK3 enhances Ca2+ influx into cells via increased surface expression of transient receptor potential cation channel subfamily V members 5 and 6 (Leng et al., 2006; Zhang et al., 2008).
Contrary to WNK2, WNK3 is implicated as potentially promoting cancer. Decreased WNK3 expression is correlated with an increased apoptotic response by a caspase-3–dependent mechanism (Verissimo et al., 2006). WNK3 has also been shown to promote neurologic injury from stroke. WNK3 knockout mice had decreased infarct volume, axonal demyelination, cerebral edema, and accelerated neurobehavioral recovery, and in this same study WNK3 inhibition in primary cultured neurons and oligodendrocytes was associated with increased tolerance to in vitro ischemia (Begum et al., 2015). The WNK3 knockout mice had less activating phosphorylation of OSR1/SPAK and NKCC1, suggesting WNK3 signaling through OSR1/SPAK promoted stroke-related injury. Since OSR1/SPAK activation of NKCC1 is dependent on interactions between the CCT domain and a binding motif on WNK1, inhibitors targeting this interaction have recently been developed and have showed promise in various models of brain injury (discussed below) (Gagnon et al., 2007a; Zhang et al., 2020).
WNK4
WNK4 is a 1243 amino acid protein, and to our knowledge, no experimentally determined isoforms have been identified, although several isoforms have been predicted (Uniprot Consortium, 2015). Similar to mutations that increase WNK1 expression, missense mutations in a short acidic region shortly after the kinase domain of WNK4 are also associated with increased expression and familial hypertension, PHAII (Wilson et al., 2001). In this case, the mutations disrupt interactions with kelch-like 2–cullin 3 (KLHL2-CUL3) and kelch-like 3–cullin 3 (KLHL3-CUL3) ubiquitin ligase complexes, thereby diminishing WNK4 degradation (Mori et al., 2013b; Ohta et al., 2013; Takahashi et al., 2013; Uchida et al., 2014). WNK4 is expressed in multiple regions of the kidney where it is localized to the cytoplasm and apicial membranes (Ohno et al., 2011). It is also enriched in secretory tissues of the brain, biliary ducts, epididymis, pancreas, and colon (de los Heros et al., 2018).
Similar to other WNKs, and particularly WNK1, WNK4 activates SPAK and OSR1 via a CCT domain interaction to modulate cotransporter activity (Gagnon et al., 2006; Garzon-Muvdi et al., 2007; Golbang et al., 2006). WNK4 is also localized to WNK bodies as discussed above (Boyd-Shiwarski et al., 2018; Thomson et al., 2020). Multiple ion channels are also regulated by WNK4 such as ROMK and the cystic fibrosis transmembrane conductance regulator Cl− channel (Kahle et al., 2003; Mendes et al., 2011).
OSR1 and SPAK
OSR1 and SPAK are the best known effectors of WNK pathway function. Mechanistic insights on WNK function have come in large part from identifying proteins that bind to the CCT domains of OSR1 and SPAK (discussed above and below).
Domain Organization and Conservation
OSR1 and SPAK are closely related paralogs and members of the GCK-VI subgroup of the mammalian Ste20-related protein kinase family. SPAK arose from gene duplication during vertebrate evolution (Gagnon and Delpire, 2012). As a result, OSR1 and SPAK share a common domain organization with an N-terminal kinase domain connected by a variable region to the CCT domain (Fig. 1B). Overall, the two kinases have 74% sequence identity, and the kinase and CCT domains have 87% and 78% identity, respectively. Additionally, SPAK contains an N-terminal proline-alanine–rich region of unknown function.
Dimerization by Activation Loop Domain-Swapping
Structural studies from our laboratory and others have shown that both OSR1 and SPAK crystallize as activation loop domain-swapped dimers (Fig. 1C) (Lee et al., 2009; Taylor et al., 2015; Villa et al., 2008). Residues important for catalysis, including the activation loop threonine phosphorylated by WNKs, are shared by the opposing monomer. This suggests that the dimer is the catalytically active form. However, we determined a double point mutation in the kinase activation loop that impairs the capacity of SPAK to dimerize and yet retained kinase activity (Taylor et al., 2015). The arrangement is suggestive of an activation mechanism that involves trans-autophosphorylation. Transactivation has yet to be shown unequivocally, but by artificially linking two monomers together in the presence of overexpressed activating protein calcium-binding protein 39/mouse protein 25 (Cab39/Mo25) and substrate NKCC1, K+ influx was dramatically increased in Xenopus oocytes, which is highly suggestive (Ponce-Coria et al., 2012). Another possibility is that kinase dimerization serves a scaffolding function whereby proteins interacting with each monomer are brought into close proximity, potentially via interactions with the kinase CCT domains. In this case scaffolding by domain-swapping could also possibly be regulated by kinase activation (activation loop phosphorylation).
CCT Domain
Structures of both the apo OSR1 CCT domain (residues 434–527) and the domain bound to the short peptide derived from WNK4, GRFQVT (residues 1003–1008), were solved to resolutions of 1.95 and 1.70 Å, respectively (Villa et al., 2007). Although the apo form was not fully refined, the partially refined structure suggests that peptide motif binding does not dramatically alter the fold of the domain but does result in stabilization of the loops that help form the peptide binding pocket. The domain consists of a single four-stranded β-sheet with two α-helices stacked onto the sheet, as well as multiple loops, one of considerable length containing an additional small α-helix (Fig. 1D).
Together these structural elements form a primary peptide binding pocket with an overall negative charge that binds the peptide Arg1004 side chain through multiple electrostatic interactions and H-bonds (Fig. 1D; left panel of Fig. 2A). Within this same pocket the Phe1005 side chain binds a shallow hydrophobic groove through multiple interactions with residues from different secondary structural elements. Gly1003 and the Gln1006 side chain do not interact with the domain. A secondary pocket also exists, and Val1007 binds on the periphery, making contacts with the adjacent β-strand and an α-helix. The structure suggests that sequences C-terminal to core RFxV motifs might engage this pocket. Also of considerable importance is the fact that the backbone atoms between Gln1006 and Thr1008 form H-bonds with the backbone atoms of the adjacent β-strand. Referred to as β-strand addition, this is commonly found in linear binding motif interactions as a means to provide additional binding energy to weak interactions (Remaut and Waksman, 2006). The consequence of the β-strand addition is that the backbone atom positions between Phe1005 and Thr1008 are spatially constrained owing to the directionality of H-bonds. The important feature of this directionality is that the side chains around this area are pointing in opposing directions and therefore are less likely to influence neighboring residue side chain interactions. The side chain hydroxyl of the peptide Thr1008 makes backbone hydrogen bonding interactions as well, but the relevance is unclear as it is the terminal residues of the peptide, whereas most motifs are not at the very C termini of proteins. The length of the interacting β-strand suggests that a longer peptide might engage additional backbone H-bonds C-terminal to the core motif. In an NMR study analyzing chemical shift perturbations of the OSR1 CCT domain in the presence of a WNK4-derived peptide that encompasses additional residues compared with that used in the OSR1 CCT crystal structure (SEEGKPQLVGRFQVTSSK), peptide binding altered the chemical shift of the primary pocket as expected. However, additional backbone amide resonances all along the β-strand that interacts with the peptide were also shifted, further suggesting that more residues C-terminal to the core motif engage in β-strand addition (AlAmri et al., 2019).

Structural characteristics of CCT and CCT-like domains. (A) Comparison of the primary motif binding pocket of the OSR1 CCT domain with the WNK1, WNK2, and WNK3 CCTL1 domains and with the WNK1 CCTL2 domain. Left panel depicts the crystal structure (PDB: 2V3S) of the OSR1 CCT domain (gray) bound to a GRFQVT peptide (blue). Motif residues are labeled. R, F, and V interact with the CCT primary binding pocket. Primary binding pocket residues are numbered. Right panel depicts structural alignment of the OSR1 CCT (gray), WNK1 CCTL1 (red, PDB: 2LRU), WNK2 CCTL1 (orange, PDB: 6FBK, peptide omitted), WNK3 CCTL1 (cyan, PDB: 5O2C, kinase domain omitted), and WNK1 CCTL2 (green, PDB: 5G3Q) with primary binding pocket residues labeled. The binding pocket is identical, and the pocket residues are mostly conserved or similar, as shown in the middle panel. (B) Crystal structure of the WNK2 CCTL1 (orange) bound to the WNK1-derived peptide LTQVVHSAGRRFIVSPVPESRLR (blue). The peptide contains the sequence R-R-F-x-V, and the first Arg interacts such that the interacting motif is R-x-F-x-V. The peptide folds back on itself making an additional β-strand that also interacts with the CCTL1 domain. (C) Crystal structure of the tandem WNK3 kinase (pink) and CCTL1 (cyan) domains. The interaction interface and interacting residues are shown in the enlarged view. The interface is weakly packed together, and the majority of the contacts are electrostatic interactions.
WNKs each contain two domains, originally identified as autoinhibitory, that are related to OSR1/SPAK CCT domains (Fig. 1A; Fig. 2). A crystal structure [Protein Data Bank (PDB): 6FBK] of the more N-terminal WNK2 CCT-like domain (residues 454–549) bound to a WNK1-derived peptide with the sequence LTQVVHSAGRRFIVSPVPESRLR (residues 1247–1269) was solved by the Structural Genomics Consortium (Fig. 2, A and B) (Pinkas et al., 2017a). WNK CCTL domains, discussed in more depth later, have an overall structure similar to CCT domains, and the binding pocket residues that interact with the core motif are also highly conserved (Moon et al., 2013). In the case of this WNK2 CCTL1 structure, the peptide backbone makes an additional H-bond before being disrupted by a Pro residue in the peptide, and unlike the GRFQVT peptide, the Ser hydroxyl side chain of the WNK1-derived peptide that follows the core motif no longer engages the backbone, suggesting that additional residues might bind through backbone H-bonds.
In the recent study in which we identified that R-x-F-x-V/I sequences are an alternative CCT binding motif, a molecular model of the motif interaction maintained the position of the Arg and Phe side chains while still being able to accommodate the insertion of an additional residue between them (Taylor et al., 2018). Interestingly, although contiguous Arg residues were present in the core motif of the peptide (R-R-F-x-V), the WNK2 CCTL1 structure showed that the R-x-F-x-V interaction was preferred in the crystallized complex over R-F-x-V, and the observed conformation around the R-x-F strongly resembled our modeled interactions, further substantiating the physiologic relevance of the alternative motif (Fig. 2B) (Pinkas et al., 2017a; Taylor et al., 2018).
SPAK and OSR1 orthologs, in multicellular organisms as distantly related as Caenorhabditis elegans, have conserved residues in the primary CCT peptide binding pocket, implying an evolutionarily conserved CCT-motif interaction specificity. As stated, based on structural data, CCTL1 domains found in WNKs share many of these conserved residues with CCT domains, and sequence alignments along with limited structural data indicate that CCTL2 domains also share most conserved binding pocket residues (Fig. 2A) (Bartual et al., 2017; Moon et al., 2013; Pinkas et al., 2017a; Pinkas et al., 2017b). Based on structural information, it appears as though the core motif interactions are likely conserved but that additional specificity determinants as dictated by the surrounding sequence of the motifs would likely be different owing to the decreased conservation outside the primary binding pocket in CCTL domains, and to a lesser extent between the OSR1 and SPAK CCT domains (Pinkas et al., 2017a; Villa et al., 2007). This topic is discussed in depth later.
CCT Domain Functions in WNK Signaling
Canonical Functions
The initial finding that the C-terminal regions of OSR1 and SPAK interacted with motifs with the consensus sequence R-F-x-V/I came from yeast two-hybrid analyses (Piechotta et al., 2002). In that study, the Delpire laboratory found that CCT regions encompassing OSR1 446–527 and SPAK 460–556 were sufficient for a positive interaction with KCC3a. Subsequent work in that study found that both SPAK and OSR1 interacted with multiple members of the cation-chloride cotransporter family (KCC3a, KCC3b, NKCC1, NKCC2) and that nine-residue fragments with the consensus sequence R-F-x-V were capable of maintaining the interaction. In a follow-up two-hybrid study, the authors showed that the SPAK CCT interacts with a variety of proteins [apoptosis-associated tyrosine kinase (AATYK), also known as LMTK1; gelsolin; WNK4; heat shock protein 105 (hsp105)] harboring motifs with the consensus R-F-x-V/I (Piechotta et al., 2003). In the same year expression of dominant-negative SPAK was shown to disrupt N-terminal regulatory domain phosphorylation of NKCC1 and inhibit NKCC1 activity. NKCC1 function in the presence of dominant-negative SPAK was restored by inhibition of protein phosphatase 1, implying that the effect was due to kinase/phosphatase dynamics (Dowd and Forbush, 2003). Shortly after, several studies reported that WNKs bind to and phosphorylate both OSR1 and SPAK, and this binding is mediated by interaction with WNK RFxV motifs (Anselmo et al., 2006; Moriguchi et al., 2005; Vitari et al., 2005). Phosphorylation of OSR1 and SPAK by WNKs is at conserved sites in the activation loop and regulatory region after the kinase domain (S-motif). Disruption of WNK or OSR1/SPAK activity led to decreased activation of NKCCs, providing the first clear depiction of the canonical WNK pathway, in which WNKs are activated by osmotic stress and phosphorylate and activate OSR1 or SPAK, which then regulate cation-chloride cotransporters via phosphorylation.
The findings that delineated the canonical WNK pathway suggested that CCT-RFxV/I motif interactions were important for proper kinase-substrate interactions both upstream and downstream of OSR1 and SPAK. Seminal work from Alessi’s group showed that the CCT domain was required for efficient phosphorylation of both OSR1 by WNK1 and NKCC1 by OSR1 (Vitari et al., 2006). CCT deletion prevented the interactions and disrupted phosphorylation of NKCC1. Inhibition that was observed using a competing WNK4-derived RFxV peptide (SEEGKPQLVGRFQVTSSK) was relieved when peptide core motif residues Arg, Phe, and Val were mutated to Ala. However, phosphorylation of a generic substrate lacking an RFxV motif did not depend on CCT domains, which implied that the loss of activity was due to a loss of interaction with WNK1 and NKCC1 and not a direct effect of the CCT domain on OSR1 kinase activity. Subsequent work found that RFxV/I motifs in other WNKs, NCC, NKCC2, and KCCs were all important in mediating OSR1 and SPAK activation and cotransporter phosphorylation (de Los Heros et al., 2014; Gagnon et al., 2007a; Pacheco-Alvarez et al., 2012; Ponce-Coria et al., 2008; Richardson et al., 2008; Richardson et al., 2011; Sengupta et al., 2012; Vitari et al., 2006). SPAK CCT domain function was also linked to the control of blood pressure in binding-deficient SPAKL502A/L502A mice (Zhang et al., 2015). L502 makes hydrophobic contact with another structural element in the domain based on comparison with the OSR1 CCT crystal structure (Villa et al., 2007). As a result, the mutation may also have a destabilizing effect on the domain.
Aside from WNK activation and cotransporter phosphorylation, CCT-RFxV motif interactions are also required for OSR1 and SPAK deactivation. AATYK, a single-pass transmembrane protein kinase, and NKCC1 both contain CCT and protein phosphatase 1 interaction motifs. These interactions help facilitate deactivation of OSR1 and SPAK and thus downregulate cotransporter phosphorylation (Darman et al., 2001; Gagnon and Delpire, 2010; Gagnon et al., 2007b). AATYK activity was dispensable, suggesting that AATYK and NKCC1 also serve scaffolding roles required for OSR1 and SPAK deactivation at the plasma membrane through CCT-RFxV motif interactions.
Beyond the canonical pathway, we recently showed that eight of 16 members of the Kir K+ channel family harbor an alternative CCT interaction motif (RxFxV). This alternative motif facilitates NaCl-induced plasma membrane localization and thus increased activity of Kir2.1 and Kir2.3 (Taylor et al., 2018). We found that mutation of either arginine residue in the WNK1-derived peptide SAGRRFIVSPVPE to alanine retained binding affinity similar to wild type. The Ki of the R-A-F-x-V peptide was only two to three times higher than the A-R-F-x-V, which in terms of binding affinity is quite similar, whereas the A-A-F-x-V peptide Ki was 45-fold higher for SPAK and 79-fold higher for OSR1. This led us to investigate proteins harboring this alternative motif. Both Kir2.1 and Kir2.3 activity increased in the presence of constitutively active OSR1 (T185E/S325D), whereas the activity of Kir4.1, which does not contain a similar motif, remained constant. Both mutation of the motif in Kir2.3 and deletion of the OSR1 CCT domain abolished the OSR1 activity–dependent increase in channel activity implying the motif interaction was essential for proper regulation of channel activity by OSR1. Furthermore, WNK activity was also found to be required for this regulation (Taylor et al., 2018).
WNK-Independent Regulation
CCT-RFxV motif interactions have also been implicated in cellular processes regulated by events other than the WNK pathway. SPAK was shown to be a downstream substrate of protein kinase C (PKC) θ in the activation of T cells via activator protein 1. The CCT domain of SPAK was sufficient for interaction with PKCθ, and PKCθ does have an RFxV motif. PKCα was also tested and did not phosphorylate SPAK and, as might be expected, does not have an RFxV motif (Li et al., 2004). In another study involving a PKC, this time PKCδ, the CCT domain of SPAK was again sufficient for an interaction with PKCδ (Smith et al., 2008). In this case the endogenous interaction was detected in human airway epithelial cells, and interestingly the interaction was enhanced dramatically upon treatment with high osmolarity (sucrose). PKCδ also has an RFxV motif. The results suggest that PKCδ can function upstream of SPAK in the regulation of NKCC1 activity (Smith et al., 2008).
Receptor expressed in lymphoid tissues (RELT), a member of the tumor necrosis factor receptor (TNFR) family, does not act in a manner consistent with standard TNFR signaling. Initially by yeast two-hybrid it was shown that RELT interacts with SPAK, which was confirmed by co-immunoprecipitation in HEK293 cells, and the RFxV motif was implicated because mutation of Phe to Ala disrupted binding. Additionally, overexpression of kinase-dead SPAK inhibited RELT signaling and thus inhibited activation of the kinases p38 and c-Jun N-terminal kinase (JNK) (Polek et al., 2006a). A related study of the TNFR family member, tumor necrosis factor–related apoptosis-inducing ligand (TRAIL), indicated that TRAIL-induced caspase cleavage led to cleavage of the CCT domain of SPAK. CCT cleavage decreased NKCC1 phosphorylation in an in vitro kinase assay, as might be expected, and cleaved SPAK was unable to bind RELT as well. Interestingly, small interfering RNA knockdown of SPAK enhanced HeLa cell sensitivity to TRAIL-induced apoptosis (Polek et al., 2006b). These studies, whether on canonical WNK pathway signaling or on additional signaling pathways, point to the importance of CCT domain interactions in mediating signaling through OSR1 and SPAK.
Potential Autoinhibitory Function
Using small-angle X-ray scattering it was found that inclusion of an RFxV-containing peptide decreased the overall size of OSR1, suggesting a direct regulatory role for the CCT domain on OSR1 itself (Villa et al., 2008). The two most obvious explanations are that either bound CCT domain dissociates from the kinase domain upon peptide binding or that dimerization is impacted. One study suggests an autoinhibitory function of the OSR1 CCT domain, and likely SPAK, owing to the high sequence conservation in the kinase and CCT domains (Li et al., 2014). In an in vitro assay, inactive OSR1 lacking the C-terminal region (1–338) phosphorylated glutathione S-transferase (GST)–NCC to a greater extent than full-length OSR1 (1–527), and inclusion of separately purified CCT domain in a kinase assay decreased phosphorylation of GST-NCC by OSR1. In addition, separately purified OSR1 kinase and CCT domains interact, as detected by both an in vitro pull-down assay and chemical crosslinking. Interestingly, in this same study it was found that the OSR1- and SPAK-activating protein Cab39/Mo25 interacts more strongly with OSR1 lacking its C-terminal region, and the kinase activity toward GST-NCC was virtually identical between inactive OSR1 1–338 and constitutively active OSR1 1–527 T185E when Cab39/Mo25 was included in the reaction. We routinely observe a 10–20-fold increase in activity, depending on the substrate, between SPAK wild type and the analogous activating mutation T243E (unpublished data). Together, these results imply that the CCT domain of OSR1 serves an autoinhibitory function either alone or in conjunction with Cab39/Mo25. A confounding factor is that substrates with RFxV motifs were used, which complicates the analysis of the data, as the substrate would also interact with the CCT domain. Such an interaction could have unanticipated consequences and certainly would affect the results of any assay where there is competition between a purified CCT domain and full-length OSR1 (Li et al., 2014). Another earlier study also provides contradictory evidence to autoinhibition, as OSR1 T185E lacking the CCT domain has approximately 10-fold lower activity toward GST-NKCC1 compared with wild type, but the two have approximately the same activity toward a generic peptide substrate that lacks an RFxV motif (Vitari et al., 2006).
Pharmacological Compounds Targeting OSR1 and SPAK
Considerable efforts have been made toward targeting WNKs, OSR1, and SPAK. A highly selective ATP-competitive pan-WNK inhibitor (low nanomolar IC50 values) and then an allosteric non–ATP-competitive WNK1/WNK3-selective inhibitor (low nanomolar IC50 values, 1000-fold selectivity WNK1/WNK4 and 57-fold selectivity WNK1/WNK2) were discovered by Novartis (Yamada et al., 2017; Yamada et al., 2016). Multiple strategies, in large part undertaken by the Uchida laboratory, focused on SPAK and yielded compounds that target the CCT domain. The first compounds, STOCK1S-50699 and STOCK2S-26016, were identified through a screen that reported on loss of interaction between the SPAK CCT domain and a WNK-derived RFxV motif–containing peptide (Mori et al., 2013a). These compounds prevented binding and were shown to target the CCT domain and prevent interactions with RFxV motifs in interaction partners. However, both STOCK1S-50699 and STOCK2S-26016 were not optimized (mid-micromolar IC50 values) and are quite hydrophobic. Interestingly, in a study that has implicated WNK1 and WNK4 in the regulation of β-catenin protein levels, and thus Wnt signaling, treatment of SW480 cells with STOCK2S-26016 caused a dramatic decrease in β-catenin (Sato et al., 2020). β-Catenin is partly degraded through the GID (in S. cerevisiae it is defined as glucose induced degradation deficient) ubiquitin ligase complex, and WNK1 and WNK4 attenuate the interaction between the GID complex and β-catenin. STOCK2S-26016 and a derivative (compound #13) both inhibited the interactions between both WNK1 and WNK4 and a component of the GID complex in cell culture, implying that WNK1 and WNK4 compete with β-catenin for GID complex interactions and that this competition protects β-catenin from degradation (Ishigami-Yuasa et al., 2017; Sato et al., 2020). In this same study both inhibitors reduced β-catenin in two colorectal cancer cell lines, but due to toxicity of STOCK2S-26016, only the derivative compound showed efficacy in reduction of tumor size in a mouse xenograft model. Presumably these compounds are acting to disrupt the interaction between WNKs and OSR1 or SPAK, which suggests that WNK activation of the kinases is part of the mechanism of regulation, but more work will be required to clarify the role of these kinases and the mechanism by which these inhibitors are acting.
A screen designed to detect inhibition of SPAK kinase activity toward NKCC2 identified two compounds, STOCK1S-14279 (260 nM IC50) and closantel (770 nM IC50), as compounds that inhibited SPAK in a non–ATP-competitive manner (Kikuchi et al., 2015). STOCK1S-14279 showed moderate specificity, more so than closantel, but some off-target effects, such as inhibition of Aurora A kinase, were observed; although it was more selective, STOCK1S-14279 was also more toxic. In silico docking onto the OSR1 CCT domain crystal structure using STOCK1S-50699, STOCK2S-26016, STOCK1S-14279, and closantel suggests these all bind in the secondary pocket of the domain (AlAmri et al., 2017). This same study identified the US Food and Drug Administration–approved compound rafoxanide as also binding in this same pocket through an in silico screen. As seen with the compounds that inhibited SPAK, rafoxanide was found to inhibit OSR1 in a non–ATP-competitive manner with an IC50 of 8.1 µM. Interestingly, the study found that OSR1 lacking the CCT domain was no longer inhibited by STOCK1S-50699, closantel, or rafoxanide, but only STOCK1S-50699 competed for RFxV peptide binding. The results suggest that all three compounds require the CCT domain for inhibition but only STOCK1S-50699 sterically blocks access to the primary RFxV binding pocket (AlAmri et al., 2017). Recent work utilizing pharmacophores from previously known inhibitors identified ZT-1a, which also targets the CCT domain and disrupts the interaction between SPAK and WNK1 more strongly than STOCK1S-50699 or closantel based on co-immunoprecipitation experiments (Zhang et al., 2020). Although ZT-1a has an IC50 of ∼40 µM, it was nonetheless shown to have therapeutic potential as a modulator of brain cation-chloride cotransporters in several models of brain injury. Taken together, these studies highlight the importance of the CCT domain, both in terms of WNK pathway signaling and as a target for non–ATP-competitive kinase inhibitors of SPAK and OSR1.
CCT Domain Interaction Specificity
Since the initial discovery that the conserved C-terminal domains of OSR1 and SPAK bind linear sequence motifs with the consensus sequence R-F-x-V/I, numerous studies have identified or validated a variety of motifs in OSR1 and SPAK signaling partners (Anselmo et al., 2006; de Los Heros et al., 2014; Delpire and Gagnon, 2007; Gagnon et al., 2007a; Li et al., 2004; Mori et al., 2013a; Moriguchi et al., 2005; Pacheco-Alvarez et al., 2012; Piechotta et al., 2002; Piechotta et al., 2003; Polek et al., 2006a; Polek et al., 2006b; Ponce-Coria et al., 2008; Richardson et al., 2008; Richardson et al., 2011; Sengupta et al., 2012; Smith et al., 2008; Taylor et al., 2018; Villa et al., 2007; Vitari et al., 2005; Vitari et al., 2006; Zagorska et al., 2007). By our count, there are 25 motifs identified for which there is experimental evidence to support an interaction with the OSR1 or SPAK CCT domain, two of which are the more recently identified RxFxV variant (Fig. 1E) (Taylor et al., 2018). However, a complete count of every motif in every human protein in the UniProt database indicates there are 1723 RFxV motifs and 2016 RxFxV motifs (Obenauer et al., 2003; Uniprot Consortium, 2015). Obviously, all of these motifs cannot be relevant, and therefore additional specificity determinants dictated by the sequence around the core motif as well as the cellular location and accessibility of motifs will assist in identifying bona fide CCT partners.
Sequence alignment of the known motifs provides some insight into additional factors affecting specificity (Fig. 1E). A Val-Gly, Gly, or small residue directly preceding the Arg is found in most motifs. A small residue also usually directly follows the Val/Ile, and in some cases Ser-Pro is seen. Three residues after the Val/Ile, a small hydrophobic or Ser/Thr is enriched. No prolines are found directly adjacent or within the core motif, which would make sense in light of the fact that the motif is making peptide backbone H-bonds with the CCT domain. However, there is no broadly generalizable consensus sequence that is well enough defined to be able to predict with confidence which of the many motifs found in the proteome are relevant (bottom of Fig. 1E). Based on the fact that the motif binds through β-strand addition, as discussed earlier, it seems plausible that some residues, specifically those involved in backbone interactions and their adjacent residues, may discretely contribute to the overall binding energy without being influenced much by adjacent residues owing to the alternating position of the side chains (left panel of Fig. 2A) (Remaut and Waksman, 2006; Villa et al., 2007). Therefore, if multiple residues each contributed varying degrees of binding energy to the interaction without being influenced by the identity of adjacent residues, then a variety of sequence combinations could potentially lead to a physiologically relevant interaction.
In addition to sequence alignments, multiple studies have reported on interaction affinities. Surface plasmon resonance data using WNK1- and WNK4-derived motifs indicate low nanomolar dissociation constants for OSR1 (Villa et al., 2007; Vitari et al., 2006; Zagorska et al., 2007), whereas solution-based fluorescence measurements for SPAK indicate high nanomolar to low micromolar dissociation constants using WNK1-, WNK4-, and NCC-derived peptides (Mori et al., 2013a; Taylor et al., 2018; Zhang et al., 2015). We have conducted a systematic analysis of motif binding affinities using fluorescence polarization, and we find low micromolar dissociation constants to be the norm for both OSR1 and SPAK (unpublished data). As discussed earlier, we also investigated the affinity of RxFxV motifs and found that both OSR1 and SPAK CCT domains bound a mutant WNK1-derived peptide containing an R-A-F-x-V motif with low micromolar dissociation constants (Taylor et al., 2018). In addition, it has been shown that phosphorylation of Ser/Thr residues directly after the core motif decrease binding affinity by orders of magnitude, which could suggest a regulatory role for these residues (Villa et al., 2007). Interestingly, approximately 25% of the experimentally verified motifs contain a Ser-Pro after the Val/Ile in the core motif. Ser-Pro is a hallmark of MAPK phosphorylation sites (Fig. 1E).
Systematic Studies
Two systematic studies have been undertaken to better understand which motifs might be relevant. A genomewide search for motifs containing the sequence S/G/V-R-F-x-V/I-x-x-V/I/T/S found 215 such motifs in the human genome (Delpire and Gagnon, 2007). This early work used an alignment of 12 motif sequences to derive the search sequence, and based on this, the authors were able to analyze the distribution of amino acids in the surrounding sequence. The caveats to this valuable study are that the results are heavily biased toward the input search sequence, and motifs that would be inaccessible to interaction with CCT domains (i.e., buried motifs, extracellular) were not excluded. Another study used in silico analysis based on the OSR1:GRFQVT crystal structure to derive the specificities for individual residues in the motif (Austin et al., 2014). The study provided potentially interesting insights such as Lys and His being able to replace Arg in the core motif, but some of the results are inconsistent with data on known CCT-RFxV interactions (e.g., Asp, Tyr, and Phe having higher predicted binding energies than Ile in replacement of Val in the core motif of the hexapeptide).
WNK CCT-Like Domains
As mentioned above, each of the four WNKs contains two CCT-like domains. CCTL1 immediately follows the kinase domain, and CCTL2 is found several hundred residues past the kinase domain (Fig. 1A). The WNK1 CCTL1 domain has autoinhibitory activity toward the adjacent kinase domain (Xu et al., 2002). However, the potential physiologic relevance of this finding is uncertain, as the fragment used for the study truncated the C-terminal helix of the domain which may have been destabilized (Moon et al., 2013).
Several NMR and X-ray crystal structures are available now of multiple WNK CCTL domains (Fig. 2). The WNK1 CCTL1 domain structure (PDB: 2LRU) was determined by NMR (Moon et al., 2013). Topologically, the structure of WNK1 CCTL1 is distinct from that of the OSR1 CCT domain since the secondary structural elements are arranged in a different order, but the overall structure is remarkably similar with a Cα root mean square deviation of 1.2 Å , whereas the primary binding pocket residues have a Cα root mean square deviation of 0.7 Å (right panel of Fig. 2A) (Moon et al., 2013; Villa et al., 2007). The crystal structure of the WNK3 fragment including both the kinase and CCTL1 domains (PDB: 5O2C), the WNK2 CCTL1 bound to the peptide LTQVVHSAGRRFIVSPVPESRLR (PDB: 6FBK), and the WNK1 CCTL2 (PDB: 5G3Q) all have virtually identical topologies and overall structures as compared with the WNK1 CCTL1, but residue identities and loop regions differ to varying extents (Fig. 2) (Bartual et al., 2017; Pinkas et al., 2017a; Pinkas et al., 2017b). The relatively few and almost entirely electrostatic contacts between the kinase and CCTL1 domains in the WNK3 structure suggest that CCTL1 is flexibly tethered to the kinase core (Fig. 2C) (Bartual et al., 2017). The fact that WNK3 CCTL1 interactions with the kinase are mediated almost entirely by electrostatics could suggest a regulatory role for the interactions since WNKs are salt-sensitive. Also of structural interest is the fact the WNK1 CCTL1 domain ends with a long helix that is predicted to be a coiled-coil (Fig. 1A). This coiled-coil extends outward further and might participate in oligomerization. More structural studies, such as by cryo-electron microscopy, could help elucidate the tertiary structure and physiologic relevance of such oligomeric complexes.
The WNK CCTL structures also provide clues as to the specificities of the domains. The acidic residues that interact with the guanidinium group of the motif Arg side chain appear to be conserved between CCT and CCTL1 domains, as are some other residues around the primary binding pocket, whereas the WNK1 CCTL2 domain lacks one of the two acidic residues (Fig. 2A). However, other residues, such as OSR1 L473 that interacts with the Val/Ile of motifs, are replaced by Phe or Tyr in the CCTL1 structures suggesting the possibility of alternative core motif specificities for the CCTL1 domains (Villa et al., 2007). In fact, the WNK1 CCTL1 domain was shown to interact with several RFxV motif–containing peptides with mid-micromolar dissociation constants, which have reduced affinities compared to the CCT domains, and this could be due to suboptimal binding motifs (Moon et al., 2013). The most telling structural information regarding CCTL1 specificity comes from the structure of the WNK2 CCTL1 bound to a motif peptide (Fig. 2B) (Pinkas et al., 2017a). In this case the WNK1-derived peptide contains both motif variants at once (R-R-F-x-V) yet crystallizes utilizing the RxFxV motif interaction, further indicating that these motifs are physiologically relevant alternative motifs (Taylor et al., 2018). When aligned to the OSR1 CCT-GRFQVT peptide structure, the positions of the Arg guanidinium moiety and the Phe and Val side chains are virtually identical between the two structures, which strongly suggests that WNK CCTL1 domains bind these motifs in a similar manner as CCT domains (Villa et al., 2007). Finally, and quite strikingly, the much longer motif-containing peptide (as compared with the CCT structure with the GRFQVT peptide) folds back onto, itself making an additional β-strand. The loop and terminal β-strand of the peptide make multiple additional interactions with the α-helix and subsequent loop that make up the primary binding pocket. Therefore, the peptide does not bind linearly across the WNK2 CCTL1 and provides evidence for a more complex set of interactions with CCTL1 domains and probably CCT and CCTL2 domains for at least some interaction motifs (Pinkas et al., 2017a).
Discussion
Functions of the OSR1 and SPAK CCT and WNK CCT-like protein interaction domains are an area of rich and understudied biology within the WNK pathway. OSR1 and SPAK CCT binding partners have revealed much about WNK-dependent ion transport regulation. Nevertheless, the sheer number of motifs in the proteome and the structural evidence for multifunctionality of CCTs suggest that much more remains to be discovered about how CCT and CCTL interactions guide pathway function.
Important questions for future work on the simplest level will address the diversity of motifs in proteins. For example, can unrecognized variations on the motif substitute for the canonical RFxV motif, much like the newly found RxFxV motif? What are the specificity determinants outside of the core motif, and how might these determinants, such as sites for post-translational modifications as discussed above, impact physiologic consequences of the interactions?
Far less is known about the WNK CCT-like (CCTL1 and CCTL2) domains. Although it is clear that they have a structure similar to OSR1 and SPAK CCT domains, have similar or identical binding pocket residues, and in the case of the WNK1 and WNK2 CCTL1, can bind peptides with RxFxV or RFxV motifs, it is unclear whether they interact preferentially with these motif types or have unique core motif specificity. Furthermore, because there are eight of these CCTL domains in WNKs with varying sequences, some or all may have unique sequence preferences that have yet to be determined. In spite of compelling structural data, no physiologically relevant interactions with CCTL domains have yet been demonstrated to begin to fill in this gap.
A good first step toward answering some of these questions is to better understand what drives specificity in the CCT domains to enhance our ability to predict what proteins are interacting. We recently undertook a large-scale biochemical and biophysical study of CCT motif specificity (unpublished data). Our preliminary data suggest that outside of the core motif some positions have a clear preference for certain amino acids, but overall, each position outside the core motif only contributes a small amount to the binding energy, suggesting that a wide variety of different sequences could generate physiologically relevant interactions.
On a larger scale, how do CCT-motif interactions influence the assembly of WNK/OSR1/SPAK complexes? Given the domain-swapping propensity of OSR1/SPAK, OSR1 or SPAK kinase dimers may work as adapters bringing two proteins that both interact with CCT domains in close proximity. If so, then these interactions may be spatially regulated depending on the ability of SPAK or OSR1 to dimerize. Furthermore, findings with Kir imply interactions with not fully assembled oligomeric complexes. Additional factors may also be relevant for complex formation such as three-way interactions with WNKs, compartmentalization, and dynamics of competition among motif-containing proteins.
CCT interactions have also been suggested to have mechanistic regulatory impact on the pathway as discussed above. Conformational changes upon motif binding may affect kinase function by relieving autoinhibition. Either high or low pathway activity may be necessary to enable interactions. The accessibility of some motifs may also be regulated, such as by phosphorylation of other domains of the binding partner. WNK CCT-like (CCTL1 and CCTL2) domains may serve other structural or regulatory functions, such as through the coiled-coil that extends from the WNK1 CCTL1 C terminus or through salt disruption of electrostatic interactions at the interface between the CCTL1 and kinase domain of WNK3.
The available data suggest that CCT and CCTL domains in the WNK pathway have the potential to provide a diverse array of signaling outputs either through domain-specific motif interactions or through mechanisms outside of their function as modular protein interaction domains. This is a fertile area for future discovery in the WNK pathway.
Acknowledgments
The authors thank members of the Cobb laboratory for valuable suggestions, especially UT Southwestern STARS student Justin Li, and Dionne Ware for administrative assistance.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Taylor, Cobb
Footnotes
- Received April 19, 2021.
- Accepted July 14, 2021.
Recent work from the Cobb laboratory discussed here was supported by National Institutes of Health [Grant R01-HL147661], Welch Foundation [Grant I1243], and Cancer Prevention and Research Institute of Texas (CPRIT) Training [Grant RP160157] for support of C.A.T.
Abbreviations
- AATYK
- apoptosis-associated tyrosine kinase
- CCT
- conserved C-terminal
- CCTL
- conserved C-terminal–like
- GID
- glucose induced degradation deficient
- GST
- glutathione S-transferase
- KCC
- K+-Cl− cotransporter
- Kir
- inward-rectifying K+ channel
- KS-WNK1
- kidney-specific WNK1
- NCC
- Na+-Cl− cotransporter
- NKCC
- Na+-K+-2Cl− cotransporter
- OSR1
- oxidative stress responsive 1
- PDB
- Protein Data Bank
- PHAII
- pseudohypoaldosteronism type II
- PKC
- protein kinase C
- RELT
- receptor expressed in lymphoid tissues
- ROMK
- renal outer-medullary K+ channel
- SPAK
- SPS/STE20-related proline-alanine–rich kinase
- TNFR
- tumor necrosis factor receptor
- TRAIL
- tumor necrosis factor–related apoptosis-inducing ligand
- WNK
- with no lysine (K)
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}