


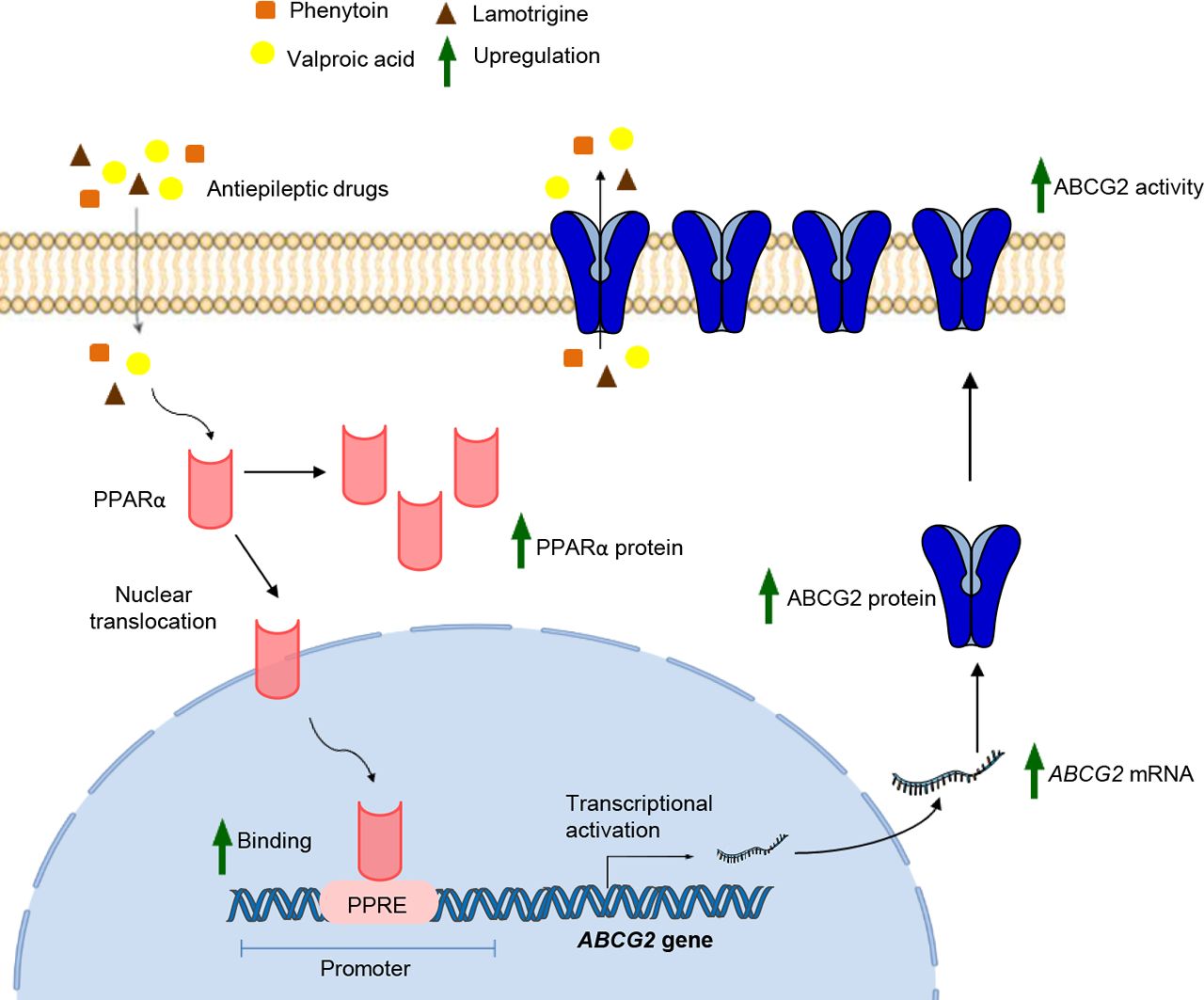
Visual Overview

Abstract
Despite the progress made in the development of new antiepileptic drugs (AEDs), poor response to them is a rising concern in epilepsy treatment. Of several hypotheses explaining AED treatment failure, the most promising theory is the overexpression of multidrug transporters belonging to ATP-binding cassette (ABC) transporter family at blood–brain barrier. Previous data show that AEDs themselves can induce these transporters, in turn affecting their own brain bioavailability. Presently, this induction and the underlying regulatory mechanism involved at human blood–brain barrier is not well elucidated. Herein, we sought to explore the effect of most prescribed first- and second-line AEDs on multidrug transporters in human cerebral microvascular endothelial cells, hCMEC/D3. Our work demonstrated that exposure of these cells to valproic acid (VPA) induced mRNA, protein, and functional activity of breast cancer resistance protein (BCRP/ABCG2). On examining the substrate interaction status of AEDs with BCRP, VPA, phenytoin, and lamotrigine were found to be potential BCRP substrates. Furthermore, we observed that siRNA-mediated knockdown of peroxisome proliferator-activated receptor alpha (PPARα) or use of PPARα antagonist, resulted in attenuation of VPA-induced BCRP expression and transporter activity. VPA was found to increase PPARα expression and trigger its translocation from cytoplasm to nucleus. Findings from chromatin immunoprecipitation and luciferase assays showed that VPA enhances the binding of PPARα to its response element in the ABCG2 promoter, resulting in elevated ABCG2 transcriptional activity. Taken together, these in vitro findings highlight PPARα as the potential molecular target to prevent VPA-mediated BCRP induction, which may have important implications in VPA pharmacoresistance.
SIGNIFICANCE STATEMENT Induction of multidrug transporters at blood–brain barrier can largely affect the bioavailability of the substrate antiepileptic drugs in the brains of patients with epilepsy, thus affecting their therapeutic efficacy. The present study reports a mechanistic pathway of breast cancer resistance protein (BCRP/ABCG2) upregulation by valproic acid in human brain endothelial cells via peroxisome proliferator-activated receptor alpha involvement, thereby providing a potential strategy to prevent valproic acid pharmacoresistance in epilepsy.
Introduction
Antiepileptic drug (AED) monotherapy is the initial standard mode used worldwide for epilepsy management (Park et al., 2019). However, one of the biggest concerns to date is that a major percentage (40%–50%) of patients do not respond to first-line monotherapy consisting of 25% to 30% refractory cases (Rawat et al., 2018; Park et al., 2019; Le et al., 2021), which is majorly attributed to the inadequate AED concentration reaching the epileptogenic brain (Kwan and Brodie, 2005; Remy and Beck, 2006). One prominent hypothesis explaining this is excessive AED efflux across the blood–brain barrier (BBB) (Löscher et al., 2011). The BBB is the most essential component of a healthy central nervous system, which allows selective entry of substances into the brain (Haddad-Tovolli et al., 2017). The presence of transport proteins such as members of the ATP-binding cassette (ABC) efflux transporters and solute carrier proteins at the brain endothelial cell membrane controls this transport. Indeed, these transporters have the potential to impede transport of pharmacologically relevant substrate drugs across BBB (Girardin, 2006).
Expression studies performed in the brains of refractory patients with epilepsy (PWE) and in an animal model of pharmacoresistant epilepsy revealed overexpression of multidrug transporters (MDTs) belonging to ABC transporter family such as P-glycoprotein (ABCB1), multidrug resistance-associated proteins (MRP1-2/ABCC1-2, MRP4-6/ABCC4-6), and breast cancer resistance protein (BCRP/ABCG2) as the potential phenomenon for epilepsy pharmacoresistance (Dombrowski et al., 2001; Volk and Loscher, 2005; Banerjee Dixit et al., 2017). Multiple causal factors have been put forward to explain this elevated expression (Leandro et al., 2019). High transporter levels in surgically resected brain tissues of PWE associated with a known structural brain abnormality such as cortical dysplasia or brain tumors suggest disease etiology as the underlying cause (Leandro et al., 2019). An alternative explanation comes from the data obtained from animal models with experimentally induced seizures, showing recurrent seizure activity and the associated neuronal stress as the responsible factor (van Vliet et al., 2005, 2007; Bankstahl and Loscher, 2008). However, these studies are few in number, and the focus of these experiments has been ABCB1 with very little research on other MDTs. Yet another plethora of studies supports the concept that chronic use of AEDs promotes the excessive expression of transporters, possibly indicating a defensive response of body against xenobiotics. This has been widely explored in the literature both in vitro and in vivo. While multiple AEDs have depicted to exert inductive effect on ABCB1 (Wen et al., 2008; Yang et al., 2008; Ke et al., 2019), this effect on other MDTs is limited and confined to either rat BBB (Lombardo et al., 2008) or other tissue barriers such as intestine, placenta, liver cells, etc. (Rubinchik-Stern et al., 2015; Grewal et al., 2017). Furthermore, there are only two reports available on the human BBB discussing the AED-mediated regulation of MDTs, and again the transporter investigated is ABCB1. The first report by Alms et al. (2014) in hCMEC/D3 cells demonstrated an upregulatory effect of carbamazepine (CBZ) on ABCB1 activity, albeit the concentration of the CBZ used (100 µM) exceeded its therapeutic plasma concentration range. Recently, our group led by Rawat et al. showed downregulation of ABCB1 in hCMEC/D3 cells by valproic acid (VPA) (Rawat et al., 2020). There is no evident result of MDT induction by AEDs and the underlying molecular basis at human BBB, which necessitates further exploration. Besides, induction of the functional expression of MDTs may reduce the bioavailability of AEDs to enter the brain when these drugs are also the substrate of MDTs. Hence, it becomes imperative to systematically investigate the substrate status of AEDs to explain the importance of MDTs in AED efflux.
In the present study, we evaluated the effect of widely prescribed first-line AEDs [phenytoin (PHT), VPA, and CBZ] and second-line AEDs [lamotrigine (LTG), topiramate (TPM), and levetiracetam (LEVI)] on MDTs in hCMEC/D3 cells and determined whether these AEDs show substrate interactions with MDTs. Further, we explored the molecular basis of AED-mediated transporter regulation. Our data revealed that VPA upregulates expression and activity of BCRP in hCMEC/D3 cells via peroxisome proliferator‐activated receptor alpha (PPARα)-dependent mechanism. In addition, we found VPA, PHT, and LTG show substrate interaction with BCRP. Multiple evidences in mouse models show PPARα to be an important regulator of ABC transporters, suggesting that PPARα may alter the disposition of substrate toxicants/drugs of these transporters, thus influencing either the drug toxicity or bioavailability (Moffit et al., 2006; Hoque et al., 2015; Wang et al., 2018). Our data substantiates the importance of PPARα in VPA pharmacoresistance.
Materials and Methods
Chemicals and Reagents
Endothelial Basal Medium-2 (EBM-2) was purchased from Lonza (Walkersville, MD, USA). Collagen I (rat tail) and chemically defined lipid concentrate were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Dulbecco’s modified Eagle’s medium, hydrocortisone, ascorbic acid, HEPES, DMSO, PHT, CBZ, VPA, LTG, TPM, LEVI, bovine serum albumin (BSA), Ko143, clofibrate, and GW7647 were procured from Sigma-Aldrich (St. Louis, MO, USA). MK886 was from Cayman Chemical Co. (Ann Arbor, MI, USA). Basic fibroblast growth factor, FBS, penicillin-streptomycin, and BODIPY FL Prazosin (BPZ) were purchased from Invitrogen (Carlsbad, CA, USA).
Cell Culture, Drug, and Inhibitor Treatment
Immortalized human cerebral microvascular endothelial cell line, hCMEC/D3, was purchased from Cedarlane Laboratories (Burlington, Canada) and was used within passages 25 to 35. They were maintained at 37°C with 5% CO2 in EBM-2 media supplemented with 1/100 chemically defined lipid concentrate, 1.4 μM hydrocortisone, 5 μg/ml ascorbic acid, 10 mM HEPES buffer, 1 ng/ml basic fibroblast growth factor, 2.5% FBS, and 1% penicillin-streptomycin and cultured on collagen I coated flasks and plates. Human embryonic kidney HEK293 cells were grown in Dulbecco’s modified Eagle’s medium high glucose (4.5 g/l) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS and were maintained at 37°C in 5% CO2. PHT, CBZ, VPA, LTG, TPM, MK886, BPZ, and Ko143 were dissolved in DMSO, and LEVI was dissolved in water. The concentration of AEDs used for cell culture treatments were consistent with the recommended serum therapeutic range for individual AEDs in epilepsy patients (Supplemental Table 1). For PPARα antagonist studies, MK886 was used as a nuclear receptor PPARα antagonist (Kehrer et al., 2001; Mogilenko et al., 2013). hCMEC/D3 cells were treated with VPA in the presence or absence of MK886, and then BCRP expression and activity were determined using western blot and BPZ efflux assay, respectively. MK886 was added to the cells at a final concentration of 8 μM. DMSO (0.1% v/v) was used as vehicle control (VC) in all experiments unless specified.
Assessment of Cell Viability
To ensure that the concentrations of the test chemicals did not affect the hCMEC/D3 cell viability, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was performed. Briefly, cells were plated on collagen-coated 96-well plates for 24 hours and subsequently treated with varying concentrations of each chemical. Assay was performed after 72-hour treatment with PHT, CBZ, VPA, LTG, TPM, and LEVI and 48-hour treatment with MK886. MTT (100 μl; final concentration of 0.5mg/ml; Ameresco, Fountain Parkway, Solon, OH, USA) dissolved in media was added to each well, and the plates were incubated at 37°C for 3 hours. The formed formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm with a reference filter of 630 nm using Infinite 200 PRO multimode plate reader (Tecan, Mannedorf, Zurich, Switzerland). The results were expressed as a percentage ratio of absorbance of treated cells to the absorbance of control cells.
Reverse Transcription and Quantitative Real-Time PCR
Total RNA was extracted from treated hCMEC/D3 cells using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol. RNA quantity (absorbance 260 nm) and purity (absorbance 260 nm/absorbance 280 nm) were assessed by Infinite 200 PRO NanoQuant (Tecan). One microgram of total RNA was reverse transcribed using cDNA synthesis components (Thermo Fisher Scientific). Quantitative real-time PCR (qPCR) was performed using PowerUp SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific) and gene-specific primers. Beta-2 Microglobulin (B2M) was used as an internal standard for normalizing gene expression values. Analysis of data were done using the ΔΔCt method (Yuan et al., 2006). Results were expressed as fold change of the control group. Gene-specific primer sequences used for qPCR are mentioned in Supplemental Table 2.
Cytoplasmic, Nuclear, and Total Protein Extraction
Nuclear and cytoplasmic fractions from hCMEC/D3 cells were extracted using a NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. The total cell protein was obtained using radioimmunoprecipitation assay lysis buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS] supplemented with protease inhibitor cocktail.
Western Blot Analysis
The protein concentration in the respective lysates was quantified using a bicinchoninic acid (BCA) protein estimation kit (Pierce Thermo Fisher Scientific Inc., Waltham, MA, USA). Protein sample preparation for PPARα detection was done by heating samples at 95°C for 5 minutes. For detecting BCRP, samples were deglycosylated using peptide-N-glycosidase F (New England Biolabs, UK) during preparation. Equal amount of protein from each sample were separated on 10% SDS-PAGE gel and transferred onto nitrocellulose membrane. After blocking with 5% BSA at room temperature for 1 hour, the membranes were incubated at 4°C overnight with primary antibodies to detect BCRP protein (4477, rabbit polyclonal, 1:1000; Cell Signaling Technology, Danvers, MA, USA) (Sridharan et al., 2019), PPARα (ab24509, rabbit polyclonal, 1:2000; Abcam, Cambridge, MA, USA) (Wang et al., 2020), heat shock cognate protein 70 (HSC, sc-7298, mouse monoclonal, 1:2000; Santa Cruz Biotechnology, Dallas, TX, USA) (Ahmad et al., 2022), glyceraldehyde-3-phosphate dehydrogenase (GAPDH, ab8245, mouse monoclonal, 1:10,000; Abcam, Cambridge, MA, US) (Lu et al., 2021), and lamin (sc-376248, mouse monoclonal, 1:1000; Santa Cruz Biotechnology, Dallas, TX, USA) (Rosain et al., 2022). Subsequently, blots were incubated with respective horseradish peroxidase-conjugated bovine anti-rabbit secondary antibody and goat anti-mouse antibody, and bands were detected using enhanced chemiluminescence reagent (Pierce Thermo Fisher Scientific Inc.) on the Chemi Doc MP Imaging chemiluminescence system (Bio-Rad, Hemel Hempstead, UK). Protein expression was analyzed with the AlphaImager 3400 (Alpha InnoTech corporation, San Leandro, CA, USA) software. Bands of HSC were used to normalize the intensity of the target total protein. Bands of lamin and GAPDH were used to normalize intensities of nuclear and cytoplasmic proteins, respectively.
Functional Studies
A functional assay for BCRP was performed using established BCRP substrate, BPZ, as described earlier (Hori et al., 2004) with modifications. The assay quantifies a decrease in intracellular BODIPY fluorescence as a result of transporter activity. hCMEC/D3 cells were seeded in a 12-well plate one day prior to drug treatment. Cells were treated with VC or test AED for various time points under study. On the day of assay, cells were washed with 1X PBS to remove drug and incubated with medium containing 250 nM BPZ for 30 minutes with or without 1 μM Ko143 (BCRP-selective inhibitor) at 37°C in 5% CO2 (uptake period). Subsequently, cells were washed three times with ice-cold 1X PBS and incubated in BPZ-free media in presence or absence of 1 μM Ko143 for 90 minutes to allow BPZ efflux to occur (efflux period). Efflux was terminated by washing cells with ice-cold 1X PBS thrice. Cells were lysed by adding 1% triton X-100 followed by incubation at 37°C for 15 minutes. Twenty microliters of this lysate was aliquoted into 384-well black assay plates and subjected to plate reader for fluorescence detection (excitation: 488 nm; emission: 530nm). The remaining lysate was used for protein estimation by the BCA method to normalize the obtained fluorescence with total protein concentration.
ATPase Assay
Transport of compound by ABC transporters is coupled with ATP hydrolysis; hence, this assay measures the amount of inorganic phosphate generated during ATP hydrolysis by the substrate drug of the transporter by colorimetric means. The assay was performed using SB BCRP M PREDEASY ATPase Kit (Solvo Biotechnology, Budaors, Hungary) according to the manufacturer’s instructions. Isolated membranes from MCF-7 cells overexpressing BCRP (5 mg/ml) were incubated with 2% v/v DMSO or water as the VC or the test AED in the presence or absence of sodium orthovanadate. MgATP was added to initiate the reaction. The results were expressed as vanadate-sensitive ATPase activity. In the activation assay, the ability of the test drug to stimulate the baseline ATPase activity is calculated while results of the inhibition assay measures the test drug-mediated decrease in maximal transporter activity associated with the presence of sulfasalazine (known BCRP activator).
Competitive Cellular Efflux Assay
The assay is based on the potential of the test drug to compete with the transporter’s known fluorescent substrate for its efflux out of the cell via the transporter. The resulting increase in intracellular fluorescence is estimated. The assay determines the potential substrate interaction of the test drug with the transporter (Terashi et al., 2000; Kim et al., 2010; Grewal et al., 2017; Wu et al., 2020). In our study, the ability of the AED to compete with the efflux of BPZ through BCRP was measured. Briefly, seeded cells were preincubated for 30 minutes at 37°C in a medium containing BPZ with VC or the AEDs or reference inhibitor, Ko143. Cells were washed with 1X PBS and further incubated for 90 minutes in BPZ-free media continuing with or without Ko143. Termination of assay and fluorescent detection was done as mentioned in the section Functional Studies. Obtained fluorescent intensity was normalized with total protein concentration. The results were represented as the fold increase in intracellular BPZ fluorescence over VC.
siRNA Downregulation Studies
Gene-specific siRNA and negative control scramble (catalog no., assay IDs of the siRNA used and the manufacturer are listed in Supplemental Table 3) were transiently transfected into hCMEC/D3 cells using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, transfection reagent and siRNA were diluted in Opti-MEM (Gibco, Thermo Fisher Scientific) medium, and then diluted transfection reagent was gently added to the diluted siRNA. After incubation for 20 minutes at room temperature, the transfection complex was added to the cells. After 6 hours of transfection, the medium was replaced with fresh Opti-MEM medium and kept further for 18 hours. Following this, cells were washed with 1X PBS and treated with VPA or VC in EBM-2 complete media and further cultured for indicated time periods to check changes in the gene expression and activity.
Immunofluorescence
hCMEC/D3 cell monolayer was grown onto collagen-coated coverslips in six-well plates in the EBM-2 complete medium. After treatment with VPA or VC, cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature. Following this, cells were permeabilized with 0.1% Triton X-100 for 20 minutes. Nonspecific sites were blocked with 1% BSA in PBS for 30 minutes. Cells were then incubated overnight at 4°C with PPARα primary antibody diluted (1:250) in 1% BSA. After washing with 1X PBS, cells were incubated with the Alexa488-conjugated anti-rabbit secondary antibody (1:1000) for 2 hours at room temperature. Nuclei were counterstained with 0.5 µg/ml 4’,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 15 minutes at 37°C and mounted on glass slides using ProLong Diamond Antifade Mountant (Invitrogen). Images were acquired using Nikon confocal A1R HD with Ti2-E with 60X Nikon objective (1.4 NA). Fluorescence images were obtained by sequential z stage scanning in two channels (DAPI, Alexa Fluor-488).
To quantify the PPARα fluorescence signals in the nucleus and cytoplasm, the Image J software was used. Whole cell masks and nuclear masks were generated by applying Huang’s thresholding method on PPARα-stained channel and on the DAPI-stained channel. Total area and PPARα fluorescence intensity corresponding to each of the cell was measured. Similarly, the nuclear area and fluorescence intensity corresponding to the nuclear region were measured. The cytoplasmic area and fluorescence signal were obtained by subtracting the nuclear area and fluorescence from total cellular area and fluorescence, respectively. Nuclear and cytoplasmic fluorescence intensities were then normalized to corresponding areas and the obtained values were divided to determine the nuclear/cytoplasmic ratio of PPARα fluorescence intensity. The average fluorescence intensity for each treatment group per experiment was the mean of all measurements taken from at least 80 cells from 12 random fields.
Chromatin Immunoprecipitation Assay and Quantitative Real–Time PCR
Treated hCMEC/D3 cells were cross-linked by adding 1% formaldehyde for 10 minutes at room temperature, followed by quenching using 125 mM glycine solution. Fixed cells were scraped in 1X PBS and centrifuged to obtain the cell pellet. The pellet was resuspended in immunoprecipitation buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, NP-40 (0.5% vol/vol), Triton X-100 (1.0% vol/vol)]. The samples were kept on ice for 20 minutes and then centrifuged at 12000 rpm for 1 minute at 4°C to obtain the nuclear pellet. This pellet was resuspended in 1% SDS lysis buffer, and sonication was performed to obtain the sheared chromatin. One-tenth of the sonicated lysates were taken out as the input control. Immunoprecipitation was performed overnight at 4°C with 5 µg anti-PPARα antibody chromatin immunoprecipitation grade (ab227074, rabbit polyclonal; Abcam, Cambridge, MA, USA) (Murphy et al., 2021). Rabbit IgG was used as a nonspecific control. The resultant chromatin precipitates were captured by Protein A dynabeads (Invitrogen). After washing the whole complex in different buffers as described previously (Gade and Kalvakolanu, 2012), the protein–DNA complex from the antibody was eluted by adding freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3). Cross-linking was reversed by adding proteinase K to the samples and heating the eluate and input DNA at 65°C for 2 hours. Purified DNA was amplified using qPCR with primers flanking the PPARα binding sites in the human ABCG2 promoter regions. The binding sites have previously been verified in the report by Hoque et al. (2012). Primer sequence used were forward primer 5′-TGGAAGGCTGTGAGTCACTT-3′ and reverse primer 5′-AGGACCTTCCTCATTAGGTCAGA-3′. Recruitment was calculated as % of total input.
Plasmid Construction
A 345 bp fragment of the human ABCG2 promoter (−4037bp/−3692bp) containing the PPARα response elements (PPRE) was PCR amplified using the forward primer 5′-CAACGGTACCCTGGTGCACAGGCATTCA-3′ (underlined nucleotides indicate the KpnI site) and the reverse primer 5′-ATAACTCGAGGTTCAGATTAAAGCCAGC-3′ (underlined nucleotides indicate the XhoI site). The amplified fragment was cloned into pCR2.1 TA vector (3929 bp) and then subcloned upstream to the luciferase reporter gene driven by the SV40 promoter into pGL2-promoter vector (5789 bp) linearized with XhoI and KpnI restriction enzymes. The desired final construct (PGL2-prom-ABCG2) was confirmed through sequencing.
Transient Transfection and Dual–Luciferase Assay
For luciferase assay, HEK293 cells (105 cells) were cultured in six-well plates and grown for 24 hours. Cells were then co-transfected with pRenilla plasmid (20 ng/well) and pGL2 reporter plasmid vectors (3 µg/well) that expressed Renilla and Firefly luciferase activities, respectively. The pGL2 reporter plasmids were pGL2–prom–ABCG2 and pGL2–control. Transfections were performed using polyethylenimine. VPA or VC was added for 24 hours after transfection and cells were immediately harvested by scrapping. Cells were lysed and the Firefly and Renilla luciferase activity was measured using the Dual-Luciferase Reporter Assay System. Firefly luciferase reporter activity of each data set was normalized to Renilla luciferase activity.
LanthaScreen TR-FRET PPARα Competitive Binding Assay
Interaction of VPA with PPARα nuclear receptor was studied using a cell-free Lanthascreen TR-TRET ligand-binding assay (Invitrogen). The assay is based on the principle of competitive displacement of the reference fluorescent ligand (Tracer) from its recombinant terbium-labeled PPARα LBD. VPA and a selective PPARα agonist, GW7647, were diluted in DMSO and assay was performed according to the manufacturer’s protocol. Results are displayed as percent displacement of the tracer.
Statistical Analysis
Data were expressed as mean ± S.D. Statistical significance was assessed by unpaired t test or one-way ANOVA followed by Dunnett’s or Tukey’s post hoc test or two-way ANOVA followed by Tukey’s post hoc test as indicated against each experimental result. The 95% CI accompanying the percent changes was calculated using standard error of the mean between the groups. A P value < 0.05 was considered to be statistically significant. The results of the tests have been summarized in Supplemental Table 4.
RESULTS
Effect of First- and Second-Line AEDs on mRNA Expression of MDTs in hCMEC/D3 Cells
After confirming the absence of any cytotoxicity of AEDs on hCMEC/D3 cells at doses that represent the therapeutic plasma concentration for individual AEDs (Supplemental Fig. 1), we studied the effect of therapeutic doses of AEDs on mRNA expression of ABCC1, ABCC2, ABCC4, ABCC5, and ABCG2. Cells were treated with first-line AEDs (PHT: 40 µM, 80 µM; CBZ: 21 µM, 42 µM; VPA: 300 µM, 600 µM) and second-line AEDs (LTG: 15 µM, 60 µM; TPM:15 µM, 60 µM; LEVI: 40 µM, 120 µM) for 24 hours. Reverse transcriptase (RT)-qPCR analysis showed a dose-dependent increase in ABCG2 mRNA expression in response to VPA treatment (1.71-fold increase at 300 µM, 2.43-fold increase at 600 µM) (Fig. 1). However, no effect of PHT, CBZ, LTG, TPM, and LEVI was found on expression of transporters under study (Supplemental Fig. 2). We then checked for any time-dependent variations in this induction. It was found that VPA at both the doses induced ABCG2 mRNA at all the time points under study (6, 12, 24, and 48 hours), although the maximum fold change observed was at 24 hours (Fig. 2).

Effect of VPA on mRNA expression of MDTs in hCMEC/D3 cells. Cells were treated with VPA (300 μM, 600 μM) for 24 hours. Total RNA was extracted and subjected to RT-qPCR to determine changes in mRNA levels of ABCC1, ABCC2, ABCC4, ABCC5, and ABCG2. The mRNA levels of these genes were normalized to those of B2M and expressed as fold change over VC (0.1% DMSO). The data shown are the means ± S.D. of 3 independent experiments. *P < 0.05, **P < 0.01 (one-way ANOVA with Tukey's post hoc test performed on log-converted values).

Effect of VPA on mRNA expression of ABCG2 in hCMEC/D3 cells at different time points under study. Cells were treated with VPA (300 μM, 600 μM) for 6, 12, 24, and 48 hours. Total RNA was extracted and subjected to RT-qPCR to determine changes in mRNA levels of ABCG2. The mRNA level of ABCG2 was normalized to those with B2M and expressed as fold change over VC (0.1% DMSO). The data are the mean ± S.D. of three independent experiments. *P < 0.05, **P < 0.01 (two-way ANOVA with Tukey’s post hoc test performed on log-converted values).
VPA Increases the Protein Expression and Functional Activity of BCRP in hCMEC/D3 Cells
Similar to the effect on mRNA expression, exposure of hCMEC/D3 cells with VPA (300 μM, 600 μM) displayed upregulation of BCRP protein levels starting from 12 to 72 hours (Fig. 3A). The increase was found to be maximum at 48 hours (1.74-fold at 300 µM, 2.09-fold at 600 µM). A representative blot for 48-hour protein data is shown in Fig. 3B. To test whether an increase in protein expression is also reflected in its functional activity, we performed a BPZ efflux assay in the presence of BCRP inhibitor, Ko143. BCRP activity was determined as the ratio between the mean intracellular fluorescence of cells incubated with Ko143 (BCRP inhibited) and cells incubated without Ko143 (noninhibited) in the presence of VPA, normalized to the corresponding ratio in the absence of VPA. We found that cells treated with VPA (300 μM, 600 μM) for over 48 and 72 hours resulted in a decrease in BPZ intracellular fluorescence indicating elevated BCRP-specific BPZ efflux (Fig. 3C).

Effect of VPA on BCRP expression and functional activity in hCMEC/D3 cells. (A) Whole cell lysates from hCMEC/D3 cells treated with VC (0.1% DMSO) or VPA (300 μM, 600 μM) for 6, 12, 24, 48, and 72 hours were subjected to SDS-PAGE and western blot to measure BCRP expression. Bands of HSC were used to normalize the results. Mean densitometric values of the bands obtained from three independent experiments was used to calculate fold change over the VC. (B) Representative blot for 48-hour VPA treatment is shown. (C) hCMEC/D3 cells were treated with VC (0.1% DMSO) or VPA (300 μM, 600 μM) for 12, 24, 48, and 72 hours prior to the assay. Intracellular BPZ fluorescence was measured in the presence and absence of the BCRP inhibitor Ko143. Fluorescence intensities were normalized with total protein content. BCRP activity was determined as the ratio between the mean intracellular fluorescence of cells incubated with Ko143 and cells incubated without Ko143 in the VPA treated group, normalized to the corresponding ratio in the control (VC) group. Data are the means ± S.D. of four independent experiments. *P < 0.05, **P < 0.01 (two-way ANOVA with Tukey’s post hoc test performed on log-converted values).
PHT, VPA and LTG Stimulates the ATPase Activity of BCRP
To determine whether VPA or other AEDs (PHT, CBZ, LTG, TPM, LEVI), which are also co-administered with VPA are substrates of BCRP, ATPase assay with the BCRP-overexpressing membrane vesicles was performed for different AEDs over a concentration range, and the amount of Pi released from ATP hydrolysis by the test AED was measured by a colorimetric reaction. As seen from the Fig. 4 A, C, and D, PHT, VPA, and LTG stimulated the vanadate-sensitive baseline ATPase activity in the activation assay, indicative of their substrate interaction with BCRP. For CBZ, TPM, and LEVI, no difference from baseline activity was observed (Fig. 4 B, E, and F). The inhibition assay was done in the presence of a test drug and known activator of BCRP-sulfasalazine, which maximally stimulates the ATPase activity. At higher doses of LTG (120 µM) and CBZ (80 µM), a reduction in maximally stimulated ATPase activity was observed.

Effect of increasing concentrations of AEDs (A) PHT, (B) CBZ, (C) VPA, (D) LTG, (E) TPM, and (F) LEVI on vanadate-sensitive BCRP ATPase activity. The ATPase activity is expressed as nmol phosphate generated/mg protein/min. Lower dotted line represents baseline activity (in the presence of 2% v/v DMSO or water), and upper dotted line represents activity of fully activated membrane (in the presence of known BCRP activator sulfasalazine). Activation study measures stimulation of baseline ATPase activity indicating drug as BCRP substrate. Data are presented as means ± S.D from three independent experiments. Statistically significant differences between the baseline and drug-stimulated activity in the activation assay (*P < 0.05, **P < 0.01) were determined using one-way ANOVA with Dunnett’s post hoc test performed on log-converted values. Inhibition study measures decrease in maximally stimulated BCRP activity indicating interaction of drug with BCRP. Data are presented as means ± S.D from three independent experiments. Statistically significant differences between the maximal membrane activity and drug-treated activity in the inhibition assay (*P < 0.05) were determined using one-way ANOVA with Dunnett’s post hoc test performed on log-converted values.
PHT, VPA and LTG Increases the Intracellular Accumulation of BPZ
The substrate interaction of PHT, VPA, and LTG with BCRP detected in the activation assay was further verified in competitive substrate efflux assay in which BPZ was included as a competing substrate. Fig. 5, A–C shows that co-incubation of PHT (80 μM), VPA (300 μM, 600 μM), and LTG (15 μM, 60 μM) with the assay substrate BPZ increased cellular accumulation of BPZ, providing a more definitive evidence of these AEDs as potential substrates of BCRP.

Competitive substrate efflux assay to study substrate relationship of PHT, VPA, and LTG with BCRP. hCMEC/D3 cells were co-incubated with either the assay substrate BPZ (250 nM) and VC (0.1% DMSO) or test AED [(A) PHT, (B) VPA, and (C) LTG] or an inhibitor of BCRP (1 µM Ko143), and intracellular accumulation of BPZ was measured. Results are reported as the fold increase in intracellular fluorescence in treated cells over control fluorescence. The data are the mean ± S.D. of five independent experiments. Statistical significance was calculated using one-way ANOVA with Dunnett’s post hoc test performed on log-converted values (**P < 0.01; BPZ+VC vs. BPZ+PHT/VPA/LTG or BPZ+VC vs. BPZ+Ko143).
Since the VPA-mediated changes in BCRP expression and activity were observed to be maximum at the higher dose (i.e., 600µM), all the experiments related to the understanding of mechanistic of this regulation were performed at this dose.
PPARα Silencing/Antagonism Attenuates VPA-Induced BCRP Expression and Activity
To identify the molecular factors involved in VPA-induced ABCG2 expression, a list of 17 important factors reported to regulate ABCG2 was obtained from the literature (Kukal et al., 2021) (Factors and their primer sequences for qPCR are shown in Supplemental Table 2.) Constitutive mRNA expression level for each factor was examined in hCMEC/D3 cells by qPCR (data not shown). Xenobiotic receptors, pregnane X receptor, and constitutive androstane receptor were undetectable in these cells. Thus, siRNA experiments were performed for the remaining factors at mRNA level. Validation data of PPARα siRNA is shown in Fig. 6A, whereas the data for other siRNAs are shown in Supplemental Fig. 3. Of these factors, a 35% (95% CI: 20.66%, 48.33%) decrease in VPA-induced ABCG2 mRNA was observed only when PPARα was silenced, suggesting the regulation to be PPARα dependent (Fig. 6B). Further, PPARα knockdown suppressed VPA-induced BCRP protein and activity by 38.9% (95% CI: 28.68%, 47.92%) and 30.5% (95% CI: 23.76%, 36.82%), respectively (Fig. 6, C–E). For the remaining factors, siRNA knockdown did not affect VPA-induced ABCG2 mRNA (Supplemental Fig. 4).

Effect of PPARα knockdown on VPA-induced ABCG2 expression and activity. Expression of PPARα was silenced by transient transfection of siPPARα (siRNA specific to human PPARα) in hCMEC/D3 cells. Then, cells were treated with VC (0.1% DMSO) or VPA (600 μM) for 24, 48, and 72 hours to check ABCG2 mRNA, protein and activity, respectively. SCR was used as nontargeting control. (A) RT-qPCR analysis of PPARα expression. mRNA level of PPARα was normalized to those with B2M. The data are the mean ± S.D. of three independent experiments. **P < 0.01, VC (SCR vs. siRNA); #P < 0.01, VPA (SCR vs. siRNA) (unpaired t test). (B) RT-qPCR analysis of ABCG2 expression. mRNA level of ABCG2 was normalized to those with B2M (C) Representative blot of PPARα and BCRP is shown; bands of HSC were used to normalize the protein expression. (D) Western blot analysis of BCRP expression and (E) analysis of BCRP functional activity. The data are the mean ± S.D. of the independent experiments. **P < 0.01, N.S., not significant (two-way ANOVA with Tukey’s post hoc test).
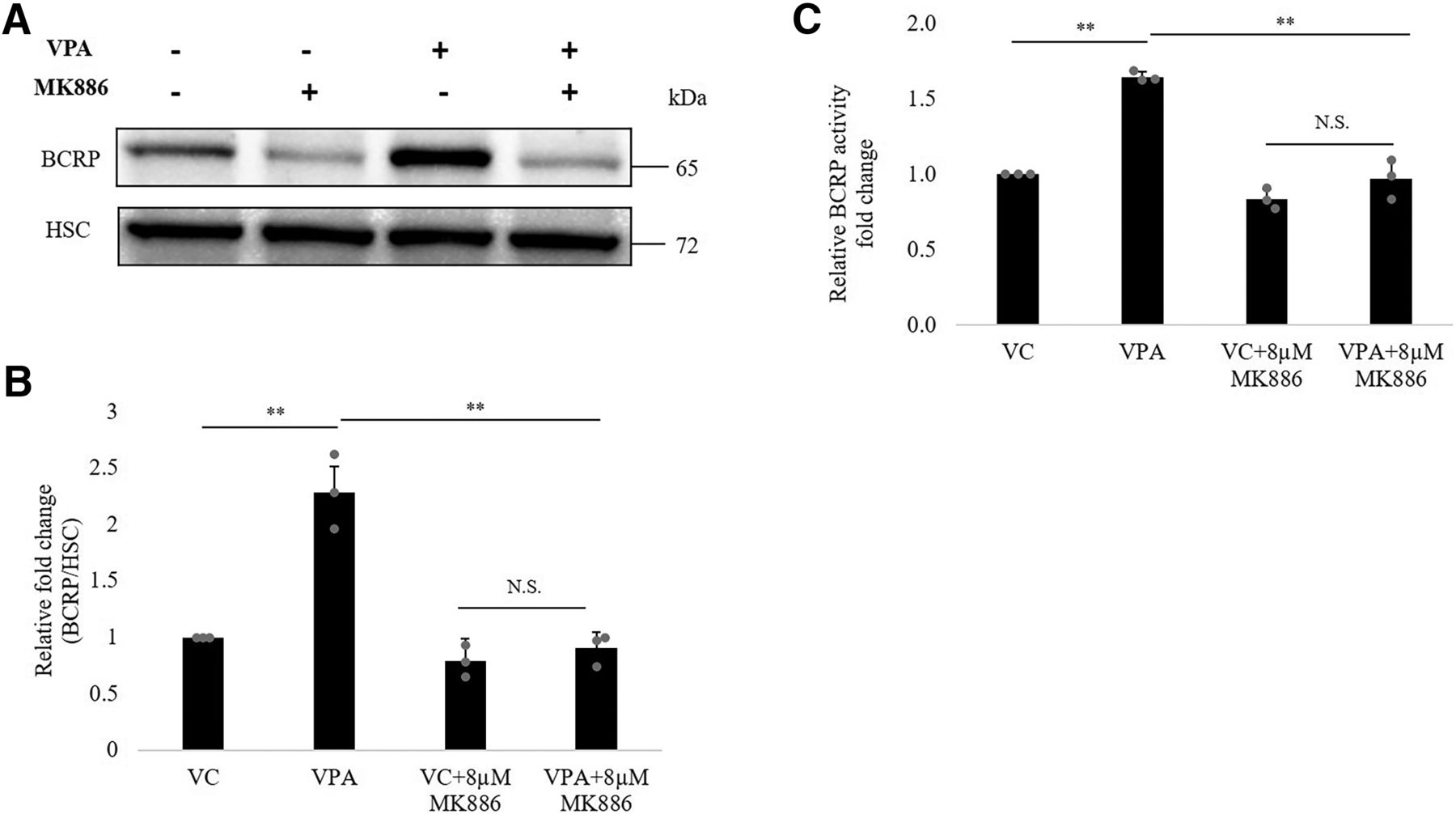
To further examine the regulatory role of PPARα in VPA-mediated ABCG2 upregulation, we incubated hCMEC/D3 cells with VPA in the presence or absence of PPARα antagonist, MK886 for 48 hours (MTT data for MK886 is shown in Supplemental Fig. 5). As shown in Fig. 7, the addition of MK886 (8 μM) abrogated the upregulatory effect of VPA on BCRP protein (Fig. 7, A and B) and activity (Fig. 7C) by 50% (95% CI: 30.38%, 65.49%) and 30% (95% CI: 20.1%, 38.9%), respectively.

Effect of PPARα antagonist MK886 on inductive effect of VPA on BCRP expression and activity. hCMEC/D3 cells were cultured with VC (0.1% DMSO) alone or VPA (600 μM) alone or VC/VPA in the presence of PPARα antagonist MK886 for 48 hours to check effect on ABCG2 protein and activity. (A) Representative blot of BCRP is shown; bands of HSC were used to normalize the protein expression. (B) Western blot analysis of BCRP expression and (C) analysis of BCRP functional activity. The data are the mean ± S.D. of three independent experiments. **P < 0.01, N.S., not significant (two-way ANOVA with Tukey’s post hoc test performed on log-converted values).
Treatment of hCMEC/D3 cells with known PPARα agonist, GW7647 (100 nM) and clofibrate (100 µM) also showed ABCG2 mRNA induction (Supplemental Fig. 6), validating the regulatory effect of PPARα on ABCG2.
VPA Increases PPARα Expression and Its Nuclear Translocation
To get insights into the mechanism of the VPA-mediated ABCG2 overexpression via PPARα, we first examined the effect of VPA on PPARα expression. Upon treatment of hCMEC/D3 cells with VPA, PPARα mRNA started to be induced at 3 hours (1.43-fold) and reached a maximum at 6 hours (2.04-fold). The increase was continued until 24 hours (1.76-fold) (Fig. 8A). However, at protein level, the increase was seen until 6 hours, after which no increase was observed (Fig. 8B).

Effect of VPA on expression of PPARα and its target genes. hCMEC/D3 cells were treated with VC (0.1% DMSO) or VPA (600 µM) for the indicated time points. (A) RT-qPCR was done to check the effect on PPARα mRNA expression. B2M was used as the reference gene for normalization. (B) Whole-cell lysates were subjected to western blot to measure change in PPARα protein expression. (C) Representative PPARα blot for 3 hours is shown. HSC protein bands served as a loading control. The data are the mean ± S.D. of three independent experiments. *P < 0.05, **P < 0.01; VC vs. VPA (two-way ANOVA with Tukey’s post hoc test performed on log-converted values). (D) RT-qPCR was done to check the effect of VPA on mRNA expression of PPARα target genes PDK4 and THBD at 24 hours. B2M was used as the reference gene for normalization. The data are the mean ± S.D. of three independent experiments. **P < 0.01; VC vs. VPA (unpaired t test performed on log-converted values).
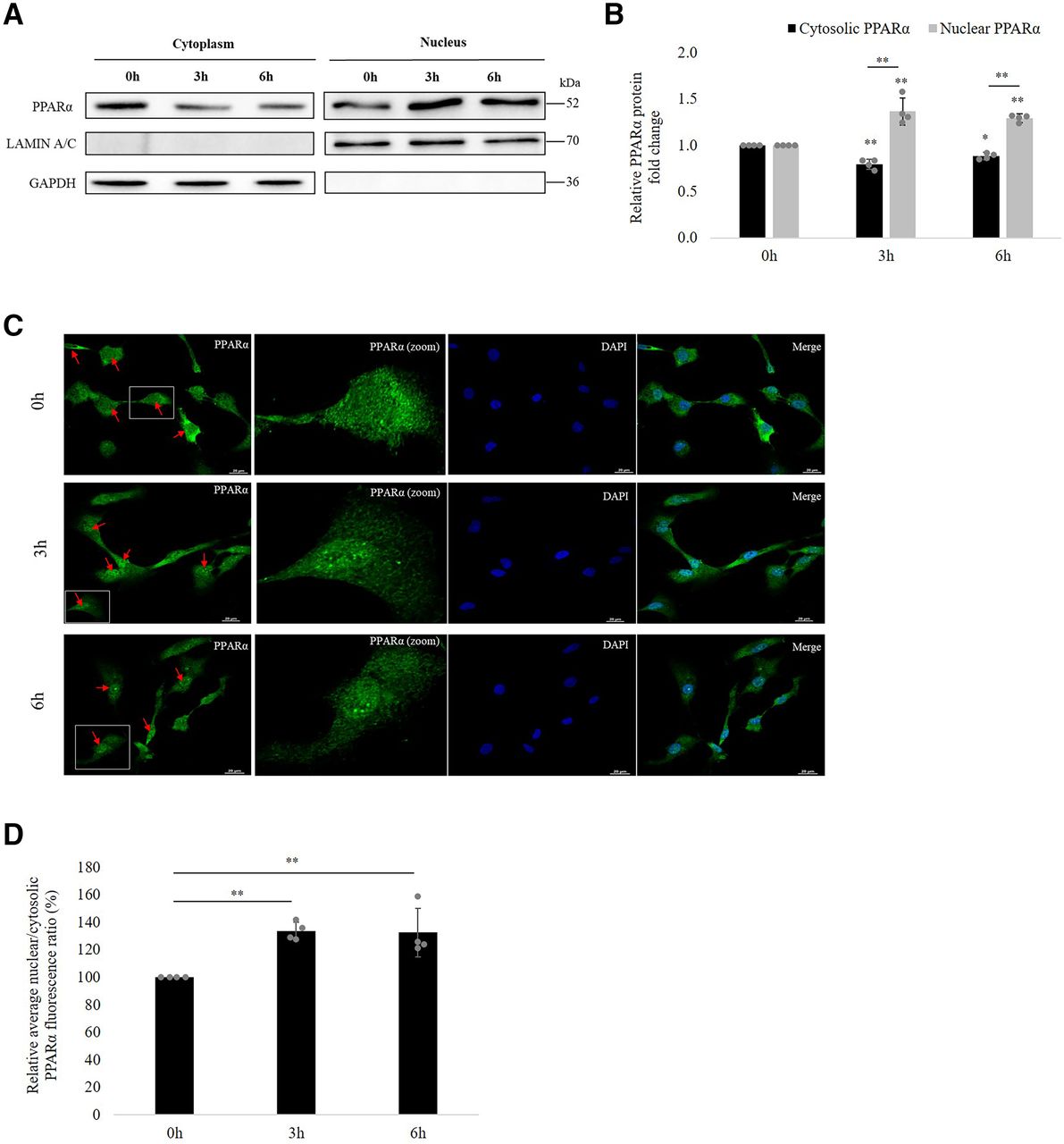
Next, we investigated the possibility that VPA affects PPARα translocation in hCMEC/D3 cells. For this, we fractionated the cytoplasmic and nuclear content and checked for the protein expression of PPARα in each fraction at 0, 3, and 6 hours of VPA treatment. GAPDH and lamin A/C were used as loading marker genes for cytoplasmic and nuclear extracts, respectively. No cross-contamination of nuclear and cytoplasmic fractions was seen. As shown in Fig. 9A, at 0 hours, PPARα was found to be expressed in both cytoplasm and nucleus, although nucleus had comparatively lesser levels. However, at 3 and 6 hours of VPA treatment, an increase in nuclear PPARα levels could be noted with a simultaneous decrease in cytoplasm, revealing that VPA induces PPARα nuclear translocation to initiate downstream signaling. Quantitative analysis of the relative nuclear and cytosolic PPARα protein fold change is shown in Fig. 9B.

Effect of VPA treatment on PPARα translocation to nucleus. (A) hCMEC/D3 cells were incubated with VPA (600 µM) for 0, 3, and 6 hours. Isolated nuclear and cytoplasmic fractions of the cell lysate were subjected to western blot with PPARα antibody. Lamin A/C and GAPDH was used as nuclear and cytosolic marker respectively. (B) The bar graph illustrates the relative nuclear and cytosolic PPARα protein expression fold change at 3 and 6 hours w.r.t expression at 0 hours. The data are the mean ± S.D. of four independent experiments. *P < 0.05, **P < 0.01 (one-way ANOVA with Tukey’s post hoc test performed on log-converted values). (C) hCMEC/D3 cells were treated with VPA (600 µM) for 0, 3, and 6 hours and subsequently fixed with 4% paraformaldehyde. Fixed cells were incubated with PPARα primary antibody and Alexa Fluor 488–conjugated secondary antibody. Nuclear staining was performed with DAPI. The intracellular distribution of the fluorescent tag was examined under a confocal microscope. Color in green (Alexa488-conjugated antibody) and blue (DAPI) represent PPARα and nucleus, respectively. Arrows indicate cells depicting the translocation. Average PPARα fluorescence ratios (nuclear to cytoplasm) at 3 and 6 hours compared with that at 0 hours were obtained from four independent experiments (80 cells per treatment group per experiment). (D) Bar graph shows relative quantification of nuclear: cytoplasmic ratios of PPARα fluorescence. **P < 0.01 (one-way ANOVA with Tukey’s post hoc test performed on log-converted values).
Immunofluorescence staining of PPARα in the fixed hCMEC/D3 cells was also carried out to evaluate the PPARα translocation. In accordance with the findings of western blot, confocal microscopic imaging and quantitative analysis of PPARα fluorescence revealed the presence of PPARα in both cytoplasm and nucleus at 0 hours. After 3 and 6 hours of VPA exposure, a 33.8% (95% CI: 27.35%, 40.22%) and 32.6% (95% CI: 15.4%, 49.98%) increase in Nuc:Cyt ratio of fluorescence was observed (Fig. 9, C–D), respectively, suggesting activation of PPARα by VPA.
In hCMEC/D3 cells, VPA also increased the mRNA level of PPARα target genes pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4) (Janssen et al., 2015) and thrombomodulin (THBD) (Shiono et al., 2020) (Fig. 8D). This further corroborated the activating effect of VPA on PPARα.
VPA Enhance the Recruitment of PPARα to ABCG2 Promotor and Results in Increased Promoter Activity
The ability of VPA to enhance the binding of PPARα in the promoter of its target gene ABCG2 was studied by immunoprecipitation assay. Chromatin preparation from cells treated with VC or VPA was immunoprecipitated with anti-PPARα antibody or negative IgG antibody. The ABCG2 promoter region in the immunoprecipitated complex DNA was amplified using qPCR primers flanking the PPARα binding region (−3946bp/−3796bp) of ABCG2 promoter. As shown in Fig. 10A, VPA treatment at 3 and 6 hours caused enrichment of PPARα to the ABCG2 promoter region. A representative gel image for recruitment at 3 hours of treatment is shown in Fig. 10B.

Effect of VPA on PPARα recruitment to the ABCG2 promoter and promoter activation (A) hCMEC/D3 cells were treated with VC (0.1% DMSO) or VPA (600 µM) for 3 and 6 hours, and PPARα recruitment to the region of interest was assessed by ChIP-qPCR. A nonspecific rabbit IgG antibody was included as negative control. Recruitment is presented as percentage of total input. The data are the mean ± S.D. of three or four independent experiments. **P < 0.01 (VPA vs. VC) (unpaired t test performed on log-converted values). (B) 1% agarose gel image showing qPCR product from 3-hour treated samples. (C) Luciferase reporter gene construct containing the PPRE sequences of the ABCG2 promoter (pGL2-prom-ABCG2) or the pGL2 control vector was cotransfected with renilla-luciferase expression vector into HEK293 cells. Cells were treated with VC (0.1% DMSO) or VPA (600 µM) for 24 hours before measurement of luciferase activities. Firefly luciferase activities were normalized to renilla luciferase activities to consider changes in the transfection variability. Firefly/renilla activity of the VPA treated cells were then normalized to VC treatment (set as1) to obtain the relative luciferase activity fold change. Data are the mean of three independent experiments. **P < 0.01 (VPA-treated cells compared with VC treatment set to1; unpaired t test performed on log-converted values).
To examine the effect of VPA on ABCG2 promoter activity driven by PPRE, a 345 bp promoter region containing the PPARα binding site was cloned upstream of the minimal SV40 promoter driving the luciferase reporter gene (PGL2-prom-ABCG2). HEK293 cells were then co-transfected with either pGL2-prom-ABCG2 and pRenilla or control vector and pRenilla followed by incubation with VPA or VC. Firefly luciferase activities were normalized to renilla luciferase activities to consider changes in the transfection variability. Firefly/renilla activity of the VPA-treated cells were then normalized to VC (set as 1) to obtain the relative luciferase activity. VPA was found to increase ABCG2 luciferase activity by around 1.42-fold compared with VC, demonstrating ABCG2 promoter activation by VPA, whereas the control vector was not affected by VPA treatment. Fig. 10C depicts the relative luciferase activity of PGL2-control and PGL-prom-ABCG2.
VPA Displayed Weak Ligand Interaction With PPARα
To determine whether VPA shows ligand interaction with the PPARα receptor, a LanthaScreen TR-FRET PPARα competitive binding assay was conducted. As depicted in the curve between percent displacement and the concentration (Supplemental Fig. 7), while GW7647 showed a strong binding to the receptor with an IC50 value of 0.0224 μM, VPA started competing with the tracer at a concentration of 556 µM (within the therapeutic range of VPA). Its IC50 value was achieved at a much higher concentration of 9690 µM, indicating weak binding of VPA with PPARα.
Discussion
Despite the availability of 35 AEDs (Jacob et al., 2019), poor response is experienced by a considerable section of PWE. The extensively studied theory behind this is the overexpression of efflux transporters at the BBB (Löscher and Potschka, 2002; Le et al., 2021). Earlier studies depict that AEDs have the potential to regulate these MDTs (Alms et al., 2014; Ke et al., 2019). However, AED-mediated regulation of MDTs implicated in drug-resistant epilepsy (other than ABCB1), at the human BBB, is largely unknown. Our results demonstrated an inductive effect of VPA on ABCG2 mRNA, protein, and functional activity in hCMEC/D3 cells. Our data are in line with a previous report where this induction was demonstrated in placental cells to check the effect of AEDs on placental carriers altering the fetal exposure to certain nutrients (Rubinchik-Stern et al., 2015). A recent report studying the effect of histone deacetylase inhibitors on MDTs in hCMEC/D3 cells showed an increase in ABCG2 mRNA at a 5 mM dose, exceeding the clinical therapeutic plasma concentration range of VPA (You et al., 2019).
Increased expression of BCRP, a well-recognized drug efflux transporter, has been implicated in drug-resistant PWE. Strong BCRP expression was reported in endothelial cells from brain tumors associated with epileptogenic pathology in refractory PWE (Aronica et al., 2005). A retrospective investigation done in epileptogenic tuberous sclerosis specimens (refractory to AEDs) revealed the presence of BCRP in brain endothelial cells (BECs) in addition to the high MDR1 and MRP1 expression (Lazarowski et al., 2006). An upregulation of ABCG2 mRNA was found in hippocampal tissues from patients with mesial temporal lobe epilepsy, demonstrating the potential of ABCG2 as prognostic marker for epilepsy pharmacoresistance (Banerjee Dixit et al., 2017). Recognizing the role of BCRP in MDR phenotype, it is important to know how its expression is regulated by AEDs. Our study reports that VPA elevates BCRP functional expression in hCMEC/D3 cells.
We next assessed the BCRP substrate status of AEDs and found PHT, VPA, and LTG as the substrates. CBZ, TPM, and LEVI did not show substrate interaction. The observations to some extent corroborated previous findings. BPZ accumulation assay performed in mouse fibroblasts transduced with human ABCG2, in the presence of VPA, demonstrated increase in accumulation of BPZ, indicating its substrate interaction (Cerveny et al., 2006). A concentration equilibrium transport assay with MDCK2 cells overexpressing BCRP identified LTG as the BCRP substrate out of seven AEDs including VPA, CBZ, TPM, and LEVI (Romermann et al., 2015). Recently, a bidirectional transport assay concluded that BCRP does not contribute in efflux of LEVI (Goncalves et al., 2021). On the contrary, a comparison of brain distribution of AEDs between Mdr1a/1b(−/−) and Mdr1a/1b(−/−)/Bcrp(−/−) mice showed involvement of BCRP in restricting brain access to LEVI (Nakanishi et al., 2013).
The finding that VPA induces BCRP and is also its substrate led us to investigate the underlying cellular mechanism driving this process at the BBB. Using siRNA knockdown and antagonist studies, the role of PPARα signaling was revealed in this induction. While it is previously demonstrated that PPARα regulates BCRP expression in mouse intestine and liver (Hirai et al., 2007; Eldasher et al., 2013), hCMEC/D3 cells (Hoque et al., 2012), and rat brain capillaries (More et al., 2017), the involvement of this receptor in VPA-mediated BCRP expression at the BBB is a novel finding.
PPARα is a ligand-activated nuclear receptor that has a major involvement in the regulation of lipid metabolism. The physiologic ligands of PPARα includes fatty acids and its derivatives (Rakhshandehroo et al., 2010) and is demonstrated to translocate into nucleus upon its activation (Li et al., 2016; Xue et al., 2018). The receptor is shown to be activated by xenobiotics and regulates various genes involved in metabolism and efflux (Omiecinski et al., 2011; Chen et al., 2012). PPARα activation with clofibrate was reported to upregulate P-gp, Mrp3, Mrp4, and Bcrp in mice liver (Moffit et al., 2006). Mrp3/4 induction along with the decrease in Mrp2 was observed in mice liver in response to anti-rheumatic drug leflunomide, which involved increase in PPARα expression. The outcome of this regulation was enhanced hepatic exposure to the co-administered drug methotrexate (substrate of Mrp2/3) and liver toxicity (Wang et al., 2018). PPARα-mediated upregulation of Bcrp was also demonstrated in mouse and rat brain capillaries, indicating PPARα as a therapeutic target to improve brain drug delivery (Hoque et al., 2015). Thus, these studies substantiate the role of PPARα in influencing the toxicity/bioavailability of drugs that are substrates of such efflux transporters.
We next checked whether VPA has an effect on PPARα in hCMEC/D3 cells and found an increase in expression and nuclear translocation of PPARα. This inductive effect has been previously reported in human hepatic cells (Rodrigues et al., 2016; Xu et al., 2019). Moreover, VPA is suggested to be a pan-PPAR activator (Cullingford et al., 2002) and is demonstrated to activate PPARα LBD in Chinese hamster ovary cells (Lampen et al., 2001).
We speculated that VPA being a fatty acid may behave as ligand for PPARα receptor to activate it. LanthaScreen TR-FRET assay results showed a weak ligand interaction of VPA (at its therapeutic dose) with PPARα. There might be certain essential proteins involved in enhancing ligand-receptor interaction within the biologic system, an effect that cannot be seen in cell-free TR-FRET assay; therefore, performing cell-based assays are warranted to confirm the ligand interaction. In addition to ligand-mediated regulation of PPARα activity, post-translational modifications of the receptor are also reported to modify its activity, suggesting the possibility that VPA may activate PPARα via this additional mechanism.
We further showed that exposure of hCMEC/D3 cells to VPA augmented PPARα binding at PPRE in the ABCG2 promoter. A luciferase assay in HEK293 cell lines depicted that VPA-mediates ABCG2 promoter activation via these response elements.
Taken together, evidence in the present study demonstrates that VPA upregulates expression and activity of BCRP in human BECs. This involves an increase in PPARα expression, nuclear translocation, and its enhanced binding to the ABCG2 promoter, thus activating the gene’s transcriptional activity.
It is worth noting that although VPA activated PPARα to upregulate BCRP, it did not affect expression of MRP2, despite the reported increase of both these transporters by PPARα agonists in rat brain capillaries (More et al., 2017). A possible explanation could be the varied species-specific response of human and rat BECs cells toward PPARα activation (Lawrence et al., 2001). These responses can be attributed to differences in transacting factors in human and rodent cells, affecting PPARα target gene expression or the difference in the human ABCC2 promoter sequence, rendering the gene unresponsive to PPARα activation. Further studies are required to determine the mechanism behind these differences.
To date, conventional AEDs remain the major prescriptions due to their cost-effectiveness and superior efficacy. Among these, VPA is the drug of choice for the first-line treatment because of its effectiveness against a broad spectrum of seizure types (Perucca, 2002; Rawat et al., 2020). However, similar to other AEDs, VPA fails to work in one-third of the population (Chen et al., 2019). According to a recent report, resistance to VPA is suggested to be a clinical marker for declaring patients with genetic generalized epilepsy as drug-resistant (Gesche et al., 2017). Considering the significance of VPA in epilepsy treatment, it becomes important to investigate molecular mechanism underlying VPA resistance. Our study highlights PPARα/BCRP axis as one of the possible mechanisms that could lead to higher VPA efflux from BECs and decrease its intracellular concentration. Future validation of these findings in PPARα CRISPR knockout cell line will be of much importance.
Our study has some limitations. First, the AEDs as a substrate of BCRP were evaluated through indirect assays. Future experiments should directly quantify the transport of AEDs via BCRP. One such approach will be to perform high-performance liquid chromatography for intracellular drug quantification. Second, this work was performed in the hCMEC/D3 cell line, which has relatively low junctional tightness and thus an inadequate barrier function compared with primary brain endothelial cells. This limits its use in vectorial drug transport studies for small molecules (Helms et al., 2016). However, despite the limitation of hCMEC/D3 cells to fully recapitulate the human BBB, these cells are reported to retain the expression of most transporters (including ABCG2) and receptors expressed at human BBB and are so far the most widely used human brain endothelial cell line to perform gene regulation studies (Weksler et al., 2013). Third, the approach used in this study was in vitro. To determine the in vivo relevance of the study, data validation utilizing animal models of epilepsy is warranted. PPARα knockout in such models and evaluating the brain levels of VPA and response to the drug will actually provide the direct evidence of PPARα as a therapeutic target to overcome VPA pharmacoresistance.
In conclusion, these findings demonstrate that VPA upregulates BCRP in human BECs via PPARα activation. Moreover, PHT, VPA, and LTG displayed substrate interactions with BCRP. Hence, BCRP induction by VPA may likely lead to increased efflux of VPA itself or efflux of PHT/LTG if administered concomitantly. Therefore, targeting of PPARα may represent a potential therapeutic strategy to address resistance to VPA and possibly other AEDs used in conjunction with VPA.
Acknowledgments
We thank, Dr. Anurag Agrawal, the director of the CSIR-Institute of Genomics and Integrative Biology (IGIB), for his scientific vision, motivation, and support. We would like to acknowledge Dr. Ankit Srivastava for his support and help in data interpretation. We thank the anonymous reviewers for their helpful suggestions in improving the manuscript.
Authorship Contributions
Participated in research design: R. Kukreti, Kukal.
Conducted experiments: Kukal, Bora, Kanojia, Sagar, Satyamoorthy.
Performed data analysis: Kukal, P. Singh, Rawat, Bhatraju.
Wrote or contributed to the writing of the manuscript: R. Kukreti, Kukal, Bora, Kanojia, Paul, Rawat, Bhatraju, Grewal, A. Singh, S. Kukreti.
Footnotes
- Received May 31, 2022.
- Accepted November 3, 2022.
This work was supported by the Council of Scientific and Industrial Research (CSIR) [grant numbers OLP1154, OLP1142, OLP2301]. S.K. acknowledges the Department of Biotechnology (DBT) and CSIR, Govt. of India; S.B., N.K., P.S., and P.R.P. acknowledges CSIR, Govt. of India; and C.R. acknowledges University Grants Commission (UGC), Govt. of India, for their financial assistance.
The authors declare that they have no conflicts of interest with the contents of this article.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ABC
- ATP-binding cassette
- AED
- antiepileptic drug
- BBB
- blood–brain barrier
- BCRP
- breast cancer resistance protein
- BECs
- brain endothelial cells
- B2M
- beta-2 microglobulin
- BPZ
- BODIPY FL Prazosin
- BSA
- bovine serum albumin
- CBZ
- carbamazepine
- DAPI
- 4’
- 6-diamidino-2-phenylindole
- EBM-2
- Endothelial Basal Medium-2
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- LEVI
- levetiracetam
- LTG
- lamotrigine
- MDT
- multidrug transporter
- MRP
- multidrug resistance-associated protein
- MTT
- 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
- PDK4
- pyruvate dehydrogenase lipoamide kinase isozyme 4
- PHT
- phenytoin
- PPARα
- peroxisome proliferator-activated receptor alpha
- PPRE
- PPARα response element
- PWE
- patients with epilepsy
- qPCR
- quantitative real-time PCR
- RT
- reverse transcriptase
- SCR
- scramble
- siRNA
- small interfering RNA
- THBD
- thrombomodulin
- TPM
- topiramate
- TR-FRET
- time-resolved fluorescence energy transfer
- VC
- vehicle control
- VPA
- valproic acid
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}