


Abstract
M4 muscarinic receptors are highly expressed in the striatum and cortex, brain regions that are involved in diseases such as Parkinson’s disease, schizophrenia, and dystonia. Despite potential therapeutic advantages of specifically targeting the M4 receptor, it has been historically challenging to develop highly selective ligands, resulting in undesired off-target activity at other members of the muscarinic receptor family. Recently, we have reported first-in-class, potent, and selective M4 receptor antagonists. As an extension of that work, we now report the development and characterization of a radiolabeled M4 receptor antagonist, [3H]VU6013720, with high affinity (pKd of 9.5 ± 0.2 at rat M4, 9.7 at mouse M4, and 10 ± 0.1 at human M4 with atropine to define nonspecific binding) and no significant binding at the other muscarinic subtypes. Binding assays using this radioligand in rodent brain tissues demonstrate loss of specific binding in Chrm4 knockout animals. Dissociation kinetics experiments with various muscarinic ligands show differential effects on the dissociation of [3H]VU6013720 from M4 receptors, suggesting a binding site that is overlapping but may be distinct from the orthosteric site. Overall, these results demonstrate that [3H]VU6013720 is the first highly selective antagonist radioligand for the M4 receptor, representing a useful tool for studying the basic biology of M4 as well for the support of M4 receptor-based drug discovery.
SIGNIFICANCE STATEMENT This manuscript describes the development and characterization of a novel muscarinic (M) acetylcholine subtype 4 receptor antagonist radioligand, [3H]VU6013720. This ligand binds to or overlaps with the acetylcholine binding site, providing a highly selective radioligand for the M4 receptor that can be used to quantify M4 protein expression in vivo and probe the selective interactions of acetylcholine with M4 versus the other members of the muscarinic receptor family.
Introduction
The five subtypes of muscarinic (M) acetylcholine receptors mediate a wide variety of physiologic functions and pathophysiological processes. The M1, M3, and M5 subtypes are preferentially coupled to Gq/11 G proteins and activate phospholipase C, which catalyzes the phosphatidylinositol trisphosphate cascade and leads to activation of protein kinase C and intracellular Ca2+ mobilization. In contrast, the M2 and M4 subtypes are coupled to Gi/o and inhibit adenylyl cyclase activity and reduce cyclic AMP production upon activation; additionally, signaling downstream of these receptors also modulates ion channels (Ashkenazi et al., 1988; Peralta et al., 1988; Offermanns et al., 1994; Ishii and Kurachi, 2006; Lebois et al., 2018). Muscarinic acetylcholine receptor subtypes are distributed in both the central nervous system and periphery. Although non-selective muscarinic receptor antagonists have shown therapeutic potential for the treatment of certain neurologic disorders, usage has been limited due to intolerable side effects resulting from their broad inhibition of all the muscarinic receptor subtypes (Fahn et al., 1990; Cloud and Jinnah, 2010). This has led to the development of subtype-selective ligands to improve efficacy while reducing side effects.
Of the muscarinic receptors, the M4 subtype is most highly expressed in striatum, cortex, and some subcortical regions, such as the thalamus (Brann et al., 1988; Buckley et al., 1988; Weiner et al., 1990). It has been reported that M4 is the primary muscarinic receptor subtype responsible for regulating dopamine signaling and release in the basal ganglia (Gomeza et al., 1999; Jeon et al., 2010; Foster et al., 2016; Moehle et al., 2017; Moehle and Conn, 2019) and M4 has been implicated in several neurologic disorders, such as Parkinson’s disease, schizophrenia, and dystonia (Katzenschlager et al., 2003; Moehle and Conn, 2019; Moran et al., 2019).
Despite these biologic observations regarding the therapeutic potential of M4 modulation, development of highly selective M4 receptor orthosteric and allosteric modulators has been challenging, and, by extension, the development of radioligands specifically targeting M4 has historically been difficult. Recently, two M4 receptor positive allosteric modulators (PAMs), [11C]-VU0467485/AZ13713945 (Deng et al., 2019) and [18F]-M4R-1911 (Deng et al., 2020) have been radiolabeled and demonstrated to specifically bind to M4 receptors. Another M4 PAM radioligand, [18F]12, has also been shown to display species’ differences in terms of its binding specificity and selectivity (Haider et al., 2023). Encouragingly, the M4 receptor PAM radioligand [11C]MK-6884 has been recently reported to specifically bind to the M4 receptor and penetrate into the brain and has been used clinically as a positron emission tomography (PET) ligand (Tong et al., 2020; Li et al., 2022). In this report, we complement these selective M4 PAM radioligands with [3H]VU6013720, a highly selective M4 antagonist radioligand.
Recently, our group successfully synthesized and characterized several highly selective M4 receptor antagonists, VU6013720, VU6021302, and VU6021625 (Moehle et al., 2021), VU6028418 (Spock et al., 2021), and VU6015241 (Bender et al., 2022). Of these compounds, VU6013720 exhibited the best in vitro functional potency (rat M4 IC50 = 20 nM, human M4 IC50 = 0.6 nM from calcium mobilization assays) as well as a good selectivity profile among the muscarinic receptor family, with IC50 values of >10,000 nM at rM3 and rM5. At rM1, VU6013720 exhibited a functional IC50 of 1700 nM, displaying ∼85-fold selectivity, while VU6013720 had an IC50 of 670 nM at rM2, displaying ∼34-fold selectivity. Additionally, the compound exhibited unbound percentages of 24% and 31% in rat and mouse plasma, as well as 5% in both rat and mouse brain tissue.
In the current report, we generated a tritiated form of this compound, [3H]VU6013720. As shown here, [3H]VU6013720 selectively binds to M4 receptors with little to no binding to the M1, M2, M3, and M5 receptor subtypes. Binding assays with brain tissue homogenates reveal that [3H]VU6013720 exhibits specific binding in extracts from cortex and striatum. As other existing radioligands bind to allosteric sites, it is anticipated that a selective antagonist radioligand that interacts with the acetylcholine binding site should provide a valuable tool to explore selective binding of acetylcholine to the M4 receptor in native tissues, in vivo, in physiologic and pathophysiological disease states and for selective M4 receptor drug development.
Materials and Methods
Cell Culture and Membrane Preparation
Chinese hamster ovary (CHO-K1) cells were purchased from the American Tissue Culture Collection (ATCC, Manassas, Virginia). CHO-K1 cells stably express rat, human, or mouse muscarinic receptors (M1–M5); for M2 and M4, cells were co-transfected with Gqi5. Cells were cultured in F12 medium containing 10% fetal bovine serum, 20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid, and 1X Antibiotic/Antimycotic (contains 10,000 units/ml of penicillin, 10,000 µg/ml of streptomycin, and 25 µg/ml of Gibco Amphotericin B; Thermo Fisher Scientific, Waltham, MA) at 37°C in the presence of 5% carbon dioxide. Cells were monitored by periodical polymerase chain reaction detection using a LookOut Mycoplasma Polymerase Chain Reaction Detection Kit (Sigma-Aldrich, St Louis, MO) to eliminate potential mycoplasma infection and cells were not mycoplasma positive for the studies described here.
Cell membranes were made as previously reported (O’Brien et al., 2018; Moehle et al., 2021). CHO-K1 cells stably expressing different muscarinic receptor subtype were seeded in 150-mm dishes and allowed to grow to approximately 80–90% confluence. Cells were washed once with ice-cold phosphate-buffered saline, scraped from the dishes, and collected using a 5-minute centrifugation at 1000 g. The supernatant was removed, and the cell pellet was stored at −80°C. To prepare membrane protein, cell pellets were thawed, re-suspended in membrane binding buffer (20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid with 10 mM MgCl2 and 10 mM NaCl, pH 7.4), and homogenized using two 15-second bursts from a TR-10 polytron (Tekmar, Vernon, BC, Canada). Cell lysates were then centrifuged at 1000g for 10 minutes at 4°C, and the pellet was discarded. The resultant supernatant was centrifuged at 20,000 g for 30 minutes at 4°C and the supernatant was discarded. The remaining pellet was re-suspended in membrane binding buffer. The protein concentration was determined by bicinchoninic acid assay (Thermo Scientific, Rockford, IL). Membranes were then stored at −80°C until use.
Radiochemistry
The synthesis of VU6013720 has been reported previously (Moehle et al., 2021). Cold VU6013720 compound was radiolabeled by RC TriTec (Teufen, Switzerland) with photocatalytic H/T exchange (Fig. 1), with the [3H] label most likely in the alpha position of the tertiary amine. The final radioligand, [3H]VU6013720, had a purity >98% with a specific activity 23.6 Ci/mmol (233.8 GBq/mmol) and a concentration of 1 mCi/ml (37 MBq/ml). [3H]NMS was manufactured on 03/08/2018, Specific activity: 80.0 Ci/mmol, Dilution: 1 mCi/ml (PerkinElmer, Boston, MA).

Structure of VU6013720. Cold VU6013720 was labeled using nonspecific hydrogen/tritium exchange with 99% tritium gas (RC Tritec, Switzerland) with a specific activity of 23.6 Ci/mmol.
[3H]VU6013720 Saturation Binding Assays
Saturation binding experiments were performed to determine the specificity of [3H]VU6013720 binding to the muscarinic receptor subtypes. Briefly, 10 µg CHO-K1 cell membranes expressing each receptor were incubated with [3H]VU6013720 in deep 96-well plates (Corning, New York) on a shaker at room temperature for 3 hours in a reaction volume of 500 μl. Saturation binding experiments were performed in triplicate or duplicate, and nonspecific binding was assessed by addition of 10 µM atropine or VU6013719 (VU6013720 structural analog, Supplemental Fig. 1). The reactions were terminated by vacuum filtration onto Unifilter-96 GF/B plate through a 96-well harvester. The filters were washed three times with cold membrane harvesting buffer (50 mM Tris-HCl and 0.9% NaCl, pH 7.4). Plates were then dried overnight and 40 µl of MicroScint 20 (PerkinElmer) was added to each well. The membrane-bound radioactivity was determined using a TopCount NXT microplate scintillation and luminescence counter (PerkinElmer). Calculation of specific binding versus total counts revealed that radioligand depletion was not a factor in these studies using this assay protocol.
Competition Binding Assays with [3H]NMS and [3H]VU6013720
Radioligand competition binding assays were performed as previously described (Moehle et al., 2021) using concentrations of radioligand that were within 2-fold of the calculated equilibrium dissociation constant (Kd). Briefly, M4 antagonists were 1:3 serially diluted into membrane binding buffer and added to each well of a 96-well plate, along with 10 μg/well cell membrane and approximately 100 pM [3H]-N-methylscopolamine ([3H]NMS, PerkinElmer, Boston, MA) or 100 - 400 pM [3H]VU6013720 in a reaction volume of 500 μl. Following a 3-hour incubation period on a shaker at room temperature, the membrane-bound ligand was separated from free ligand by filtration through glass fiber 96-well filter plates (Unifilter-96, GF/B; PerkinElmer, Boston, MA) using a 96-well Brandel harvester. The filters were washed three times with cold membrane harvesting buffer (50 mM Tris-HCl and 0.9% NaCl, pH 7.4). Plates were then dried overnight and 40 µl of MicroScint 20 (Perkin Elmer) was added to each well. The membrane-bound radioactivity was determined using a TopCount NXT microplate scintillation and luminescence counter (PerkinElmer). Nonspecific binding was determined using 10 μM atropine.
Kinetic Studies
Kinetic studies of [3H]VU6013720 association/dissociation from M4 receptors were performed at room temperature. For association assays, membranes were incubated with [3H]VU6013720 at concentrations 2-fold over the Kd value for 0, 2, 5, 10, 20, 30, 45, 60, 90, 120, 150, and 180 minutes. For dissociation assays, membranes and [3H]VU6013720 were allowed to equilibrate for 120 minutes and then 10 µM of cold competitor was added at 120, 90, 60, 45, 30, 20, 15, 10, 5, 2, and 1 minute before termination of the assay. Following these pretreatment times, membranes were immediately harvested by quick filtration to a GF/B plate using ice-cold harvesting buffer and a 96-well Brandel harvester. Buffer and temperature conditions for all binding studies were the same.
Animals
Male and female wildtype (two male and two female) or Chrm4 knockout (two male and two female) mice (bred in-house) and male Sprague Dawley (n = 4) rats (Envigo, Indianapolis, IN) were used for this study. They were housed in the animal care facility certified by the American Association for the Accreditation of Laboratory Animal Care under a 12-hour light/dark cycle (lights on: 7 AM; lights off: 7 PM). The experimental protocols performed during the light cycle were approved by the Institutional Animals Care and Use Committee of Vanderbilt University and conformed to the guidelines established by the National Research Council Guide for the Care and Use of Laboratory Animals.
Brain Tissue Membrane Preparation and In Vitro Saturation Binding
Rats and mice (wild-type and Chrm4 knockout) were anesthetized by continuous isoflurane (5%) and killed by decapitation, and striatum and cortex were dissected on ice. The dissected brain regions were weighed and homogenized at a concentration of 1 mg of tissue per 25 volumes of membrane binding buffer with a polytron for two 20-second bursts. The homogenates were centrifuged at 20,000 g for 20 minutes at 4°C, and the supernatant was discarded. The pellet was homogenized in membrane binding buffer and centrifuged under the same conditions as above. The final homogenate was re-suspended in membrane binding buffer and incubated at 37°C for 10 minutes and centrifuged as above. The resultant pellet was re-suspended in membrane binding buffer, and the protein concentration was determined by bicinchoninic acid assay. The brain homogenate was stored at −80°C until use. To determine the specific binding of [3H]VU6013720 using brain tissue membranes, saturation assays were performed as above with CHO cell membranes but using 150 µg of brain homogenate protein per well.
Data Analysis
All experiments were carried out in duplicate or triplicate and were performed at least three times (unless stated otherwise), which aligns with our historical and standard assay protocol. Studies presented in this manuscript are exploratory in nature and were performed to characterize a new radioligand of general interest to scientists interested in the biology of the muscarinic receptors. Data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA) with the following equations and were not constrained at the top or bottom.
Saturation Binding
Nonspecific binding at each radioligand concentration was subtracted from total binding and the data were fit to the following equation:
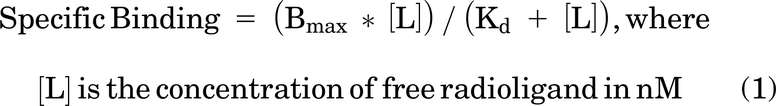
Competition Binding
Data were analyzed using the Cheng Prusoff equation:
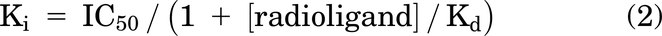
Association Kinetics
Y=Y0 + (Plateau-Y0)*(1-exp(-K*x)), where Y0 is the Y value when time (X) is 0, expressed as % [3H]VU6013720 binding; plateau is the Y value at infinite time, expressed as % [3H]VU6013720 binding; K is the rate constant, expressed as inverse minutes; Tau is the time constant, expressed in minutes; half-time is calculated as ln(2)/K, expressed as minutes, and span is the difference between Y0 and Plateau, expressed as % [3H]VU6013720 binding.
Dissociation Kinetics: One-Phase or Two-Phase Exponential Decay
One phase: Y=(Y0-NS)*exp(-K*X) + NS, where Y0 is the binding at time zero, expressed as % [3H]VU6013720 binding; NS is nonspecific binding at infinite time, expressed as % [3H]VU6013720 binding; K is the rate constant in inverse minutes; half-time equals ln(2) divided by K.
 where Y0 is the Y value when X (time) is zero, expressed as % [3H]VU6013720 binding; plateau is the Y value at infinite time, expressed in minutes; Kfast and Kslow are the rate constants, expressed as inverse minutes; time units; TauFast and TauSlow are the two time constants, expressed as time; Half-life (fast) and Half-life (slow) are computed as ln(2)/K and expressed as minutes; PercentFast is the fraction of the span (from Y0 to Plateau) accounted for by the faster of the two components.
where Y0 is the Y value when X (time) is zero, expressed as % [3H]VU6013720 binding; plateau is the Y value at infinite time, expressed in minutes; Kfast and Kslow are the rate constants, expressed as inverse minutes; time units; TauFast and TauSlow are the two time constants, expressed as time; Half-life (fast) and Half-life (slow) are computed as ln(2)/K and expressed as minutes; PercentFast is the fraction of the span (from Y0 to Plateau) accounted for by the faster of the two components.
Data from individual experiments were fit independently and then presented as the pIC50 or pKi/pKd as mean ± standard deviation. Data shown in the figures represents either combined data from multiple experiments or, in some cases, as a data set from a typical experiment as noted in the various figure legends.
Results
VU6013720 is a Selective M4 Antagonist
VU6013720 (Fig. 1) binding affinities and subtype selectivity at the five rat muscarinic receptor subtypes were assessed using binding competition against the non-selective orthosteric radioligand [3H]N-methylscopolamine (NMS) in membranes made from CHO-K1 cells expressing each receptor subtype (Table 1; Fig. 2). VU6013720 fully displaced [3H]NMS binding from rat M4 receptors with a pKi of 8.8 ± 0.1 (n = 4), while equilibrium inhibitory constant (pKi) values were 6.9 ± 0.2 (n = 4) for M2 and affinity values were above a micromolar for M1, M3, and M5. The ratio of the nanomolar Ki value at the receptor with the closest binding affinity, M2, versus M4 was 80-fold. These competition binding data, together with IC50 values from calcium mobilization assays (Moehle et al., 2021), demonstrate that VU6013720 is a highly selective M4 antagonist with single-digit nanomolar affinity, and possesses the characteristics needed to move to further characterization as a radioligand.
Characterization of the binding specificity of VU6013720 using the orthosteric muscarinic antagonist radioligand [3H]-N-methylscopolamine
pKi values were obtained from [3H]-NMS competition binding assays with CHO-K1 cell membranes expressing different rat muscarinic receptor subtypes. Data represent the mean ± S.D. of four independent experiments performed in triplicate and refer to the curve fits in Fig. 2.

Binding selectivity of VU6013720 at muscarinic acetylcholine receptor subtypes using [3H]NMS. VU6013720 and atropine competition binding curves at various muscarinic acetylcholine receptor subtypes were performed using the orthosteric muscarinic radioligand, [3H]-N-methylscopolamine. Data are the mean ± S.D. from a representative of four independent experiments performed in triplicate.
[3H]VU6013720 Binds Specifically to M4 Receptors with High Affinity
We radiolabeled VU6013720 and determined the interaction of this novel radioligand across the muscarinic receptor family using saturation binding assays performed with cell membranes expressing the various receptor subtypes. Nonspecific binding was determined in the presence of a structurally related antagonist, VU6013719 (Supplemental Fig. 1; synthesis provided in Supplemental Methods), or the nonselective muscarinic antagonist, atropine. [3H]VU6013720 bound specifically to the rat M4 receptor with a pKd of 9.7 ± 0.2 (Fig. 3A; Table 2) and a Bmax of 4800 ± 500 fmol/mg using VU6013719 as the nonspecific binding control (n = 4) and a pKd of 9.5 ± 0.2 and a Bmax of 4100 ± 500 fmol/mg using atropine (Fig. 3B; Table 2, n = 3). These values were slightly different when compared with the pKi determined with [3H]NMS (8.8 versus 9.7/9.5), which may suggest differential interaction of [3H]VU6013720 and [3H]NMS with the receptor. In contrast, [3H]VU6013720 exhibited no specific binding at the rat M1, M2, M3, and M5 receptor subtypes (Fig. 3, C–F, n = 3 per receptor). These data suggest that [3H]VU6013720 binds specifically to the M4 receptor with high affinity. We performed a comparison of the pKi value of atropine with both [3H]NMS and [3H]VU6013720 and found the pKi to be statistically different between the two radioligands (8.7 ± 0.1 with [3H]NMS and 9.0 ± 0.1 with [3H]VU6013720, mean ± S.D., **P = 0.009). As these two radioligands are of different structure, this would suggest that the binding sites of the two, or the binding pose adopted by atropine in the presence of each radioligand, are not exactly the same. We also compared Bmax values for saturation binding studies of [3H]NMS and [3H]VU6013720 and determined Bmax values of 4500 ± 700 and 4100 ± 500 fmol/mg (Supplemental Fig. 2, paired Student’s t test, P = 0.219, n = 3), respectively, suggesting that the two radioligands label a similar number of binding sites on the receptor. To assess cross-species binding of [3H]VU6013720, saturation binding was also assayed with membranes expressing the human and mouse M4 receptors. Similar to the rat M4 receptor, [3H]VU6013720 bound to these receptors with high affinity, with pKd values of 10 ± 0.1 (n = 3) at the human M4 receptor and 9.7 at the mouse M4 receptor, respectively (n = 1, Supplemental Fig. 3; Table 2).

Among the five muscarinic receptors, [3H]VU6013720 binds specifically to the rat M4 receptor. Total and specific binding for [3H]VU6013720 was determined using cells expressing each of the rat muscarinic receptors. [3H]VU6013720 bound specifically to the rat M4 receptor in a saturable manner using either VU6013719 (A) or atropine (B) as nonspecific binding controls, whereas no detectable binding was observed at rat M1, M2, M3, or M5 using atropine to determine nonspecific binding (C–F). Data shown are the mean ± S.D. of a representative of at least three independent experiments performed in triplicate.
Summary of binding properties of [3H]VU6013720 at multiple species of M4 receptor
Data represent mean ± S.D. from at least four experiments performed in quadruplicate (human and rat) and one experiment (mouse) performed in triplicate. # and @ denote the use of VU6013719 and atropine, respectively, for non-specific binding. Data refer to saturation binding curves shown in Fig. 3 and Supplemental Fig. 3.
Kinetic Evaluation of [3H]VU6013720
To fully characterize this novel radioligand, kinetic binding studies examining association and dissociation were next conducted using a concentration of radioligand calculated to be within 2-fold of the Kd in each experiment. Association experiments demonstrated that [3H]VU6013720 binding reached equilibrium within 10 minutes (Fig. 4A) with an association on-rate of 1.7 ± 0.1 nM−1min−1 (n = 3). [3H]VU6013720 dissociated completely by adding 10 µM VU6013719 or atropine, although with distinct time courses (Fig. 4B; Table 3, n = 3). The data for both of these compounds was better fit to a two-site decay model versus a single site. We observed faster dissociation with the structural analog VU6013719 than atropine (Table 3), suggesting that [3H]VU6013720 and VU6013719 may share a binding site that is distinct or overlapping that of atropine. We also examined dissociation kinetics induced by the positive allosteric modulator, VU0467154 (Bubser et al., 2014). VU0467154 induced partial but saturable displacement of [3H]VU6013720, similar to what we had previously observed with [3H]NMS (Bubser et al., 2014). This suggests that VU0467154 is binding to an allosteric site but induces negative cooperativity with [3H]VU6013720 by partially displacing the ligand.

Kinetic characterization of the [3H]VU6013720 radioligand. Time course experiments defining association and dissociation of [3H]VU6013720 at rat M4 membranes were performed at room temperature. (A) Association was initiated by addition of [3H]VU6013720 to membranes at the indicated time points before filtration. (B) Dissociation experiments were performed by allowing [3H]VU6013720 to equilibrate with membranes for 2 hours; at this point, a 10 μM final concentration of VU6013719 (white), atropine (black), or VU0467154 (red) was added at designated times before terminating the reaction by filtration. Data are the mean ± S.D. of three independent experiments performed in triplicate.
Dissociation kinetics of [3H]VU6013720 induced by multiple ligands
Dissociation was performed by adding 10 μM of atropine, VU6013719, or VU0467154 at designated times. Data were fit with both one- and two-phase decay curves. For atropine and VU6013719, two-site fits were the preferred model; for VU0467154, a one-site model was preferred. *P < 0.05, **P < 0.01, ***P < 0.001 between atropine and VU6013719.
[3H]VU6013720 Specifically Binds to M4 in the Rodent Brain
One of the main advantages of a highly specific radioligand is sensitivity for detection of receptors in native tissues. To determine the utility of [3H]VU6013720 for detecting M4 receptors in brain tissue, membranes were prepared from the striatum and cortex of rats. As shown in Fig. 5, saturation binding assays detected specific binding of [3H]VU6013720 to homogenates from rat striatum and cortex with high affinities (pKd = 9.0 ± 0.2 in cortex, (n = 3, A)) and 9.3 ± 0.2 in striatum, (n = 3, B)). Bmax values of 260 ± 110 (cortex) and 350 ± 60 (striatum) fmol/mg protein were also determined (n = 3). We would note that, when the pKd values for rat M4 cells, cortex, and striatum were compared using a one- way ANOVA, there was a significant difference (P < 0.05) between rat M4 cells and the cortex, but not the striatum. This may reflect distinct microenvironments between an in vitro cell system versus the brain.

[3H]VU6013720 binds specifically to rat brain cortical and striatal tissue as well as to cortical membranes from WT but not from Chrm4 knockout mice. Rat cortical (A) and striatal (B) homogenates were incubated with [3H]VU6013720 in the presence and absence of 10 μM atropine to determine total and nonspecific binding. [3H]VU6013720 bound to rat cortical membranes with a pKd of 9.0 ± 0.2 and a Bmax of 260 ± 110 fmol/mg of protein and to rat striatal membranes with a pKd of 9.3 ± 0.2 and a Bmax of 350 ± 60 fmol/mg of protein. Data are mean ± S.D. and representative of three independent experiments performed in triplicate. Cortical homogenates from WT (C) and Chrm4 knockout mice (D) were incubated with [3H]VU6013720 in the presence and absence of 10 μM atropine to determine total and nonspecific binding. [3H]VU6013720 binds to cortical membranes from WT mice with a pKd of 9.0 ± 0.4 and a Bmax of 240 ± 90 fmol/mg of protein, whereas little specific [3H]VU6013720 binding was detected in cortical homogenates from Chrm4 knockout mice. Data are the mean ± S.D. and a representative of three independent experiments performed in duplicate or triplicate.
To further confirm that these binding sites were indeed M4 receptors, saturation binding was also performed with membranes made from both wild-type (WT) and Chrm4 knockout mouse cortex. As shown in Fig. 5, C and D, [3H]VU6013720 specifically bound to WT mouse cortex and exhibited essentially no binding to cortical membranes from Chrm4 knockout mice. These data demonstrate that [3H]VU6013720 is a useful tool for quantification of M4 receptors in brain tissue.
Discussion
M4 muscarinic receptors are highly expressed in the brain (e.g., striatum, cortex, thalamus) and have been demonstrated to be involved in several central nervous system diseases (Pancani et al., 2015; Felder et al., 2018; Moehle and Conn, 2019; Teal et al., 2019; Dean and Scarr, 2020; Foster et al., 2021; Moehle et al., 2021). Lack of selective muscarinic receptor antagonists has prevented the utilization of anti-muscarinic therapy due to intolerable adverse effects, and tools that assist with drug discovery efforts, such as highly specific radioligands, may open new avenues for exploration. Here, we demonstrate that [3H]VU6013720 is a novel, high-affinity, highly specific antagonist radioligand for the M4 receptor. [3H]VU6013720 selectively binds to the M4 receptor with pKd values of 9.5 at rat, 10 at human, and 9.7 at mouse M4 receptors. Based on this profile, we used this high-affinity radioligand to quantify M4 receptors in brain tissue by saturation binding with brain tissue homogenates.
There are some interesting, subtle findings from the current studies. While the majority of our data point to an orthosteric or overlapping binding site for VU6013720, the pKi for cold VU6013720 is 8.8 at rat M4 using [3H]NMS binding while the pKd was calculated to be 9.5 with atropine and 9.7 with VU6013719. Additionally, competition binding studies using VU6013720 itself revealed a pKi of 9.5 (data not shown), which is close to the Kd value. When considered in the context of the significant shift of the pKi of atropine between [3H]NMS and [3H]VU6013720, it suggests that these two radioligands might make distinct contacts within similar, but not identical, binding sites. This is further supported by the dissociation kinetics experiments in which the dissociation of [3H]VU6013720 differs in the presence of atropine versus VU6013719. Other factors may impact these results, such as differences in residence time. The discrepancy between pKis for atropine with the two radioligands at M4 also suggested the possibility that, due to the high sequence conservation, a similar leftward affinity shift might exist for M2 which might have resulted in specific binding at M2; however, we did not observe any saturable binding to this receptor subtype.
We previously reported a 30-fold selectivity difference for the human and rat M4 receptors for VU6013720 in a chimeric G protein assay that allows the M4 receptor to couple to calcium mobilization (Moehle et al., 2021). In contrast, our data here show that there is only a 3-fold difference in affinity between the human and rat M4 receptors. We currently do not have an explanation for this difference, although there are several possibilities. First, the functional assays include acetylcholine, whereas the binding assays do not. If VU6013720 is truly orthosteric, this should not be a confound; however, if ACh/NMS and VU6013720 have subtly different binding sites and these binding sites are slightly different between the two species, this could contribute to these discrepancies. Additionally, if VU6013720 is actually an inverse agonist, its functional potency may be dissociated from its binding affinity due to G protein interactions or other protein/protein interactions that are induced in the cells when the receptor is functionally assessed. We also do not currently know if there is a species difference in terms of contact with the chimeric G protein used in these studies, Gqi5, and whether Gqi5 impacts receptor conformation in binding studies. Future experiments around this issue could include assessment of VU6013720 effects in an assay in which constitutive activity of the receptor might be observed, such as inositol phosphate accumulation or cAMP generation, to determine if there is a distinction in inverse agonist activity, and further exploration of the effects of various G proteins or signaling partners.
To our knowledge, this is the first development of a broadly useful, highly specific antagonist radioligand with high affinity for M4 receptors, and the development of [3H]VU6013720 is anticipated to be a powerful tool for M4 receptor studies. Previous approaches to investigate the tissue distribution of the M4 receptor include RNA detection techniques, or by antibody, with confirmation using knockout tissue (Brann et al., 1988; Buckley et al., 1988; Weiner et al., 1990; Levey et al., 1991; Levey 1993; Vilaró et al., 1993; Wolfe and Yasuda, 1995; Tice et al., 1996; Gomeza et al., 1999). Although these studies provide important insight into relative distribution, detected RNA levels are not always consistent with protein expression levels, and antibodies do not provide the same level of quantitation that is possible using specific radioligand binding. As we have shown in this report, our studies using [3H]VU6013720 were able to confirm and quantify the M4 receptor protein expression pattern and receptor density levels in native brain tissues. For this reason, we anticipate that [3H]VU6013720 will be an important tool for investigating the M4 receptor distribution in native tissues and may also be useful for interrogating potential differences that may occur in different disease states or models.
In the past several years, several M4 receptor radioligands have been reported, the M2/M4 PAM [3H]LY2119620 (Schober et al., 2014), and the selective M4 PAMs [11C]-VU0467485/AZ13713945 (Deng et al., 2019), [18F]-M4R-1911 (Deng et al., 2020), [11C]MK-6884 (Tong et al., 2020), and [18F]12 (Haider et al., 2023). However, these radioligands have been developed from M4 PAM scaffolds and bind to allosteric sites on the M4 receptor. [3H]VU6013720 is the first reported selective radiolabeled M4 receptor antagonist. Therefore, it is a unique tool that could be paired with PAM radioligands to study compound binding sites as well as for the development of future M4 antagonists and modulators. Additionally, the compound could potentially be used to map acetylcholine levels in the brain relevant for M4 function and be useful for in vitro screening of M4 antagonists for therapeutic treatment of movement disorders and may serve as a starting point for development of antagonists with higher affinity and favorable drug metabolism and pharmacokinetic properties to eventually lead to PET ligands.
In conclusion, we have developed the first highly selective M4 receptor antagonist radioligand with high affinity for the rodent and human M4 receptors, [3H]VU6013720. A reliable tool will lead to a better understanding of the physiology and pathophysiology associated with this important signaling protein and facilitate the support of drug development for neurologic diseases for which M4 modulators are predicted to exhibit efficacy, such as Parkinson’s disease, essential tremor, and schizophrenia.
Acknowledgments
The authors thank Jeff Conn and members of the Warren Center for Neuroscience Drug Discovery for stimulating and formative discussions. The authors also thank the William K. Warren Foundation for endowing the WCNDD.
Data Availability
The authors declare that all processed data supporting the findings of this study are available within the paper and its Supplemental Materials. Raw data are available on request from the corresponding author.
Authorship Contributions
Participated in research design: Qi, Rodriguez, Dickerson, Bender, Moehle, Lindsley, Rook, Niswender.
Conducted experiments: Qi, Kling, Billard, Peng, Dickerson, Bender.
Contributed new reagents or analytic tools: Engers, Bender, Lindsley.
Performed data analysis: Qi, Rodriguez, Dickerson, Rook, Niswender.
Wrote or contributed to the writing of the manuscript: Qi, Rodriguez, Dickerson, Engers, Bender, Lindsley, Rook, Niswender.
Footnotes
- Received November 2, 2022.
- Accepted August 14, 2023.
Research was supported by the National Institutes of Health [Grants R01-MH073676 and R00-NS110878], CDMRP [Grant W81XWH-19-1-0355], and Ancora Innovation, LLC.
A.M.B. and C.W.L. are inventors on applications for composition of matter patents that protect several series of M4 antagonists.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- Bmax
- maximal binding
- CHO-K1
- Chinese hamster ovary K1
- Kd
- equilibrium dissociation constant
- Ki
- equilibrium inhibitory constant
- M
- muscarinic
- NMS
- N-methyl scopolamine
- PAM
- positive allosteric modulator
- PET
- positron emission tomography
- WT
- wild-type
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}