


Abstract
High serum levels of asymmetric dimethyl arginine (ADMA) are associated with cardiovascular disease and mortality. Pharmacological agents to specifically lower ADMA and their potential impact on cardiovascular complications are not known. In this study, we aimed to investigate the effect of specific lowering of ADMA on myocardial response to ischemia-reperfusion injury (I/R) and direct effects on cardiomyocyte function. Effects of recombinant dimethylarginine dimethylaminohydrolase (rDDAH)-1 on I/R injury were determined using isolated mouse heart preparation. Respiration capacity and mitochondrial reactive oxygen species (ROS) generation were determined on mouse cardiomyocytes. Our results show that lowering ADMA by rDDAH-1 treatment resulted in improved recovery of cardiac function and reduction in myocardial infarct size in mouse heart response to I/R injury (control 22.24 ±4.60% versus rDDAH-1 15.90 ±4.23%, P < 0.01). In mouse cardiomyocytes, rDDAH-1 treatment improved ADMA-induced dysregulation of respiration capacity and decreased mitochondrial ROS. Furthermore, in human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes with impaired contractility under hypoxia and high ADMA, rDDAH-1 treatment improved recovery and beating frequency (P < 0.05). rDDAH-1 treatment selectively modified I/R-induced myocardial cytokine expression, resulting in reduction in proinflammatory cytokine IL-17A (P < 0.001) and increased expression of anti-inflammatory cytokines IL-10 and IL-13 (P < 0.01). Further in vitro studies showed that IL-17A was the predominant and common cytokine modulated by ADMA-DDAH pathway in heart, cardiomyocytes, and endothelial cells. These studies show that lowering ADMA by pharmacological treatment with rDDAH-1 reduced I/R injury, improved cardiac function, and ameliorated cardiomyocyte bioenergetics and beating activity. These effects may be attributable to ADMA lowering in cardiomyocytes and preservation of cardiomyocyte mitochondrial function.
SIGNIFICANCE STATEMENT The pathological role of asymmetric dimethyl arginine (ADMA) has been demonstrated by its association with cardiovascular disease and mortality. Currently, pharmacological drugs to specifically lower ADMA are not available. The present study provides the first evidence that lowering of ADMA by recombinant recombinant dimethylarginine dimethylaminohydrolase (rDDAH)-1 improved postischemic cardiac function and cardiomyocyte bioenergetics and beating activity. Our studies suggest that lowering of ADMA by pharmacologic treatment offers opportunity to develop new therapies for the treatment of cardiovascular and renal disease.
Introduction
High level of asymmetric dimethyl arginine (ADMA) accumulated during the conditions of ischemia, hypoxia, or oxidative stress has emerged as a risk factor for cardiovascular disease. Patients with pre-existing atherosclerotic disease, diabetes, hypertension, and heart failure exhibit high levels of plasma ADMA that is associated with disease severity and mortality (Cavusoglu et al., 2010; Xuan et al., 2016; Dowsett et al., 2020; Gać et al., 2020). Meta-analysis of results from 10 studies with 2195 patients suggested a strong correlation between plasma ADMA levels and mortality of patients with heart failure (Pan et al., 2020). In addition, patients with chronic kidney disease show increasing plasma ADMA levels with the progression of kidney disease (Reddy et al., 2015). Thus, ADMA may be a biomarker as well as a risk factor for cardiovascular disease.
ADMA is a competitive inhibitor of nitric oxide synthase (NOS) and arginine transport (Leiper and Nandi, 2011). A large number of early studies have focused on the effect of ADMA on vascular endothelium and vasoconstriction. Further studies in cellular systems and preclinical animal models have shown that high levels of ADMA may contribute to diverse pathologic responses (Druhan et al., 2008; Mihout et al., 2011). In particular, ADMA induces the uncoupling of eNOS and generation of reactive oxygen species (ROS) (Druhan et al., 2008). These pathophysiological activities of ADMA may have considerable effects on vascular as well as cardiac function, such as during heart attack, progression of chronic vascular disease, and heart failure. Thus, it has been recognized that lowering of pathologic ADMA may be a novel therapeutic target to reduce the risk of cardiovascular and kidney disease (Leiper and Nandi, 2011; Schwedhelm and Böger, 2011).
The major pathway modulating ADMA levels is through its metabolism by the enzyme dimethylarginine dimethylaminohydrolase (DDAH) (Leiper and Nandi, 2011; Schwedhelm and Böger, 2011). Two isoforms, DDAH-1 and DDAH-2, encoded by separate genes have been identified in mammalian species (Leiper and Nandi, 2011). Both enzymes catalyze the degradation of ADMA to citrulline and dimethylamine (Teerlink, 2005). Reduced expression or activity of DDAH has been found in animal models of cardiovascular and renal ischemia (Stühlinger et al., 2007; Nakayama et al., 2014). More recently, it was discovered that the expression of DDAH-2 was reduced in human diabetic kidney (Barwinska et al., 2021). The role of DDAH as a modulator of ADMA in physiologic and pathologic states has been confirmed using rodent genetic models. Deletion of the DDAH-1 gene leads to increased ADMA levels and vascular dysfunction (Leiper et al., 2007). To the contrary, overexpression of DDAH-1 in transgenic animals or by adenoviral gene transfer leads to reduction in ADMA, which is associated with improved vascular responses and reduced disease severity (Stühlinger et al., 2007; Jacobi et al., 2010; Nakayama et al., 2014; Xu et al., 2017). DDAH gene transfer studies have suggested that the reduction of ADMA by increasing DDAH represents a new therapeutic approach to treat cardiovascular and renal disease (Arrigoni et al., 2010; Leiper and Nandi, 2011; Xu et al., 2017). At the present time, however, pharmacological drugs to specifically lower ADMA are not available.
In the present study, we have used recombinant DDAH (rDDAH)-1, which specifically metabolizes ADMA, as a pharmacological agent to investigate its potential therapeutic actions in myocardial ischemia-reperfusion (I/R) injury. We show that postischemic infusion of rDDAH-1 in a mouse model protected the heart against I/R-induced myocardial damage. rDDAH-1 treatment also improved beating frequency and recovery of human induced pluripotent stem cell derived cardiomyocytes (iCMs) from hypoxia- and ADMA-induced stress. Cell culture studies showed that rDDAH-1 improved ADMA-dysregulated cardiomyocyte bioenergetics and reduced ADMA-induced inflammatory cytokines and mitochondrial ROS production. These results suggest that a rDDAH-1 based biotherapeutics may represent a novel approach for the treatment of ischemic heart disease.
Methods and Materials
Generation and Characterization of rDDAH-1
A human DDAH-1 homologous gene from Pseudomonas Aeruginosa-DDAH was cloned and expressed in E. coli. Coding sequence of DNA was synthesized and ligated into a pE-SUMO vector. Plasmid containing DDAH genes was transformed into BL21 (DE3) E. coli cells (EMD Millipore, United States), plated on LB agar plate with 50 µg/mL kanamycin, and grown overnight. For preparation of purified DDAH, 50 mL culture were inoculated in 1 L LB with 50 µg/mL kanamycin and grown to 0.8 optical density at 600 nm and then induced with isopropyl β-D-1-thiogalactopyranoside (IPTG). After 16 hours, cells were collected by centrifugation, resuspended, and lysed by sonication. DDAH in the supernatant was then purified using a Ni-sepharose column. DDAH purity was determined using SDS gel electrophoresis. DDAH activity was determined by generation of L-citrulline from ADMA using a colorimetric assay (Knipp and Vasak, 2000). The purified recombinant DDAH used in this study is designated as rDDAH-1.
ADMA Measurement
ADMA in perfusate was analyzed by modification of previously published high performance liquid chromatography (HPLC) based method (de Jong and Teerlink, 2006; Teerlink, 2007). Test samples were prepared by solid-phase extraction using an Oasis MCX Cartridge (Waters, United States). Briefly, the column was conditioned with 30% ammonium hydroxide:water:methanol (10:40:50 by volume) followed by ultrapure water. Plasma was mixed, vortexed, and spun with cold methanol for extraction, mixed with PBS, and loaded on to the column. The column was rinsed with 0.1 M hydrochloride and methanol two times. Analytes were eluted with elution buffer and dried under nitrogen. Samples were reconstituted in ultrapure water and derivatized with ortho-phthaldialdehyde reagent (Sigma-Aldrich, United States) (4:1 by volume). After mixing, the analytes were heated at 30°C for 1 minute and injected into the HPLC system, equipped with a fluorescent detector RF-10AXL (ex:340, em:455) and Chromolith performance RP-18e column (100 × 4.6 mm) (EMD Millipore, United States). Mobile phase A consisted of 25 mM potassium phosphate buffer (pH 6.5), and mobile phase B was methanol/tetrahydrofuran (97/3 by volume). Chromatographic separation was performed at room temperature at a flow rate of 2 mL/min (10%–20% solvent A).
ADMA Measurement in Cardiomyocyte
Cardiomyocytes prepared from adult mouse hearts were plated in 12 well plates at a density of 28,000 cells/well. Groups with ADMA (10 µM) were incubated for 30 minutes and washed afterward. rDDAH-1 (10 µg/mL) was then added, and cells were incubated for 1 hour at 37°C. After that, medium was removed and cells were washed once in medium without ADMA. Collected cells were lysed using M-PER lysis buffer and mixed, vortexed, and spun with cold methanol for extraction and proceeded for ADMA measurement using HPLC.
Animals
Male C57BL/6J mice were purchased from the Jackson Laboratories (Bar Harbor, United States) and acclimated in the LARC facility for at least 5 days with a standard diet. Eleven- to 18-week old mice were employed for experiments. The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Indiana University. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, revised 1996).
Isolated Mouse Heart Ischemia/Reperfusion Injury (Langendorff Model)
Mouse hearts were isolated and subjected to Langendorff ischemia/reperfusion (I/R) as we previously described (Wang et al., 2009; Wang et al., 2014). Briefly, mice were heparinized (100 IU i.p.) and anesthetized with isoflurane, and hearts were rapidly excised. The aorta of the isolated heart was canulated, and the heart was then perfused with oxygenated perfusion buffer containing ADMA (1 µM) in an isovolumetric Langendorff mode (70 mmHg). Left ventricular developed pressure (LVDP) was continuously recorded using a PowerLab 8 preamplifier/digitizer (AD Instruments Inc., Milford, MA). The maximal positive and negative values of the first derivative of pressure (+dP/dt and -dP/dt) were calculated using Labchart software. The coronary flow rate was measured by assessing the volume of pulmonary artery effluent per minute. Isolated mouse hearts were subjected to the I/R by at least 15 minutes equilibration with perfusion buffer followed by 30 minutes global ischemia (37°C) and 40 minutes reperfusion, and were randomly allocated to different experimental groups. Vehicle or rDDAH-1 was perfused during postischemic treatment. A dose-response study (0, 0.03, 0.1, and 0.3 µg/mL) was performed to determine the optimum dose of rDDAH-1 for protecting I/R-induced myocardial dysfunction. Since the studies on all rDDAH-1 doses are not feasible in a single day, the experimental protocol was designed such that untreated controls (0.0 µg/mL) were included with each dose tested. This has led to more data points in the 0 µg/mL group than other groups. Also, 0.1 µg/mL of rDDAH-1 was the optimum concentration, and we performed an additional study to confirm it along with a control group.
Infarct Size Measurement
The left ventricle (LV) from one set of mouse hearts +/− rDDAH-1 (0.1 µg/mL) after Langendorff I/R was transversely sectioned into ∼1mm-thick frozen slices using a mouse heart slicer. The heart slices were then stained with 1% 2,3,5-triphenyltetrazolium chloride (TTC). Images were taken, and the infarct area were analyzed using ImageJ (NIH). The infarct size (percentage) was calculated as total infarct weight per total LV weight from all slices as we previously reported (Wang et al., 2019). Infarct size measurement was conducted by an independent researcher blinded to the sample ID and the experimental groups.
Adult Mouse Cardiomyocyte Isolation
Cardiomyocytes were isolated from adult male mouse hearts as we previously described (Wang et al., 2019). Briefly, after mice were injected with heparin (100 IU, i.p.) and euthanized, the hearts were removed rapidly. Hearts were retrogradely perfused and digested with collagenase II. The cells were sequentially restored in calcium-contained buffer (100, 250, 500, or 1000 µmol/L CaCl2), adult mouse cardiomyocytes were seeded into laminin-precoated plates with cardiomyocyte plating medium (MEM with glutamine + 2.5% FBS, 10 mM BDM, and 1% Pen/Strep) and used for experiments.
Measurement of Mitochondrial Superoxide Production
Cardiomyocytes were treated with various concentrations of ADMA in the absence or presence of rDDAH-1 (20 µg/mL) for 2 hours. After that, cardiomyocytes were loaded with 5 µM MitoSOX Red (Thermo Fisher Scientific, United States) and incubated for 20 minutes at 37°C. Intracellular MitoSox Red selectively targets mitochondria and is oxidized by mitochondrial superoxide but not by other ROS. After a 20-minute incubation, cells were washed once. The live-cell images of cardiomyocytes were taken using an Axio Observer Z1 motorized microscope (Zeiss, Germany) with a 10X objective. Red fluorescence intensity in an individual cardiomyocyte was quantified using ImageJ (NIH) as we previously stated (Wang et al., 2019).
Mitochondrial Respiration Capacity by Seahorse XF Cell Mito Stress Test
Oxygen consumption rate (OCR) measurements were performed using a Seahorse Bioscience XF-96 instrument (Seahorse Biosciences, United States). One day before the assay, the sensor cartridge was hydrated overnight using the calibration buffer supplied by the manufacturer. Isolated mouse cardiomyocytes were plated in laminin-precoated XF96 cell culture microplate at 2500 cells/well. The cells were cultured overnight. On the day of the experiment, the cells were treated with ADMA in the absence or presence of rDDAH-1 (20 µg/mL) in supplemented XF medium and incubated at 37°C without CO2 for 2 hours. The bioenergetics profile of cardiomyocytes was measured sequentially as baseline OCR, ATP-linked production by injection of 1 μM oligomyocin, maximal uncoupled respiration by adding 0.75 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and nonmitochondrial respiration by injection of 0.5 μM rotenone and antimycin A.
Human Induced Pluripotent Stem Cell and Human Cardiomyocyte Culture
DiPS 1016 SevA human induced pluripotent stem cells (hiPSCs) (Harvard Stem Cell Institute, United States) derived from human skin fibroblasts (Passage number 40–50) were seeded and kept in culture on 1% Geltrex (Invitrogen, USA)-coated culture flasks using mTeSR (StemCell Technologies, Canada) supplemented with 1% penicillin (VWR, United States). At 80% confluency, hiPSCs were detached using Accutase (StemCell Technologies, Canada), and seeded into culture well plates in mTeSR1 media supplemented with 5 µM Rho-associated, coiled-coil containing protein kinase inhibitor (StemCell Technologies, Canada). Cells were maintained with daily mTeSR1 media changes until 95% confluency was reached. iCM differentiation from hiPSCs was adapted from a previously established protocol (Zhao et al., 2019). Briefly, at 95% confluency, hiPSCs were treated with RPMI Medium 1640 (Life Technologies, United States) supplemented with 2% B27 without insulin (Invitrogen), 0.1 mM beta-mercaptoethanol (Promega, United States), and 1% penicillin [BDM (2,3-butanedione monoxime) (-)] with the addition of 10 µM wingless-related integration site (Wnt) activator, CHIR99021 (CHIR) (Stemgent, United States) for exactly 24 hours. Afterward, media was replaced with BDM (-) supplemented with 2 µM CHIR on days 2 and 3. On day 4, iCMs were treated with BDM (-) media supplemented with the 5 µM wingless-related integration site inhibitor, IWP-4 (Stemgent). On day 6, media was replaced with BDM (-). On day 9 of the differentiation, media was replaced with RPMI Medium 1640 supplemented with 2% B27 (Invitrogen, Carlsbad, CA), 0.1 mM beta-mercaptoethanol, and 1% penicillin [BDM (+)]. After day 9, media was changed every 3 days, and beating was observed generally by day 21 of differentiation.
In Vitro iCM Functional Analysis
After physiologic beating of iCMs was observed, the baseline beat rate was recorded via brightfield videos using an Axio Observer Z1 motorized microscope and ORCA flash 4.0 camera (Hamamatsu, United States). Culture media was removed and replaced with RPMI Medium 1640 supplemented with B27 without antioxidants (Invitrogen, Carlsbad, CA), 0.1 mM beta-mercaptoethanol and 1% penicillin that was pregassed with nitrogen to remove dissolved oxygen and the iCMs were incubated in anoxic conditions (37°C, 94.9% N2, 5% CO2, and 0.1% O2) until beating ceased (typically between 4 and 8 hours). After anoxia, media was replaced with RPMI Medium 1640 supplemented with 30 µM ADMA ±20 µg/mL rDDAH-1 and cultured in standard cell culture conditions for 72 hours with timelapse videos taken 24 and 72 hours after anoxia treatment. A previously developed block-matching algorithm (Ellis et al., 2017; Acun et al., 2019) was applied to analyze the beating velocity of iCMs. This was done for all frames of the time-lapse video to create a time series of iCM beating velocity vectors. The peak velocity for each vector determined over time was averaged with that of all other vectors within each beating cluster to yield a single value representing the beating velocity of the entire field of view.
Cytokine Changes in Mouse Heart Perfusate
The cytokine content was determined in the perfusate from control and I/R mouse heart after Langendorff experiment, using Quantibody Mouse Cytokine Array 1 according to the manufacturer’s instructions (RayBiotech, United States).
Cytokine Changes in Cell Culture
Mouse cardiomyocytes used for investigation of cytokine changes were isolated as described in methods under Adult Mouse Cardiomyocyte Isolation. To mimic the ischemia-reperfusion of the heart, cardiomyocytes were subjected to hypoxia (5% oxygen) for 30 minutes, then normoxia for 1 hour in the presence or absence of 10 µM ADMA or 10 µM ADMA + 20 µg/mL rDDAH-1. Supernatants were collected for cytokine assay. Human iCMs: Supernatants from human iCMs were collected for cytokine analysis at the end of 72 hours from the experiment described under In Vitro iCM Functional Analysis. Human umbilical vein endothelial cells (HUVECs) (1,000,000 cells/well) plated in six-well plates with serum-free defined medium. After overnight plating, cells were subjected to hypoxia (5% oxygen) for 30 minutes, then normoxia for 3 hours in the presence or absence of 10 µM ADMA or 10 µM ADMA + 20 µg/mL rDDAH-1. Supernatants were collected for cytokine assay. A custom designed cytokine kit from RayBiotech was used for cytokine determination in supernatant of human iCM and HUVEC. Slides were read in a GenePix scanner and performed analysis using Q-Analyzer application.
NADPH Oxidase Assay
HUVECs were subjected to 5% hypoxia for 30 minutes and normoxia for 3 hours in the absence or presence of 10 µM ADMA, 20 µg/mL rDDAH-1, or 10 µM ADMA + 20 µg/mL rDDAH-1. Cells were then suspended in 100 µL of Krebs-HEPES buffer containing 0.5 mM lucigenin. Fifty µL of suspension was transferred to assay plate, and 5 µL of 1 mM NADPH was added. Photo emission expressed as relative light units was measured every 1 minute for 10 minutes. The readings of the luminescence increased linearly within 5 minutes, and the slope for the trend line was defined as the relative nitric oxide activity, expressed as luminescence unit relative light units/minute.
Statistical Analysis
The reported results were means ±SD. Data were checked for distributions using Shapiro-Wilk normality test. Statistical significance was evaluated by unpaired t test when only two samples were being compared, one-way ANOVA with Dunnett’s post hoc testing (single experimental variable), or two-way ANOVA with Sidak’s comparison test (two experimental variables). Data sets not passed for normality test were evaluated using either Kruskal-Wallis test or Mann Whitney test. P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. All statistical analyses were performed using the GraphPad Prism software.
Results
rDDAH-1 Lowered Extracellular and Intracellular ADMA in Cardiomyocytes
The role of DDAH in the metabolism of ADMA and cardiovascular pathophysiology has been demonstrated using rodent models by DDAH gene deletion or DDAH overexpression. However, pharmacological agents to specifically modulate ADMA levels in cellular systems in vitro and in disease models are not available. We have recently generated rDDAH-1 by cloning and expressing in E.coli. rDDAH-1 was highly effective in lowering ADMA in plasma in vitro and when administered to rats (Lee et al., 2021). Further, treatment with rDDAH-1 protected kidney from I/R injury in a rat model. Thus, rDDAH-1 represents an important pharmacological agent for specific modulation of ADMA. To investigate ADMA lowering in heart and its potential direct effect on cardiomyocytes, we first determined if rDDAH-1 can lower ADMA levels in the extracellular as well as intracellular milieu of cardiomyocytes. Figure 1 shows that addition of rDDAH-1 to the perfusion buffer used for the study of mouse heart I/R resulted in dose-dependent lowering of ADMA. Reduction in ADMA was achieved at concentrations as low as 0.1 µg/mL of rDDAH-1 [0.88, 95% confidence interval (CI), 0.87–0.95 versus 0.96, 95% CI, 0.91–1.01 in group 0; P < 0.05] (Fig. 1A). Figure 1B shows that when added to the primary cultures of mouse cardiomyocyte, rDDAH-1 (10 µg/mL) produced about 50% lowering the intracellular ADMA within one hour. These data show that rDDAH-1 can directly modulate ADMA concentration within cardiomyocytes, providing the basis for investigating its effect on heart and cardiomyocyte function.

rDDAH-1 lowered ADMA in heart perfusate and cardiomyocytes in vitro. (A) Different concentrations of rDDAH-1 were added to the perfusate solution containing 1 µM ADMA used for mouse heart perfusion in the ischemia-reperfusion studies and incubated at 37°C for 1 hour. ADMA remaining in the perfusate was then determined. The data show *P < 0.05, ***P < 0.001 using one-way ANOVA with Dunnett’s post hoc test. (B) Reduction of intracellular ADMA in cardiomyocytes when incubated in the presence of rDDAH-1. Cardiomyocytes prepared from adult mouse hearts were treated with or without ADMA (10 µM) for 30 minutes (3 wells/group). rDDAH-1 (10 µg/mL) was then added, and cells were incubated for 1 hour at 37°C. Extracellular ADMA was removed by washing the cells with medium without ADMA. Intracellular ADMA remaining in cell extract was determined. The experiments were conducted in cardiomyocytes isolated from four mouse hearts. The data show *P < 0.05, **P < 0.01, unpaired t test. All data are expressed as mean ± S.D.
rDDAH-1 Improved Cardiac Function and Reduced Myocardial Infarction after I/R Injury
To determine the effect of rDDAH-1 on myocardial function, we performed a concentration-responsive study on isolated mouse hearts using an ex-vivo I/R model. To mimic the ADMA levels found in disease situation, especially after myocardial ischemic injury, 1 µM of ADMA was added to the perfusion buffer. Mouse hearts were subjected to 30 minutes of ischemia and then 40 minutes of reperfusion. rDDAH-1 was infused into the isolated mouse hearts postischemia. Figure 2 shows that addition of rDDAH-1 to the perfusion buffer protected myocardium against I/R, as demonstrated by increase in cardiac contractility (LVDP: 44.08, 95% CI, 32.92—55.25 versus 17.56, 95% CI, 10.91–24.22 in group 0, P < 0.01; and dP/dt: 54.21, 95% CI, 34.86–73.56 versus 20.27, 95% CI, 12.48–28.07, P < 0.01) (Fig. 2, A and B) and improved LV relaxation (-dP/dt) (56.71, 95% CI, 33.67–79.76 versus 22.86, 95% CI, 13.5–32.32, P < 0.01) (Fig. 2C). Cardiac rate pressure production (RPP = LVDP x heart rate) was increased by rDDAH-1 treatment (36.18, 95% CI, 22.54–49.82 in group 0.1 versus 13.32, 95% CI, 5.74–20.91 in group 0, P < 0.01) (Fig. 2D). 0.1 µg/mL of rDDAH-1 improved cardiac function of LVDP, ±dP/dt and RPP by 1.5- to 1.7-fold compared with group 0. rDDAH-1 treatment did not change the heart rate (Fig. 2E). An increased flow rate of coronary effluent [about 45% increase in group 0.3 (95.51, 95% CI, 75.35–115.7) versus group 0 (65.88, 95% CI, 54.03–77.72)] was observed in rDDAH-1 infused hearts (Fig. 2F), which may result from vasodilation or myocardial protection. rDDAH-1 alone did not affect cardiac function or flow rate of coronary effluent in mouse hearts without I/R injury (Supplemental Fig. 1). Postischemic usage of rDDAH-1 improved myocardial functional recovery, as demonstrated by about 150% increase of LVDP compared with vehicle group after I/R (P < 0.0001) (Fig. 3A). In addition, the recovery of cardiac function was earlier than the vehicle control group. Histologic analysis of heart also showed about 30% reduction in myocardial infarct size in rDDAH-1 treated group (15.9, 95% CI, 12.65–19.15) as compared with the vehicle group (22.24, 95% CI, 18.7–25.77) (Fig. 3, B and C). Collectively, these results showed that pharmacological treatment with rDDAH-1 protected myocardium from I/R injury and markedly improved recovery of LV function postischemia.

rDDAH-1 improved cardiac function in the mouse I/R model. LV function was determined in isolated mouse heart using the Langendorff preparation. Isolated mouse hearts were equilibrated with the perfusion buffer, and the cardiac functions were determined (values at Eq) for each animal. Each mouse heart was then subjected to 30 minutes of warm ischemia followed by 60 minutes of reperfusion. Indicated concentrations of rDDAH-1 was infused during the reperfusion period, and cardiac functions were determined. The data for the groups are expressed as percentage of cardiac function at Eq for each mouse heart. LV function was evaluated as: (A) LVDP = LV systolic pressure − LV diastolic pressure. (B) dP/dt (the maximum first derivative of LV pressure with respect to time). (C) -dP/dt (the maximum negative value of the first derivative of the pressure with respect to time). (D) RPP (rate pressure product = LVDP × heart rate). (E) Heart rate at end of reperfusion among groups. (F) The flow rate for coronary effluent was measured at 20 minutes postischemia. Each symbol indicates the individual value from an isolated mouse heart. All data are given as mean ± S.D. Significance determined by control-compared one-way ANOVA with Dunnett’s post hoc test. *P < 0.05, **P < 0.01.

rDDAH-1 improved myocardial functional recovery and reduced cardiac damage in mouse I/R injury model. (A) Shows change in LVDP over time in control and of rDDAH-1 (0.1 µg/mL) after 30 minutes warm ischemia and 40 minutes I/R. The data are ****P < 0.0001 determined by two-way ANOVA with Sidak’s comparison test. (B) Representative photographs of transverse slices with TTC staining from mouse hearts without or with postischemic rDDAH-1 treatment after I/R. (C) Myocardial infarct size as determined by TTC staining of control and rDDAH-1 (0.1 µg/mL) treated hearts. The data show **P < 0.01, unpaired t test. All data are given as mean ± S.D.
rDDAH-1 Improved Beating Frequency in Human Cardiomyocyte Culture
To further examine the direct effect of ADMA lowering on cardiomyocytes, we used iCMs. Our group has previously characterized human iCMs and their beating response in vitro in both two-dimensional and three-dimensional cardiac models (Basara et al., 2021; Ellis et al., 2021; Ozcebe et al., 2021). iCMs were subjected to hypoxia and ADMA to mimic the condition that may occur during myocardial infarction. Addition of ADMA to iCM cultures immediately after anoxia inhibited beating frequency with about 25% reduction (5.39, 95% CI, 3.43–7.35 versus 7.4, 95% CI, 5.21–9.59 in control at 24 hours; 5.92, 95% CI, 4.2–7.65 versus 7.5, 95% CI, 6.18–8.81 in control at 72 hours after anoxia) (P < 0.05) and velocity recovery with about 30% decrease (54.76, 95%, 41.21–68.31 versus 77.22, 95% CI, 62.81–91.63 in control, P < 0.01) (Fig. 4, A and B). We observed that treatment of iCMs with rDDAH-1 immediately after anoxia reduced the detrimental effects of ADMA on beating rate (7.49, 95% CI, 6.18–8.81, P < 0.05) and velocity recovery (76.25, 95% CI, 62.02–90.48, P < 0.01) within 72 hours. These data suggest that rDDAH-1 provides a protective effect on human cardiomyocytes under conditions of high ADMA after anoxic conditions.

rDDAH-1 improved contractility recovery in ADMA treated iCMs after anoxia. iCM were subjected to hypoxia by incubation in 0.1% oxygen and deoxygenated media for 6 hours until beating ceased. The cells were then transferred to normoxia, and media was replaced with oxygenated media in the presence or absence of ADMA (30 µM) or rDDAH-1 (20 µg/mL). (A) Changes in beating velocity was measured at baseline (0 hour), 24, and 72 hours after hypoxia. (B) Beating rate recovery was determined by imaging soon after transfer to normoxia and 24 and 72 hours, as described under methods. The addition of ADMA inhibited both iCM contractility velocity and rate recovery, which was rescued in the presence of rDDAH-1. The sample size for all groups was n = 3. All data are expressed as mean ± S.D. Significance determined by ADMA-compared one-way ANOVA with Dunnett’s post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
rDDAH-1 Improved Cardiomyocyte Mitochondrial Bioenergetics
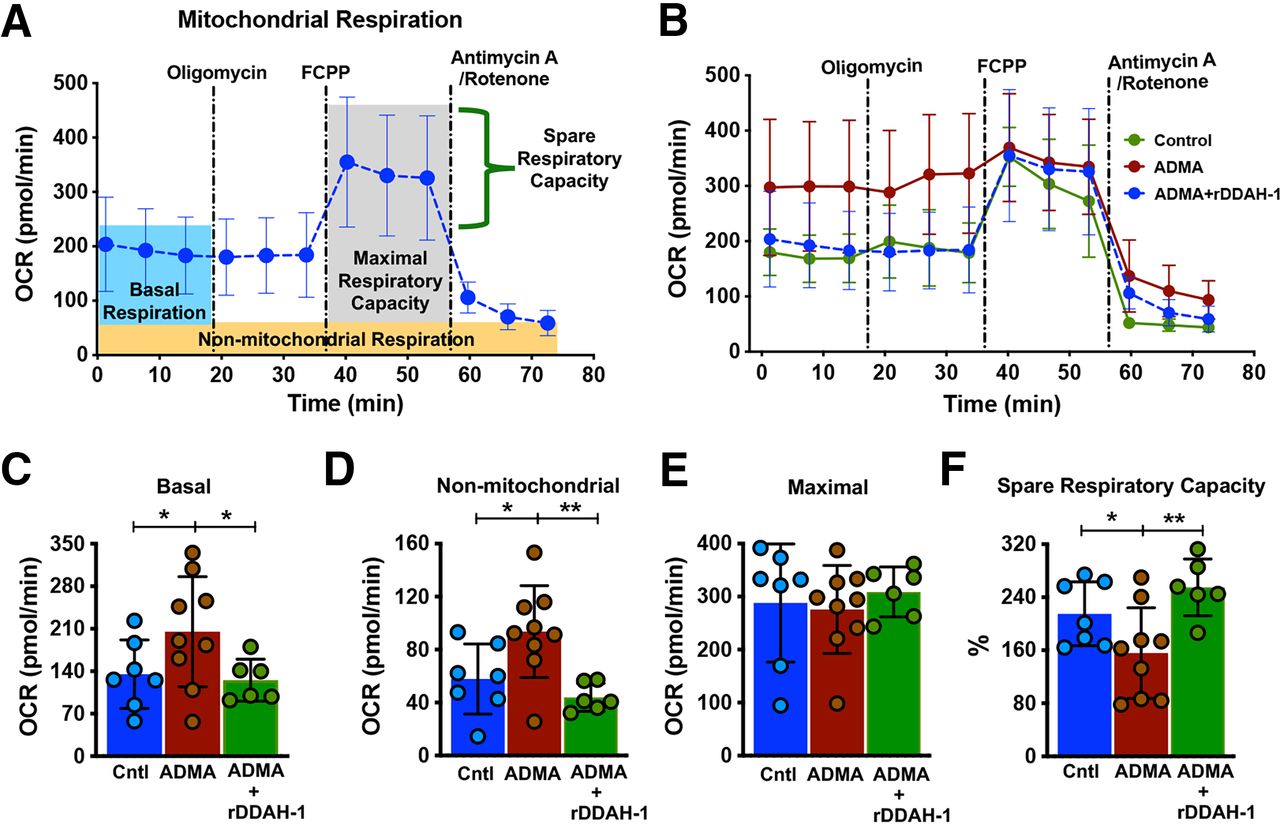
Normal cellular metabolism is a prerequisite for maintaining cardiomyocyte function and survival. Impaired mitochondrial activity alters cellular metabolism and energy production. Previous studies on the role of ADMA in cardiovascular disease have focused on its inhibition of eNOS resulting in vascular dysfunction and vasoconstriction. The potential direct effect of ADMA on cardiomyocyte has not been well studied. In this study, we investigated the effect of ADMA and rDDAH-1 on the bioenergetic profile of mouse cardiomyocyte mitochondria using the Seahorse Cell Mito Stress Test assay (Fig. 5, A and B). Treatment of cardiomyocytes with ADMA increased OCR at the basal (205.1, 95% CI, 135.4–274.8 versus 135.1, 95% CI, 82.76–187.4 in control, P < 0.05) and nonmitochondrial respiration level (93.64, 95% CI, 67.02–120.3 versus 57.84, 95% CI, 33.34–82.34 in control, P < 0.05) (Fig. 5, C and D), whereas the maximal OCR was unchanged (Fig. 5E). The maximum, uncontrolled OCR was obtained after addition of FCCP, which leads to a rapid consumption of oxygen without ATP generation. Notably, ADMA reduced spare respiratory capacity/reserve capacity (the ability of cells to respond to increased energy demand under stress) of cardiomyocytes (155.8, 95% CI, 103.2–208.4 versus 215, 95% CI, 170.5–259.6 in control, P < 0.05) (Fig. 5F). These data suggest that ADMA directly alters mitochondrial respiratory function, which could result in decreased tolerance to stress as under the conditions of hypoxia or ischemia. Importantly, treatment with rDDAH-1 reversed the ADMA-induced mitochondrial respiratory dysfunction in cardiomyocytes, including reduced basal (125.2, 95% CI, 88.87–161.5, P < 0.05) (Fig. 5C) and nonmitochondrial respiration (43.97, 95% CI, 32.92–55.02, P < 0.01) (Fig. 5D), as well as improved spare respiratory capacity (254.8, 95% CI, 210–299.7, P < 0.01) (Fig. 5F). In normal adult cardiomyocytes, treatment with rDDAH-1 alone did not impact mitochondrial energetics in (Supplemental Fig. 2). These results demonstrate that altered cardiomyocyte respiration under pathologic levels of ADMA can be reversed by treatment with rDDAH-1.

rDDAH-1 improved ADMA-induced mitochondrial respiration capacity in adult male mouse cardiomyocytes. (A) Shows Seahorse Cell Mito Stress Test assay in control cardiomyocytes from adult male mouse hearts and the calculable parameters. Dotted lines indicate addition of ATP synthase inhibitor oligomycin, electron transport chain uncoupler FCCP, and complex I and III blockers rotenone and antimycin A. (B) OCR in mouse primary cardiomyocytes treated with 2-hour vehicle (Control), ADMA (3 µM), or ADMA + rDDAH-1 (20 µg/mL), respectively. (C–F) Calculated values for respiratory parameters. The experiment was performed on cardiomyocytes from a total of three male mouse hearts. All data are given as mean ± S.D. Significance determined by unpaired t test. *P < 0.05, **P < 0.01.
rDDAH-1 Reversed the Effect of ADMA on Mitochondrial ROS Production
It is known that oxidative stress, particularly mitochondrial-generated ROS, can substantially contribute to cardiac dysfunction. Therefore, we further investigated if ADMA and rDDAH-1 directly affected the generation of ROS in mitochondria. Mitochondrial ROS production was determined in isolated mouse cardiomyocytes in the presence or absence of ADMA and rDDAH-1 using the MitoSoX Red probe. In this assay, superoxide production in mitochondria is detected by the increase in fluorescence intensity of MitoSoX Red (Fig. 6A). Addition of ADMA (3, 10, and 30 μM) to cardiomyocytes resulted in the elevation of oxygen free radical production within 1 hour (1.79, 95% CI, 1.65–1.92; 1.7, 95% CI, 1.53–1.86, and 2.18, 95% CI, 1.94–2.41 in groups of 3, 10, and 30 µM of ADMA versus 0.998, 95% CI, 0.9–1.1 in group 0, P < 0.0001) (Fig. 6B). Treatment with rDDAH-1 decreased ADMA-induced mitochondrial superoxide production (1.31, 95% CI, 1.14–1.48 and 1.15, 95% CI, 0.98–1.32 in 3 and 10 μM ADMA + rDDAH-1, respectively, P < 0.0001) (Fig. 6C). ADMA and rDDAH-1 treatment did not change NADPH oxidase activity (Supplemental Fig. 3), suggesting that the induction of ROS by ADMA may be largely due to its previously well documented effect on eNOS uncoupling (Druhan et al., 2008; Sud et al., 2008; Karbach et al., 2014; Badran et al., 2016; Janaszak-Jasiecka et al., 2021). These data show that rDDAH-1 can reduce ROS generation under the condition of high ADMA as observed after ischemia or in chronic heart failure.

rDDAH-1 reduced ADMA induced ROS generation in cardiomyocytes. Mitochondrial superoxide production in mouse cardiomyocytes was determined using MitoSOX Red. (A) Representative images in cardiomyocytes with MitoSOX Red. (B) Concentration-dependent changes ROS after 2 houra treatment with ADMA (0, 3, 10, and 30 µM. The data are ****P < 0.0001, Kruskal-Wallis test with Dunn’s comparison. (C) rDDAH-1 (20 µg/mL) treatment decreased mitochondrial superoxide production in mouse cardiomyocytes. The data are *P < 0.05, ****P < 0.0001, Mann-Whitney test. The experiment was repeated in two individual trials, and cardiomyocytes were isolated from a total of three male mouse hearts. All data are given as mean ± S.D.
rDDAH-1 Modified Inflammatory Cytokine Expression in Mouse Heart After I/R and Cardiac Cells in Vitro
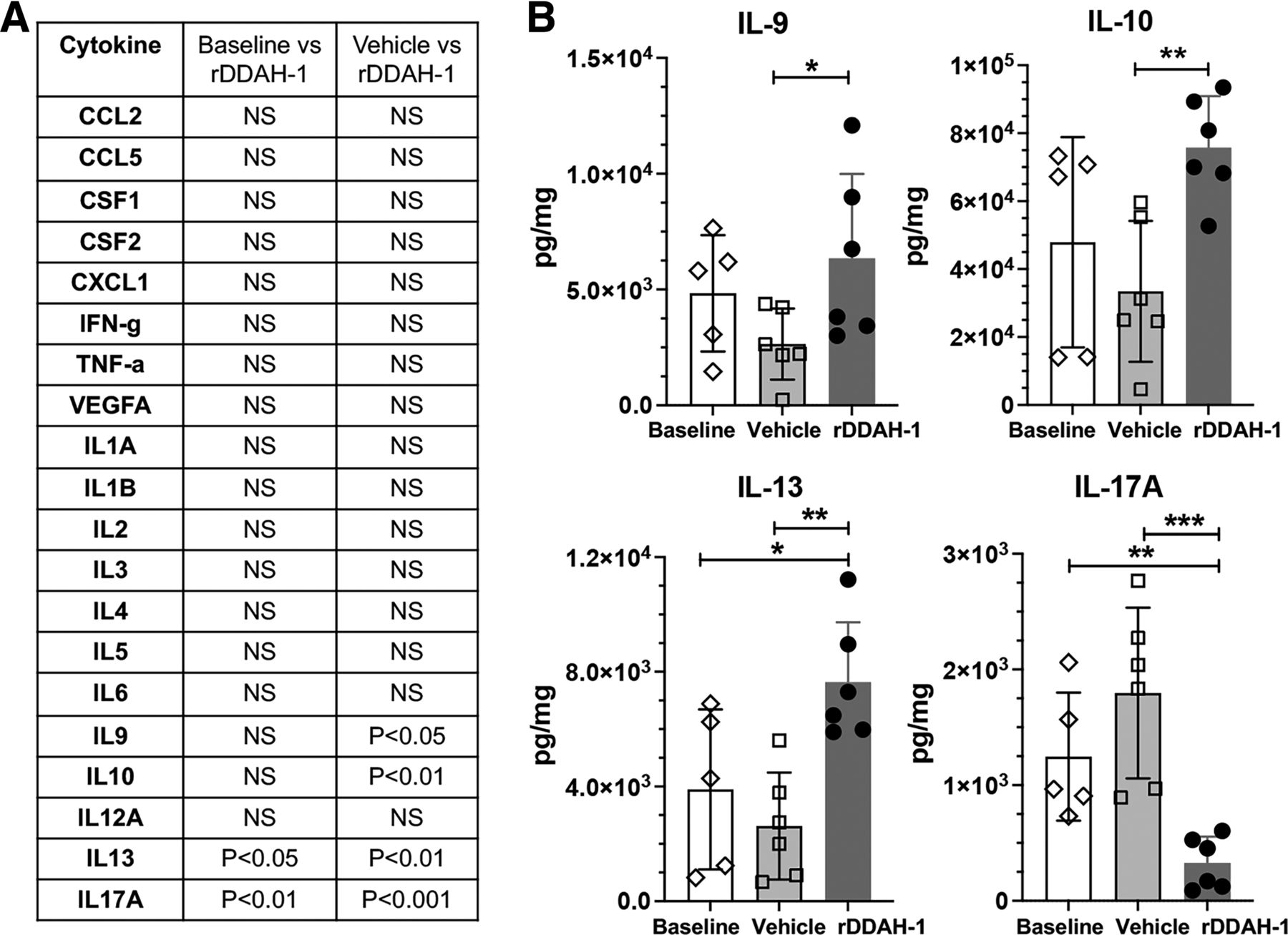
Cardiac I/R is associated with increased inflammatory cytokine expression, which can produce further injury to the heart. Previous studies have not examined the cytokine profile when the heart is subjected to high ADMA and I/R. We examined a large panel of cytokines released in the coronary effluent obtained from the isolated mouse hearts subjected to I/R with or without rDDAH-1 treatment. Our results show that in mouse I/R heart, rDDAH-1 selectively reduced proinflammatory cytokine IL-17A (327.9, 95% CI, 91.02–564.9 versus 1796, 95% CI, 1021–2571 in vehicle, P < 0.001) and increased anti-inflammatory cytokine IL-10 (75,764, 95% CI, 59,874–91,654 versus 33,437, 95% CI, 11,687–55,186 in vehicle, P < 0.01) and IL-13 (7641, 95% CI, 5451–9831 versus 2620, 95% CI, 659.7–4580 in vehicle, P < 0.01) (Fig. 7B).

rDDAH-1 modified cytokine expression after I/R in mouse heart. The perfusate from the mouse heart I/R study were collected at the end of the experiment and concentrated 10-fold by membrane centrifugation. (A) The concentration of each cytokine was determined in the samples from the effluent of mouse hearts without I/R (baseline), with I/R but no rDDAH-1 treatment (vehicle), and with I/R plus DDAH treatment (rDDAH-1). The level of each cytokine among the three groups was compared, and those with P values greater than 0.05 are listed as N.S. B. Shows data on key cytokine expression (IL-9, IL-10, IL-13, IL-17A) in the perfusate at baseline, after I/R (vehicle), and I/R with 0.1 µg/mL rDDAH-1 treatment. All data are given as mean ± S.D. Significance determined by baseline-compared one-way ANOVA with Dunnett’s post hoc test or vehicle-compared unpaired t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Further investigation of the effect of ADMA and rDDAH-1 on cytokine expression in mouse cardiomyocytes, human iCM, and endothelial cells showed that changes in IL-17A in response to ADMA or ADMA + rDDAH-1 was common to these cell types (Fig. 8). It is expected that the cytokine expression profile of the different cells subjected to ischemia-reperfusion may be different. This may be due to difference in species (mouse versus human cells), cell type, and the cell culture conditions required for maintenance of different cell types (Fig. 8A). Despite the differences, the effect of ADMA and rDDAH-1 was limited to specific cytokines. We observed that ADMA increased IL-17A production in human iCM (64, 95% CI, 31.71–96.29 versus 27.67, 95% CI, 6.98–48.35 in control, P < 0.01) (Fig. 8C), mouse cardiomyocytes (48.1, 95% CI, 18.32–77.88 versus 22.67, 95% CI, 4.36–40.98 in control, P < 0.05) (Fig. 8D), and HUVEC (141, 95% CI, 76.56–205.4 versus 59, 95% CI, 39.13–78.87 in control, P < 0.01) (Fig. 8E). Importantly, rDDAH-1 decreased ADMA-induced IL-17A levels in human iCM (39, 95% CI, 23.89–54.11, P < 0.05) and mouse cardiomyocytes (25.63, 95% CI, 17.31–33.96, P < 0.05). Our studies, for the first time, reveal that the effect of ADMA and rDDAH-1 on IL-17A was common to all cells. These results suggest that increased IL-17A may be an important mechanism of mediating the inflammatory response to ADMA.

ADMA and rDDAH-1 modulate IL-17A expression in cardiac and endothelial cells. (A) A summary of key cytokine changes (P < 0.05) in response to ADMA and rDDAH-1 in various cells in vitro. (B–E) Show changes IL-17A in mouse heart effluent (B), human iCM (C), mouse CM (D), HUVEC (E), in the presence or absence of ADMA or ADMA + rDDAH-1. All data are expressed as mean ± S.D. Significance determined by baseline- or control-compared one-way ANOVA with Dunnett’s post hoc test or vehicle- or ADMA-compared unpaired t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Patients with cardiovascular disease risk factors including hypertension, hypercholesterolemia diabetes, and heart failure exhibit high blood levels of ADMA. High ADMA levels are associated with cardiovascular disease progression and mortality (Cavusoglu et al., 2010; Xuan et al., 2016; Dowsett et al., 2020; Gać et al., 2020). The findings from clinical studies are further substantiated by the induction or exacerbation of disease in animal models in which ADMA levels are increased either by DDAH gene deletion (Leiper et al., 2007) or by ADMA administration (Achan et al., 2003). Based on the clinical and preclinical studies, ADMA is considered as a risk factor for vascular dysfunction and cardiovascular disease.
In the heart, high levels of ADMA may reduce nitrogen oxide bioavailability inducing endothelial dysfunction and vasoconstriction. In addition, increasing evidence suggests that the pathologic action of ADMA may extend beyond its vasoconstriction activity. ADMA has been found to affect mitochondrial function in type 2 diabetic rats by inducing oxygen free radicals, which may contribute to its direct effect on myocardial cell metabolism (Xiong et al., 2020). Recent studies have shown that treatment with ADMA directly inhibited the maximum contraction tension of papillary muscle (Xiong et al., 2021). Moreover, cardiac-specific deletion of DDAH-1 gene produced larger infarct size and impaired LV function in a mouse model of myocardial infarction (Hou et al., 2018). Thus, extravascular effect of ADMA, particularly in the heart, may be an important mechanism for its pathologic actions in ischemic heart disease and heart failure. In this study, we have for the first time used a pharmacological approach to specifically lower ADMA in the heart and investigated its effect on the isolated cardiomyocytes. We show that the intracellular levels of ADMA in cardiomyocytes can be directly modulated by exogenously added rDDAH-1. We show that direct treatment with ADMA metabolizing enzyme DDAH reduced myocardial ischemic injury and preserved cardiac function in mouse model of global ischemia. Treatment with rDDAH-1 also decreased myocardial infarct size after I/R. Notably, rDDAH-1 treatment reduced inflammatory cytokine IL-17A while increasing anti-inflammatory cytokines IL-10 and IL-13. These findings suggest the ADMA lowering may prevent I/R-induced inflammatory response, which may cause further myocardial damage. Further in vitro investigation of the effect of ADMA and rDDAH-1 on cardiomyocytes and endothelial cells showed that IL-17A expression was uniformly modulated by ADMA in these cells. In this regard, it is important to note that previous studies have shown that IL-17A plays an important role in ischemia reperfusion injury and cardiac remodeling. In patients with myocardial infarction, high serum levels of IL-17 have been observed (Jafarzadeh et al., 2009; Garza-Reyes et al., 2020). Also, genetic association IL-17 with carotid medial-intimal thickening (Wu et al., 2018) and coronary artery disease (Ghaznavi and Soltanpour, 2020) has been reported. These clinical and preclinical studies have suggested that IL-17 is an important player in the postinfarct inflammation and ventricular remodeling in preclinical model of ischemia (Liao et al., 2012; Zhou et al., 2014; Chang et al., 2018). Our studies are the first to reveal a link between IL-17A and ADMA, suggesting that IL-17A may play an important role in ADMA mediated inflammation and cardiovascular disease.
Our studies show that direct treatment with rDDAH-1 improved cardiomyocyte mitochondrial respiratory dysfunction. rDDAH-1 treatment improved the ADMA induced basal and nonmitochondrial respiration and impaired spare respiratory capacity. Since cardiomyocytes undergo major metabolic and biochemical shifts during myocardial I/R, the deprivation of oxygen and nutrients alters mitochondrial function and ATP production (McDougal and Dewey, 2017; Garbern and Lee, 2021). The improvement in mitochondrial respiratory capacity observed in our studies suggest that rDDAH-1 may increase the ability of myocardium to respond to the increased energy demand under stress conditions, such as ischemia and oxidative stress. Our findings suggest that in addition to its effect on vascular function, high levels of ADMA may directly induce cardiac dysfunction, which can be prevented by treatment with rDDAH-1.
We further show that addition of rDDAH-1 reduced the ADMA induced ROS in cardiomyocyte mitochondria. The role of ROS in mitochondrial damage and bioenergetics has been well documented. ROS can induce opening of the mitochondrial permeability transition pore and destroy the intracellular redox balance leading to oxidative stress, causing extensive damage to protein. Previous studies have shown that ADMA induces the generation of ROS by uncoupling of NOS (Druhan et al., 2008). Our studies show that under the condition of high ADMA, therapeutic treatment with rDDAH-1 reduced mitochondrial ROS generation. Thus, pharmacological lowering of ADMA may reduce oxidative stress and prevent ROS mediated mitochondrial dysfunction, which has been shown to contribute to ischemic heart disease and progression of heart failure.
Our studies with hiPSC derived human cardiomyocytes have provided further evidence for a direct effect of ADMA lowering on cardiomyocyte function. iCMs have been previously characterized for their cardiomyocyte-like properties and beating behavior. Transient exposure to ADMA dramatically reduced beating frequency and velocity during iCM recovery from hypoxia induced beating cessation. Treatment with rDDAH-1 (P < 0.05) improved the beating rate and the recovery from hypoxia and ADMA induced stress. These studies further support a direct effect of ADMA on cardiomyocyte and functional improvement by treatment with rDDAH-1.
A limitation of our studies is the use of recombinant PA-DDAH (rDDAH-1) for the investigation of the effect of pharmacological lowering of ADMA in vitro and in vivo. We have cloned and expressed both human DDAH-1 and PA-DDAH in E. Coli. We found that as compared with the human DDAH-1, the PA-DDAH is expressed at much higher levels and was 10- to 30-fold more active. More importantly, PA-DDAH was stable under the experimental conditions required to perform the current studies. With the proof of efficacy with PA-DDAH, further studies will be undertaken on the expression of recombinant human DDAH-1, its protein biochemistry and stability for future pharmacological studies.
In summary, our studies have shown that pharmacological modulation of cellular ADMA by rDDAH-1 can provide an important approach to investigate the pathologic effects of ADMA in various disease states. High levels of ADMA are considered a risk factor for cardiovascular disease. Therefore, the reduction of ADMA by a pharmacological agent represents an important strategy for the development of new drugs for the treatment of cardiovascular disease.
Acknowledgments
The authors thank Dr. Chandan K. Sen at Indiana University School of Medicine for allowing access to the Seahorse Bioscience XF-96 instrument and thank Dr. Kanhaiya Singh from Division of Plastic Surgery at Indiana University School of Medicine for his assistance in acquiring the Seahorse data.
Authorship Contributions
Participated in research design: Lee, Singh, Zorlutuna, Wang.
Conducted experiments: Lee, Scott, Ellis, Wang.
Contributed new reagents or analytic tools: Lee, Wang.
Performed data analysis: Lee, Scott, Ellis, Wang.
Wrote or contributed to the writing of the manuscript: Lee, Singh, Wang.
Footnotes
- Received August 30, 2021.
- Accepted January 16, 2022.
This work was supported by the Indiana Clinical and Translational Sciences Institute [Grant UL1TR001108] from National Institutes of Health, National Center for Advancing Translational Sciences, Clinical, and Translational Sciences Award; AIM grant from Indiana University Health and Indiana-Clinical and Translational Sciences Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of National Institutes of Health.
No author has an actual or perceived conflict of interest with the contents of this article.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ADMA
- asymmetric dimethyl arginine
- DDAH
- dimethylarginine dimethylaminohydrolase
- hiPSC
- human induced pluripotent stem cell
- HUVEC
- human umbilical vein endothelial cell
- iCM
- induced pluripotent stem cell derived cardiomyocytes
- I/R
- ischemia-reperfusion injury
- LV
- left ventricle
- LVDP
- left ventricular developed pressure
- NOS
- nitric oxide synthase
- OCR
- oxygen consumption rate
- rDDAH
- recombinant dimethylarginine dimethylaminohydrolase
- ROS
- reactive oxygen species
- TTC
- 2,3,5-triphenyltetrazolium chloride
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}