


Abstract
Dysfunction of p53 and resistance to cancer drugs can arise through mutually exclusive overexpression of MDM2 or MDM4. Cisplatin-resistant cells, however, can demonstrate increased binding of both MDM2 and MDM4 to p53 but in absence of cellular overexpression. Whether MDM2 inhibitors alone can activate p53 in these resistant cells was investigated with the goal to establish the mechanism for potential synergy with cisplatin. Thus, growth inhibition by individual drugs and combinations was assessed by a colorimetric assay. Drug-treated parental A2780 and resistant tumor cells were also examined for protein expression using immunoblot and reverse phase protein array (RPPA) and then subjected to Ingenuity Pathway Analysis (IPA). Gene expression was assessed by real-time polymerase chain reaction, DNA damage by confocal microscopy, cell cycle by flow cytometry, and homologous recombination (HR) by a GFP reporter assay. Our results demonstrate that Nutlin-3 but not RITA (reactivation of p53 and induction of tumor cell apoptosis) effectively disrupted the p53-MDM2-MDM4 complex to activate p53, which increased robustly with cisplatin/Nutlin-3 combination and enhanced antitumor effects more than either agent alone. RPPA, IPA, and confocal microscopy provided evidence for an “apparent” increase in DNA damage resulting from HR inhibition by cisplatin/Nutlin-3. Molecularly, the specific HR protein Rad51 was severely downregulated by the combination via two mechanisms: p53-dependent transrepression and p53/MDM2-mediated proteasomal degradation. In conclusion, Nutlin-3 fully destabilizes the p53-MDM2-MDM4 complex and synergizes with cisplatin to intensify p53 function, which then downregulates Rad51 through a bimodal mechanism. As a result, HR is inhibited and antitumor activity enhanced in otherwise HR-proficient sensitive and resistant tumor cells.
SIGNIFICANCE STATEMENT Rad51 downregulation by the combination of cisplatin and Nutlin-3 inhibits homologous recombination (HR), which leads to persistence in DNA damage but not an increase. Thus, inhibition of HR enhances antitumor activity in otherwise HR-proficient sensitive and resistant tumor cells.
Introduction
The wild-type tumor suppressor p53 is a critical transcriptional cofactor that promotes apoptotic response when cellular stress is elevated by antitumor DNA-damaging agents. Normally, p53 is kept inactive by its binding to the negative regulators MDM2 and MDM4, which reduce its half-life and inhibit transactivation functions (Shadfan et al., 2012). However, DNA damage signals after exposure to therapeutic drugs release and activate p53 through post-translational modifications, such as phosphorylation of p53 at the Ser15 residue by ataxia telangiectasia mutated and ataxia telangiestasia and Rad3-related protein (Toledo and Wahl, 2006). Unfortunately, p53 function is often attenuated or lost when the protein becomes mutated in the DNA-binding domain, and this leads to loss of p53-dependent apoptotic signal, culminating in drug-resistant tumor cells and poor survival rates in patients with cancer (Siddik, 2003; Martinez-Rivera and Siddik, 2012).
Loss of p53 function and drug resistance can also occur through mechanisms not requiring mutation. Since one critical mechanism involves overexpression of MDM2 or MDM4 (Toledo and Wahl, 2007; Wasylishen and Lozano, 2016), reactivation of p53 to resensitize tumor cells and restore therapeutic response to anticancer drugs has been pursued through the design of small molecules that disrupt the p53-MDM2-MDM4 triprotein complex (Toledo and Wahl, 2007; Tisato et al., 2017). A number of such molecules include Nutlin-3, 2-[2-chloro-4-[(1,5-dihydro-3-methyl-5-oxo-1-phenyl-4H-pyrazol-4-ylidene)methyl]-6-ethoxyphenoxy]-acetic acid methyl ester (SJ-172550), and (Z)-2-(4-((6-Chloro-7-methyl-1H-indol-3-yl)methylene)-2,5-dioxoimidazolidin-1-yl)-2-(3,4-difluorophenyl)-N-(1,3-dihydroxypropan-2-yl)acetamide (RO5963), which inhibit p53-MDM2 interaction, p53-MDM4 interaction, or both, respectively (Burgess et al., 2016; Tisato et al., 2017). The design of dual inhibitors of MDM2 and MDM4 is based on the premise that MDM2 inhibitors do not impact MDM4 binding to p53 and vice versa (Patton et al., 2006). This is supported in part by observations that MDM2 inhibitors, such as Nutlin-3, become ineffective when MDM4 is overexpressed (Hu et al., 2006).
Although a case for dual inhibitors appears rational, inhibition of MDM2 binding alone to p53 appears to be sufficient to upregulate p53 function, which supports the notion that simultaneous binding of both MDM2 and MDM4 is critical for p53 inhibition (Toledo and Wahl, 2007). This is important in context with our observations that p53 is robustly inactivated in A2780-derived resistant ovarian tumor cells by a 2- to 8-fold stoichiometric and concurrent increase in bound MDM2 and MDM4 (Xie et al., 2016). Such a characteristic profile is consistent with clinical cases based on cellular overexpression of both MDM2 and MDM4 in some tumors, including ovarian cancer (Wasylishen and Lozano, 2016). As a therapeutic strategy, therefore, inhibiting either MDM2 or MDM4 binding in these resistant tumors may be sufficient to activate p53.
Unlike MDM4 inhibitors, MDM2 inhibitors are well-advanced in their development, with several entering clinical trials (Tisato et al., 2017). However, MDM2 inhibitors at physiologic concentrations tend to robustly induce cell cycle arrest but not apoptosis (Jiang et al., 2007; Rigatti et al., 2012), and this advanced the empirical strategy of combining the MDM2 inhibitor with DNA-damaging agents in MDM2-overexpressing tumor cells (Rigatti et al., 2012; Deben et al., 2015). Therefore, in the present study, we have rationally investigated whether MDM2 inhibitors alone could also activate p53 in A2780-based resistant models concurrently harboring excessive MDM2 and MDM4 binding and restore cisplatin sensitivity. Our results demonstrate that selective inhibition of MDM2 is sufficient to activate p53 and synergize with cisplatin to restore cytotoxic potency in these resistant tumor cells. However, unlike reports ascribing such synergy to an absolute increase in DNA damage (Rigatti et al., 2012; Deben et al., 2015), our results from using reverse phase protein array (RPPA) and Ingenuity Pathway Analysis (IPA) advance our understanding by demonstrating for the first time that the increase in DNA damage is an “apparent” effect due to p53-dependent bimodal downregulation of Rad51 and resultant inhibition of the canonical homologous recombination DNA repair pathway.
Materials and Methods
Antibodies and Reagents.
Protein A beads (sc-2001) and antibodies for p53 (DO-7: sc-47698; FL-393: sc-6243), p21 (sc-6246), MDM2 (sc-13161), and β-actin (sc-47778) were purchased from Santa Cruz (Dallas, TX). Anti-MDM4 (A700-000) was purchased from Bethyl Laboratories (Montgomery, TX). Rad51 (8875) and phospho-specific antibody for p53-Ser15 (9284) were from Cell Signaling (Danvers, MA); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (19265) was from Affymetrix (Santa Clara, CA); LipofectAMINE 2000 (11668019), LipofectAMINE 3000 (L3000015), and RNAiMAX (13778150) were from Life Technologies (Brea, CA); Halt Protease & Phosphatase inhibitor (1861280) was from Thermo Scientific (Asheville, NC); and cisplatin (C2210000), MG132 (C2211), and cycloheximide (C1988) were from Sigma (St. Louis, MO). For maximal stability, cisplatin stock solutions were prepared in saline. High Capacity RNA-to-cDNA kit (4387406) from Applied Biosystems (Forster City, CA) and RNeasy Mini kit (74104) from Qiagen (Valencia, CA) were purchased. His-Ubiquitin plasmid was a kind gift from Dr. Shiaw-Yih Lin (MD Anderson Cancer Center). Apple-53BP1trunc (69531) was purchased from Addgene (Cambridge, MA). Nutlin-3 (675576-98-4) and RITA (213261-59-7) were purchased from Cayman Chemicals (Chicago, IL). These MDM2 inhibitors were prepared in DMSO, with the final concentration of this solvent in tissue cultures not exceeding 0.02%.
Cell Culture.
Tissue culture conditions and functional wild-type p53 status for sensitive A2780 and resistant 2780CP/Cl-16 and 2780CP/Cl-24 endometrioid ovarian tumor cells have been described previously (Xie et al., 2016, 2017). The wild-type p53 U2OS-DR-GFP cells, a kind gift from Dr. Guang Peng (MD Anderson Cancer Center), were cultured in McCoy’s 5A medium supplemented with 10% FBS. Control and p53 knockout cells were selected after transfection by the CRISPR system as reported (Bhatt et al., 2017; Xie et al., 2017).
MTT Cell Growth Inhibition Assay.
MTT assay was conducted as described before (Xie et al., 2016, 2017). In brief, cells were plated into 96-well plates and exposed to the drugs the next day. Growth inhibition was measured 5 days later using the MTT reagent, and the reduced formazan product was measured at 570 nm. The IC50 value was determined by fitting the concentration versus growth inhibition data to a four-parameter sigmoidal curve using the GraphPad Prism computer program. The ratios of IC50 in resistant cells to that in sensitive cells were calculated to indicate the extent of resistance (or resistance factor) in cells.
Small Interfering RNA.
The siRNA duplex and procedures for transfection were described previously (Xie et al., 2016). Briefly, A2780 cells were exposed to 100 nM of either control-siRNA or MDM2-siRNA using RNAiMAX, and cells were harvested 48 hours later for experimental studies.
Western Blot Analysis and Immunoprecipitation.
The procedures were described previously (Xie et al., 2017). In brief, cells were lysed, and 20–50 μg of protein were resolved by SDS-PAGE and transferred to nitrocellulose membrane, which were then probed overnight at 4°C with primary antibodies. Secondary antibodies conjugated to horseradish peroxidase and ECL fluorescence detection (Bio-Rad) were used to visualize the protein band. X-ray films were quantified by densitometry using the Image J software. Protein expressions normalized against β-actin were plotted by linear regression using GraphPad Prism computer program. Immunoprecipitates, isolated as described previously (Xie et al., 2016, 2017), were similarly subjected to immunoblot analysis.
Cell Cycle Analysis by Fluorescence-Activated Cell Sorting.
The assay for cell cycle analysis has already been reported by us (Kuang et al., 2001). Essentially, cells fixed in 70% ethanol were centrifuged, washed, resuspended in PBS containing 50 µg/ml PI and 20 µg/ml RNase A, and incubated at room temperature for 15 minutes before FACS analysis. For apoptosis determination, cells were analyzed by FACS using FITC Annexin V Apoptosis Detection Kit.
RPPA and IPA Analysis.
A2780 cells harvested 24 hours after drug exposures were washed three times with PBS, pelleted by centrifugation, and submitted to the RPPA Core Facility at MD Anderson for analysis, as described previously (Masuda et al., 2017). The RPPA dataset consisted of about 300 antibody probes, and protein expression data were compared with control to estimate fold change. The data were then subjected to online IPA (QIAGEN, Redwood City; http://www.ingenuity.com/) to identify canonical pathways demonstrating statistically significant changes, as indicated by Fisher exact test, after specific treatments. The results are presented as −Log(P value), with the lowest P value (or highest −Log(P value)) associated with the highest probability that the specific pathway is altered.
DNA Damage Assessment in Cells by Confocal Microscopy.
Tumor cells were transected with Apple-53BP1trunc plasmid (Wang et al., 2017) using LipofectAMINE 3000 in six-well plates, essentially as described before (Xie et al., 2017). G418 was then added 48 hours later, and stable lines were selected after sorting by flow cytometry based on Apple fluorescence. Cells stably expressing Apple-53BP1trunc plasmid were used to assess DNA damage, as described before (Yang et al., 2015). Cells exposed to drugs for 24 hours were fixed using 4% paraformaldehyde for 20 minutes at room temperature, and cells with DNA damage foci detected by confocal microscopy (LSM 510 Meta; Zeiss) were counted. DNA damage foci were also used to assess repair by exposing cells to the drugs for 24 hours and then a further 12 hours in drug-free media. Approximately 200 cells were counted in each of 10 fields, and only cells with at least 10 foci were counted as positive. Cell counts were conducted blindly, whereby the confocal images were randomly coded to avoid group or treatment identification until after the counts were completed.
Platinum Drug Uptake and DNA Adduct Studies.
Platinum accumulation and DNA adducts in cells were determined according to the procedure described previously (Arambula et al., 2011). Briefly, cells exposed to the drugs for 2 hours were split into two fractions. Cell pellet from one-third of the fraction was used for protein quantification. Cell pellet from the remaining fraction was digested overnight at 55°C in benzethonium hydroxide and acidified with HCl, and the platinum content was quantified by flameless atomic absorption spectrometry and then normalized to protein concentration. Parallel drug-exposed cultures were used to isolate and purify DNA, which was analyzed for irreversibly bound platinum by flameless atomic absorption spectrometry and normalized to DNA concentration by UV absorption at 260 nm.
Evaluation of Homologous Recombination Repair.
The assay was performed as detailed before (Peng et al., 2009, 2014). Essentially, U20S-DR-GFP cells were transfected, as described above, with SceI plasmid for 24 hours and treated with the drugs, and GFP-positive cells were quantified using flow cytometry 24 hours later.
Half-Life Determination.
The half-life of Rad51 was determined using our reported methodology (Xie et al., 2016). Briefly, drug-treated cells were exposed to 4 μM cycloheximide and then sampled at timed intervals. Cell pellets were washed and lysed with NP40 lysis buffer, and lysates were subjected to immunoblot analysis for Rad51. Band intensity was assessed using the Image J software and normalized to β-actin for loading, and protein half-life was determined by fitting the data to a monoexponential decay equation with a weighting factor of 1/y using GraphPad Prism. The half-life was calculated from the Eq. 0.693/α, in which α is the decay constant.
Ubiquitination Assay.
The assay was performed as we have reported previously but using the cell lysate directly (Xie et al., 2016). Briefly, cells transfected with His-Ubiquitin were treated with control-siRNA or MDM2-siRNA for 48 hours, and MG132 was then added for 6 hours. At this time, cells were pelleted, lysed with NP40 lysis buffer, and immunoprecipitated using anti-Rad51 antibody. Ubiquitinated Rad51 was determined by Western blot analysis using the anti-ubiquitin antibody.
Real-Time Polymerase Chain Reaction.
The assay was performed as described before (Chappell et al., 2015). A2780 cells were treated with control, cisplatin, Nutlin-3, or the combination. After 24 hours, RNA was extracted using RNeasy Mini kit and reverse transcribed to cDNA using High Capacity RNA-to-cDNA kit. Rad51 transcription level was monitored using real-time PCR using the following primers: 5′-TCTCTGGCAGTGATGTCCTGGA-3′ (forward) and 5′-TAAAGGGCGGTGGCACTGTCTA-3′ (reverse).
Analysis of Synergy.
Synergy from drug combination is defined by the combination index (CI) value, which was determined by the Compusyn software following the included detailed procedure. Briefly, fixed concentration ratios based on IC50 values in Table 1 and selected from 0.125, 0.25, 0.5, 1, 2, 4, and 8 × IC50, were used in CI determination, with % cells growth-inhibited in the MTT assay as input for fraction of cells affected by individual drug and the combination. The methodology, together with the rationale/principle of the procedure and determination of CI, has also been reported in the literature (Chou, 2010). The computed CI is based on the equation: in which (C)A and (C)B are the concentrations of Drug A and Drug B used in combination that affect (impact) x% of cells (by MTT), and (Cx)A and (Cx)B are the concentrations of Drug A and Drub B that also affect x% of cells but as single agents. In this analysis, CI value <1 indicates that the drug combination is synergistic. Synergy was also determined using the Bliss model and indicated by a statistically significant difference by t test (see below) between the observed effect and the calculated predicted (or additive) effect, as previously described (Bliss, 1939; Koh et al., 2015). Thus, the predicted effect was determined as follows:
in which (C)A and (C)B are the concentrations of Drug A and Drug B used in combination that affect (impact) x% of cells (by MTT), and (Cx)A and (Cx)B are the concentrations of Drug A and Drub B that also affect x% of cells but as single agents. In this analysis, CI value <1 indicates that the drug combination is synergistic. Synergy was also determined using the Bliss model and indicated by a statistically significant difference by t test (see below) between the observed effect and the calculated predicted (or additive) effect, as previously described (Bliss, 1939; Koh et al., 2015). Thus, the predicted effect was determined as follows: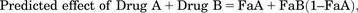 with FaA and FaB representing fraction of cells affected by drug treatment A and drug treatment B, respectively.
with FaA and FaB representing fraction of cells affected by drug treatment A and drug treatment B, respectively.
IC50 and IC50 resistance ratio of cisplatin, Nultin-3, and RITA in A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells
Results are presented as mean ± S.D. of three independent determinations and as ratios of IC50 in resistant cells to IC50 in sensitive (A2780) cells. The values in parentheses indicate the 95% confidence intervals calculated by propagation of error.
Statistical Analysis.
Unless otherwise indicated, differences between groups were evaluated using Student’s t test (two-sided), with P < 0.05 considered as statistically significant. One-way ANOVA with post-hoc Tukey was used for statistical analysis between multiple groups.
Results
Activation of p53 in Cisplatin-Resistant Tumor Cells Induces Apoptosis.
We have recently reported two cisplatin-resistant ovarian cancer models, 2780CP/Cl-16 and 2780CP/Cl-24, where elevated MDM2 and MDM4 binding to basal p53 contributes to their high resistance level (Xie et al., 2016). Thus, we rationalized that dissociation of p53-MDM2-MDM4 complex would activate p53, restore apoptosis, and circumvent cisplatin resistance. To test this, we first used validated MDM2-siRNA (Xie et al., 2016) to demonstrate that direct depletion of MDM2 is sufficient to induce p53 and induce apoptosis in sensitive and resistant cells (Fig. 1A). This indicated that p53 in the three tumor models was regulated by MDM2 and prompted us to examine whether the prototype p53-MDM2 inhibitors Nutlin-3 and RITA would similarly induce p53.

Nutlin-3 activates p53 in both sensitive and resistant cells. (A) Induction of p53 in A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells after knocking down MDM2 was visualized by Western blot analysis (upper), and resultant apoptosis was quantified by flow cytometry using Annexin V and PI double staining (lower). The results are shown as mean ± S.D. of three independent experiments; *P < 0.05. (B) Dose-response curves for Nutlin-3 and RITA were determined by the MTT assay. (C) A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells were treated with 1, 2, and 5 × IC50 of Nutlin-3 or RITA for 24 hours, and p53 and p21 were analyzed by Western blot. Ctrl, control. (D) A2780 cells treated with increasing concentrations of Nutlin-3 or RITA for 24 hours were analyzed for total p53, p21, MDM2 and MDM4, and p53-MDM2-MDM4 complex after p53 immunoprecipitation (IP). (E) Both sensitive and resistant cells were treated with increasing concentration of Nutlin-3 for 24 hours, and the proteins visualized by immunoblot analysis with the indicated antibodies. (F) IC50 of Nutlin-3 in tumor models proficient (+/+) or deficient (−/−) in p53 were determined by MTT assay. The results are shown as mean ± S.D. of three independent experiments.
To ensure use of appropriate concentrations of MDM2 inhibitors, cytotoxicity was first determined by generating sigmoidal dose-response curves, as exemplified in Fig. 1B with A2780 cells. The resulting IC50 values from these curves and corresponding cellular resistance, as indicated by IC50 ratios, are shown in Table 1 for all three cell lines. Nutlin-3 and RITA have potencies in the 1–4 µM range, with 2780CP/Cl-16 and 2780CP/Cl-24 cell lines exhibiting 26- to 27-fold resistance to cisplatin demonstrating only a 3- to 6-fold crossresistance. This suggests some overlap in the mode of action between cisplatin and the MDM2 inhibitors.
Relative Functionalization of p53 by MDM2 Inhibitors.
Equitoxic concentration (1–5 × IC50) was used to examine the ability of Nutlin-3 and RITA to functionalize p53. As shown in Fig. 1C, a 24-hour exposure to Nutlin-3 stabilized and robustly activated p53 in all three cell lines in a concentration-dependent manner, whereas RITA was less effective in activating p53, as seen from a relatively low upregulation of the target p21, especially in 2780CP/Cl-24 cells. To understand this difference, we examined the p53-MDM2-MDM4 complex after p53 immunoprecipitation after exposing A2780 cells to a range of Nutlin-3 and RITA concentrations. The “Input” in Fig. 1D confirms the difference in p53 activation by the two drugs, as indicated by relative upregulation of targets p21 and MDM2. As previously reported, upregulation of MDM2 results in proteasomal degradation of MDM4 (Xie et al., 2017), and this is also seen with Nutlin-3 in a concentration-dependent manner (Fig. 1D). In contrast, low-level increases in MDM2 by RITA had no overt effect on MDM4. More importantly, the immunoprecipitate provides evidence that binding of p53 to both MDM2 and MDM4 is simultaneously reduced after Nutlin-3 treatment, but RITA had no meaningful effect. This strongly indicates that RITA is a weak MDM2 inhibitor and, consistent with recent observations, that this agent may have a different mode of action (Wanzel et al., 2016). Thus, subsequent studies used Nutlin-3 only.
To define relative effectiveness in both sensitive and resistant cells, Nutlin-3 was examined at concentrations of 1, 2, and 5 μM. As seen in Fig. 1E, Nutlin-3 stabilized and activated p53 in all three cell lines, but the effects were attenuated in resistant cells, particularly in 2780CP/Cl-24 cells. This, however, may be due in part to higher IC50 values (lower potency) in resistant cells from crossresistance (Table 1). Interestingly, this low-level crossresistance is independent of p53 since p53 knockout increased IC50 and, therefore, resistance to Nutlin-3 in A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cell lines based on difference (∆) in IC50 between p53+/+ and p53−/− cells (Fig. 1F) to similar extents (∆IC50: 14.50 ± 0.14, 13.19 ± 0.52, and 14.75 ± 1.63, respectively). Thus, p53-dependent activity of Nutlin-3 is similar in all three tumor models, as may be anticipated with a targeted MDM2 inhibitor in tumor models with almost identical genetic background and harboring functional p53.
Activation of p53 in Resistant Cells Augments Cisplatin Cytotoxicity.
We have previously established that lack of p53 activation is a major mechanism of cisplatin resistance in 2780CP/Cl-16 and 2780CP/Cl-24 tumor cells (Xie et al., 2016, 2017; Bhatt et al., 2017). However, activation of p53 after MDM2 depletion (Fig. 1A) or Nutlin-3 treatment (Jiang et al., 2007; Rigatti et al., 2012) does not induce extensive apoptosis. Therefore, we examined Nutlin-3 in combination with DNA damage by cisplatin to enhance cytotoxicity in cisplatin-resistant models. Cisplatin or Nutlin-3 alone at the selected drug concentration induced 20%–40% growth inhibition, but the combination treatment increased inhibition to 60%–80%, which was synergistic (P < 0.05 vs. predicted value) by the Bliss model (Fig. 2A). This cytotoxic synergy was confirmed by the CI of <1 in each cell line (Fig. 2B) and validated by greater apoptosis, as determined by Annexin V staining (Fig. 2C) and immunoblot of cleaved poly (ADP-ribose) polymerase (Fig. 2D).

Combination of cisplatin and Nutlin-3 enhances cisplatin cytotoxicity in both sensitive and resistant cells. (A) Cytotoxicity of cisplatin alone, Nutlin-3 alone, or cisplatin/Nutlin-3 combination in A2780 (cisplatin, 0.1 µM and Nutlin-3, 0.2 µM), 2780CP/Cl-16 (cisplatin, 3 µM and Nutlin-3, 1 µM), and 2780CP/Cl-24 (cisplatin, 3 µM and Nutlin-3, 3 µM) was determined by MTT assay, and synergistic effect was determined. The results are shown as mean ± S.D. of six to nine independent experiments; *P < 0.05 vs. calculated predicted effect by the Bliss model. (B) CI, at the Fa from increasing concentrations of the combination between cisplatin and Nutlin-3, was determined by the Compusyn software. The results are shown as mean ± S.D. of three independent experiments. (C) Apoptotic cells after drug treatment of A2780 (cisplatin, 1 µM and Nutlin-3, 0.5 µM), 2780CP/Cl-16 (cisplatin 5 µM, and Nutlin-3, 2 µM), and 2780CP/Cl-24 (cisplatin, 5 µM and Nutlin-3, 4 µM) cells for 48 hours were determined by flow cytometry using Annexin V and PI double staining. The results are shown as mean ± S.D. of three independent experiments. *P < 0.05 vs. calculated predicted effect by the Bliss model. (D) Sensitive and resistant cells were treated for 24 hours with the drugs at the concentrations indicated in (C), then they were harvested and analyzed by Western blot using poly (ADP-ribose) polymerase antibody (PARP) as an apoptosis marker. (E) A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells exposed to a range of Nutlin-3 concentrations for 24 hours were fixed and subjected to FACS analysis to assess cell cycle distribution. (F) A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells were treated for 24 hours with cisplatin, Nutlin-3, or the combination at the same concentration as in (C) and then analyzed for cell cycle distribution by FACS.
Cell Cycle Effects of Cisplatin/Nutlin-3 Combinations.
Since Nutlin-3 induces G1 arrest (Valentine et al., 2011) and cisplatin is reported to be more effective against cells in G1 phase (Roberts and Fraval, 1980), we explored the potential of G1 arrest by the cisplatin/Nutlin-3 combination as a possible basis for enhanced cytotoxic effect. We first confirmed that Nutlin-3 arrests cells in G1 phase in all three cell lines in a concentration-dependent manner, with higher concentrations also inducing mild G2/M arrest in A2780 and 2780CP/Cl-16 cells (Fig. 2E). However, at the lower concentrations that were used in combination studies in Fig. 2, A, C, and D (A2780, 0.2–0.5 μM; 2780CP/Cl-16, 1 to 2 μM; 2780CP/Cl-24, 3 to 4 μM), the increase in G1 cells by Nutlin-3 alone ranged from minimal to moderate (Fig. 2, E and F). More importantly, cisplatin/Nutlin-3 combinations arrested A2780 and 2780CP/Cl-16 cells in G2/M and not G1 (Fig. 2F). G2/M arrest of 2780CP/Cl-24 cells, in contrast, was relatively mild. Based on earlier studies (He et al., 2013), such increases in cell numbers in G2/M phase are suggestive of an increase in DNA damage with the combination.
Combination Treatment Enhances DNA Damage in Both Sensitive and Resistant Cells.
To decipher the mechanism underlying the higher cytotoxicity from cisplatin/Nutlin-3 combination treatment, A2780 cells were treated with the drugs and 24 hours later analyzed for changes in protein levels by RPPA. The data, as fold change in protein levels versus controls, were then subjected to IPA, which identified the top 20 canonical pathways based on statistical significance (Fig. 3A). The impact of individual treatment is observed as a heat map in Fig. 3A (left panel), with Nutlin-3 generally having a relatively lower effect on each pathway. Although IPA identified several pathways of interest, the greatest effect was associated with “Molecular Mechanisms of Cancer” and “p53 Signaling,” as seen in the bar graph (Fig. 3A, right panel) representing all three treatments. Of the 36 proteins altered in these two pathways, the top 20 that were increased by the combination treatment are shown in Fig. 3B. Substantial increases in p53 are consistent with corresponding increases in p53-target proteins p21, Bax, TP53-inducible glycolysis and apoptosis regulator (TIGAR), and proapoptotic cleaved caspase-7 protein, which is in keeping with increases in apoptosis and apoptosis biomarkers in Fig. 2, C and D. That these enhanced effects are mediated by signals upstream of p53 is suggested by increases in the phosphorylated forms of Chk2 and ataxia telangiectasia mutated, which are DNA damage response proteins known to activate p53 (Meek, 2004, Toledo and Wahl, 2006). This further supports the notion garnered from G2/M data (Fig. 2F) that the combination of cisplatin/Nutlin-3 increases DNA damage. To validate this, we immunoblotted for γH2AX as a biomarker of DNA damage (Mah et al., 2010) and examined p53 activation in parallel. As shown in Fig. 3C with A2780 cells and Supplemental Fig. 1A in resistant cells, cisplatin or Nutlin-3 did indeed induce γH2AX, but γH2AX staining was substantially enhanced by the combination treatment. Similarly, robust activation of p53 with the combination was also evident from upregulation of p21 and MDM2.

Combination of cisplatin and Nutlin-3 enhances DNA damage in both sensitive and resistant cells. (A) A2780 cells treated for 24 hours with cisplatin (1 µM), Nutlin-3 (0.5 µM), or the combination were subjected to RPPA, and the top 20 canonical pathways from IPA using the RPPA data are shown. The heat map depicts −Log10(P value) for each treatment, whereas the bar graph shows −Log10(P value) for all three treatments combined. (B) Top 20 upregulated proteins grouped from the top two canonical pathways in (A) are shown as fold change vs. control and expressed as Log2. (C) A2780 cells were treated with cisplatin, Nutlin-3, or the combination using the same concentration as in Fig. 2C for 24 hours and were collected and analyzed by immunoblot with the indicated antibody. (D) A2780 cells stably transfected with a DNA damage reporter plasmid were treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as in (A) and fixed, and DNA damage foci were imaged using confocal microscopy. (E) DNA damage foci were quantified by Image J software and analyzed for synergy. The results are shown as mean ± S.D. of six independent experiments; *P < 0.05 vs. calculated predicted effect by the Bliss model. (F) A2780, 2780CP/Cl-16, and 2780CP/Cl-24 cells exposed to cisplatin (Cis-Pt) alone or in combination with Nutlin-3 (Combo) were collected, and intracellular cisplatin-induced DNA adducts were measured by atomic absorption. The results are shown as mean ± S.D. of three independent experiments. Akt, protein kinase B; AMPK, AMP-activated protein kinase; ATM, ataxia telangiectasia mutated; Chk2, checkpoint kinase 2; FAK, focal adhesion kinase; IL, interleukin; ILK, integrin-linked kinase; PCNA, proliferating cell nuclear antigen; PI3K, phosphoinositide-3-kinase; TIGAR, TP53-inducible glycolysis and apoptosis regulator.
For further evidence of enhanced DNA damage with the combination treatment, transfection of a truncated-53BP1 double-strand break reporter plasmid that localizes to γH2AX sites to form DNA damage foci (Yang et al., 2015) was explored. As shown in Fig. 3D and Supplemental Fig. 1, B and C, with quantification in Fig. 3E, synergistic increases in DNA damage foci by the Bliss model were indeed observed in A2780 and resistant cells with the combination treatment. Notably, this enhanced DNA damage with the combination treatment was not due to greater cisplatin accumulation (Supplemental Fig. 2A) or formation of cisplatin-induced DNA adducts (Fig. 3F) through possible interaction with Nutlin-3.
DNA Repair Is Inhibited by Combination Treatment.
Since cisplatin accumulation and resultant DNA adducts were not increased by Nutlin-3, it was reasonable to consider whether the increase in DNA damage by the combination was an apparent effect from reduced DNA repair. Therefore, we first examined rate of recovery from DNA damage by monitoring γH2AX, which is also a sensitive biomarker for DNA repair (Mah et al., 2010). Cells treated with drugs for 24 hours were recultured in fresh medium for another 24 hours and then harvested at different time points for immunoblot analysis of γH2AX. The immunoblot and densitometric quantification of γH2AX band clearly demonstrate that combination treatment resulted in a statistically significant increase in γH2AX protein half-life and, therefore, a reduction in DNA damage repair compared with cisplatin alone (Fig. 4A). To consolidate this observation, DNA damage foci were monitored using the truncated-53BP1 reporter plasmid to similarly assess repair. Cell imaging and bar graph in Fig. 4B indicate that, between 24 and 36 hours after initiating treatment, over twice as many cells treated with cisplatin alone repaired their DNA and became foci-negative compared with combination treatment; that is, DNA damage repair was over 2-fold slower with the combination treatment.

DNA damage repair is inhibited by cisplatin/Nutlin-3 combination. (A) A2780 cells were treated with cisplatin or the combination for 24 hours using the same concentrations as in Fig. 3A, and then they were exposed to fresh media and sampled at the indicated time points. γH2AX staining was monitored by immunoblot, and density of band was quantified by Image J software. A representative immunoblot is shown. The plot of DNA damage repair against time, however, is based on γH2AX densitometry data from three independent experiments. For valid comparisons, the plots were drawn using the long exposure for cisplatin and short exposure for the combination to ensure comparable exposure at zero time. The results are shown as mean ± S.D. *P < 0.05 vs. cisplatin. (B) A2780 cells expressing the exogenous DNA damage reporter were treated with cisplatin (Cis-Pt) or the combination (Combo) for 24 hours using the same concentrations as in Fig. 3A and then exposed to fresh media for another 12 hours. Cells were fixed, and DNA damage foci in 10 fields were visualized by confocal microscopy and quantified using Image J software. The data in the bar plot represent the change in foci-positive cells between 24 and 36 hours as a % of the 24-hour value. The results are shown as mean ± S.D. of three independent experiments. Errors were calculated by “propagation of error.” *P < 0.05 vs. cisplatin. (C) Canonical DNA damage repair pathways were analyzed by IPA using the RPPA data. BER, base-excision repair; MMR, mismatch repair; NER, nucleotide excision repair; NHEJ, nonhomologous end joining. (D) U2OS cells stably expressing DR-GFP were transfected with 2 µg of I-SceI plasmid, and 24 hours later they were treated with cisplatin (7.5 µM), Nutlin-3 (0.5 µM), or the combination for another 24 hours when GFP-positive cells were quantified by flow cytometry. The results are shown as mean ± S.D. of three independent experiments. *P < 0.05 vs. each drug. (E) U2OS cells were treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as in (D), harvested, and analyzed using immunoblot with indicated antibodies. (F) Cytotoxicity of cisplatin (0.5 µM), Nutlin-3 (0.5 µM), or the combination was determined in U2OS cells by MTT assay, and synergistic effects were determined by the Bliss model. The results are shown as mean ± S.D. of six independent experiments; *P < 0.05 vs. predicted effect.
Homologous Recombination Is Impaired by Combination Treatment.
A number of pathways are implicated in the repair of DNA damage (Hakem, 2008). Therefore, we used IPA to probe RPPA data to identify any repair pathway(s) that may be impaired after treatment of tumor cells with the cisplatin/Nutlin-3 combination. Of the five possible repair pathways, which were implicated by changes in associated proteins shown in Supplemental Fig. 2B, homologous recombination (HR) and mismatch repair in eukaryote pathways ranked high based on −Log(P value) with combination treatment rather than cisplatin or Nutlin-3 alone (Fig. 4C). Since the mismatch repair pathway, which is more important for engaging cisplatin-induced cell death machinery rather than the repair of DNA adducts (Chaney et al., 2005), is already downregulated in resistant 2780CP/Cl-16 and 2780CP/Cl-24 cells (Sumiyoshi et al., 2003), it cannot be responsible for the synergistic effect of the combination treatment in resistant cells (e.g., Figs. 2A and 3E). Thus, it is likely that the HR pathway is the major contributor of this synergy. To validate this, a specific DR-GFP system to test homologous recombination function in U2OS osteosarcoma cells was explored (Peng et al., 2009, 2014). As shown in Fig. 4D, cisplatin or Nutlin-3 alone inhibited homologous recombination to a similar level, but the combination treatment increased this inhibition compared with single drug treatment (P < 0.05). It is noteworthy that U2OS cells, like ovarian tumor cells, respond with greater increases in γH2AX, p53, p53-associated proteins, and synergistic growth inhibition (Fig. 4, E and F) with combination drug treatment than individual drugs.
Combination of Cisplatin and Nutlin-3 Downregulates Rad51.
With enhanced downregulation of HR by the cisplatin/Nutlin-3 combination treatment, it becomes important to elucidate the mechanism at the molecular level. Intuitively, downregulation of a critical component of the HR pathway would be anticipated. Since RAD51 is reportedly reduced by Nutlin-3 (Ireno et al., 2014) and is also the major component of the enzymatic HR complex (Wang et al., 2005), we examined this protein by immunoblot in A2780 cells. Figure 5A provides evidence that Rad51 is indeed severely downregulated 24 hours after combination treatment. Nutlin-3 had a relatively less intense effect, but, in contrast, cisplatin demonstrated a slight increase in Rad51 levels (Fig. 5A, left panel). Similar results were also observed in resistant cells (Supplemental Fig. 2D). The results from A2780 cells are also presented as a bar graph, which demonstrates significant difference between experimental and predicted Rad51 levels after combination treatment (Fig. 5A, right panel). Substantial downregulation of Rad51 by the combination treatment relative to single agent was also observed in U2OS cells (Supplemental Fig. 2C).

Combination treatment downregulates Rad51. (A) A2780 cells were treated for 24 hours with cisplatin, Nutlin-3, or the combination using the same concentration as indicated above in Fig. 2C. Cells were then collected, and Rad51 expression was analyzed by immunoblot using short and long exposure conditions. A representative immunoblot from three independent studies is shown (left panel). However, quantification of band density by Image J software was computed for all three experiments and is presented as a bar graph (right panel). The results are shown as mean ± S.D. *P < 0.05 vs. the calculated predicted effect by the Bliss model. (B) A2780 cells were treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as in Fig. 3A, with DMSO or MG132 (5 µM) added during the final 6 hours. Rad51 expression was determined by immunoblot. (C) A2780 cells were treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as indicated above in Fig. 3A, and cycloheximide (4 µM) was then added. Cells were sampled immediately and at the indicated time points, and Rad51 analyzed by immunoblot. A representative immunoblot is shown. (D) Rad51 half-life was determined from the plot of the densitometry data from (C) obtained using the Image J software. The results are shown as mean ± S.D. of three independent experiments. *P < 0.05 vs. each drug. (E) A2780 cells treated with cisplatin, Nutlin-3, or combination for 24 hours using the same concentration as indicated above in Fig. 3A were collected and subjected to immunoprecipitation using the Rad51 antibody. Coimmunoprecipitation of MDM2 was then analyzed by immunoblot using short and long exposure conditions. (F) A2780 cells transfected with control- or MDM2-siRNA for 48 hours were treated with cycloheximide (4 µM) and then sampled at different time points. Rad51 expression was analyzed by immunoblot. (G) Rad51 half-life was obtained from a plot of band density obtained from (F) using Image J software. (H) A2780 cells transfected with 5 µg of His-Ubiquitin plasmid for 24 hours were then treated with control- or MDM2-siRNA for another 48 hours. MG132 (5 µM) was added at this time for 6 hours. Cells were then harvested and analyzed using immunoblot with the indicated antibodies. Ub, ubiquitinated. (I) A2780 cells treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as in Fig. 3A were then sampled and analyzed by immunoblot using the indicated antibodies. (J) Correlation of MDM2 and MDM2-pS166 based on the densitometry data from (I). The values shown are densitometric units expressed as fold change relative to control. (K) Correlation between MDM2-pS166 and RAD51 from TCPA data base using information from the category “MDACC all level four cell lines” representing a variety of tumor types. The values shown are expressed as Log 2.
Combination Treatment Promotes MDM2-Mediated Rad51 Degradation.
Demonstration of downregulation of Rad51 is consistent with reduction in HR activity and may be a result of increased proteasomal degradation, as observed in the unrelated context of anchorage independence (Wang et al., 2005). Therefore, we examined this possibility by immunoblotting Rad51 after addition of the proteasome inhibitor MG132 during the final 6 hours of incubation. In absence of MG132, Nutlin-3 and, to a greater extent, cisplatin/Nutlin-3 combination downregulated Rad51, but in the presence of MG132 loss of Rad51 was attenuated, confirming degradation of Rad51 as a mechanism (Fig. 5B). In support of this, we next determined the half-life of Rad51 in A2780 cells after drug treatments for 24 hours. Cells were then exposed to cycloheximide, and samples were collected immediately and over time processed for immunoblot analysis and densitometric quantification. As shown by a representative immunoblot in Fig. 5C and the quantified data from independent experiments in Fig. 5D, cisplatin treatment alone increased Rad51 half-life compared with control, which is consistent with the increase in Rad51 levels seen in Fig. 5A. However, Rad51 half-life decreased slightly with Nutlin-3 treatment but decreased substantially with the combination treatment, and this is consistent with a more rapid reduction in Rad51 levels with the combination as compared with Nutlin-3 alone.
Considering MDM2 is an E3 ligase (Xie et al., 2016) that is elevated by cisplatin/Nultin-3 (Fig. 3C), we hypothesized that degradation of Rad51 with the combination treatment is MDM2-dependent. For this previously untested hypothesis, we first examined the interaction between MDM2 and Rad51. As shown in Fig. 5E, MDM2 binding to Rad51 increased slightly with cisplatin or Nutlin-3 but was elevated robustly after combination treatment. Next, we knocked down MDM2 and demonstrated an increase in Rad51 half-life (Fig. 5, F and G) and, conversely, decrease in Rad51 ubiquitination (Fig. 5H). Taken together, these data strongly indicate for the first time that the combination of cisplatin/Nutlin-3 downregulates Rad51, likely through MDM2-mediated proteasomal degradation.
We failed to find a human cancer data base to test the apparent inverse relationship between MDM2 and Rad51 for clinical relevance. However, MDM2 is represented as phospho-S166-MDM2 in the RPPA analysis (Fig. 3B) and also in The Cancer Proteome Atlas (TCPA) data base of human tumor cell lines (https://tcpaportal.org/tcpa/). To test phospho-S166-MDM2 as a possible surrogate for MDM2, it was first important to establish correlation between the two. Therefore, densitometry was used to quantify MDM2 and phospho-MDM2 bands in immunoblots from A2780 cells treated with cisplatin, Nutlin-3, or the combination (Fig. 5I), and the data were plotted. The plot in Fig. 5J demonstrates a strong direct relationship, and this validated the use of phospho-MDM2 as a surrogate for MDM2 to analyze TCPA data. As anticipated, levels of phospho-MDM2 correlated inversely with Rad51 across multiple cell lines representing various disease types (Fig. 5K) and specifically ovarian cancer (Supplemental Fig. 2E). Although the correlation is weak, primarily because of the heterogenous nature of the cell lines, with each cell line likely expressing an independent profile of genetic defects, these findings, nevertheless, support our conclusion that increases in MDM2 induce degradation of Rad51.
Rad51 Is Repressed by Cisplatin/Nutlin-3 Combination in a p53-Dependent Manner.
Since the data with the proteasomal inhibitor MG132 indicated incomplete recovery of Rad51 (see Fig. 5B), it implicates an additional mechanism for Rad51 downregulation. Therefore, transcriptional inhibition was also examined by real-time PCR after drug treatments. As shown in Fig. 6A, and as is supported by ANOVA/post-hoc statistical analysis (Supplemental Table 1), Rad51 mRNA level was reduced by Nutlin-3 but more robustly and synergistically reduced by the combination treatment. Cisplatin alone, on the other hand, had no effect. To probe the molecular mechanism underlying reduction in Rad51 transcripts, we reanalyzed the RPPA/IPA data by identifying the top 10 proteins that were increased or decreased by the cisplatin/Nutlin-3 combination and compared levels of these proteins to those after single drug treatments. The data are presented as a heat map in Fig. 6B, which reproduces some of the changes observed in Fig. 3B but in addition identifies proteins that were strongly decreased. Interestingly, of the 10 proteins that were reduced, hypoxia-inducible factor 1α, Chk1, polo-like kinase 1, platelet-derived growth factor receptor β, and androgen receptor are reported as targets of p53 transrepression (Riley et al., 2008; Beckerman and Prives, 2010). Therefore, the possibility of p53-dependent downregulation of Rad51 was examined in p53 knockout A2780 cells. The data in p53+/+ cells (Fig. 6C; Supplemental Table 2) were similar to those in parental A2780 cells (Fig. 6A; Supplemental Table 1), but importantly, decreases in Rad51 were prevented in p53 knockout cells (Fig. 6C). Therefore, it is evident that Rad51 is transrepressed in a p53-dependent manner.

Combination of cisplatin/Nutlin-3 induces p53-dependent transrepression of Rad51. (A) A2780 cells treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as in Fig. 3A were used to extract RNA. Samples were then reverse transcribed to cDNA, and Rad51 mRNA levels were determined by real-time PCR and analyzed by one-way ANOVA with Tukey as post-hoc. *P < 0.05 vs. all other groups; #P < 0.05 vs. all other groups and also (by t test) the calculated predicted effect by the Bliss model. (B) Top 10 upregulated and top 10 downregulated proteins from RPPA analysis are shown, based in the order from highest-to-lowest expression with the combination treatment. The data in the heat map are shown as fold change vs. control and expressed as Log 2. (C) A2780 p53-proficient (+/+) and -deficient (−/−) cells were treated with cisplatin, Nutlin-3, or the combination for 24 hours using the same concentration as in Fig. 3A, and Rad51 mRNA level was then analyzed by real-time PCR followed by one-way ANOVA with Tukey as post-hoc. *P < 0.05 vs. all other groups; #P < 0.05 vs. all other groups and also by (t test) the calculated predicted effect by the Bliss model. (D) Scheme for the disruption of homologous recombination repair of cisplatin-mediated DSBs when combined with Nutlin-3. The lighter lines and text indicate lower effects, and the dashed lines indicate potential effects, whereas the bold lines and text indicate enhanced effects. See text for additional details. AR, androgen receptor; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; Chk2, checkpoint kinase 2; FAK, focal adhesion kinase; Hif, hypoxia-inducible factor; NDRG1, N-myc downregulated 1; PAR, poly (ADP-ribose); PDGFR, platelet-derived growth factor receptor; PLK, polo-like kinase; Stat3, signal transducer and activator of transcription 3.
Discussion
Acquired resistance to cisplatin and resultant tumor progression leads to a high mortality rate in a number of cancers (Siddik, 2003; Martinez-Rivera and Siddik, 2012). Therefore, overcoming cisplatin resistance with new strategies is of great clinical significance. Previously, we have demonstrated that an increased stoichiometric and concurrent binding of MDM2 and MDM4 to p53, in absence of overexpression, contributes to substantial resistance in 2780CP/Cl-16 and 2780CP/Cl-24 tumor models (Xie et al., 2016). We have demonstrated here that Nutlin-3 efficiently disrupted p53-MDM2 interaction and activated p53 in both sensitive and resistant cells more effectively than RITA. Notably, Nutlin-3 exposure also resulted in disruption of the p53-MDM4 interaction. Moreover, the combination of cisplatin and Nutlin-3 substantially enhanced cytotoxicity by an increase in DNA damage, which is an apparent effect mediated by severe inhibition of HR activity through downregulation of Rad51.
Previous reports of elevated DNA damage with Nutlin-3 combinations with cytotoxic agents (Verma et al., 2010; Rigatti et al., 2012; Deben et al., 2015) corroborate our data. Similarly, elevated G2/M phase arrest with the combination treatment observed by us in this study and others (Deben et al., 2015) is again consistent with enhanced DNA damage, and this may possibly be due to substantial induction in p21, which may then inhibit the normally off-target cyclin-dependent kinase 1 (Harper et al., 1995; He et al., 2011). However, to our knowledge, this is the first report that enhanced DNA damage with cisplatin/Nutlin-3 combination is an apparent effect due to p53-dependent reduction in DNA repair. Repair of cisplatin DNA adducts involves several pathways, including HR, nonhomologous end joining, and nucleotide excision repair (Siddik, 2003). In the present study, the canonical HR pathway was identified as the target of severe inhibition by the combination treatment and its contribution to synergistic cytotoxicity with the cisplatin/Nutlin-3 combination. This is consistent with reports of clinical hypersensitivity to cisplatin of ovarian cancers harboring compromised HR activity (Wang et al., 2011; da Cunha Colombo Bonadio et al., 2018). Thus, combination of cisplatin with targeted MDM2 inhibition could be used effectively to enhance clinical response against cancers proficient in HR activity.
The HR pathway requires a number of proteins to affect DNA repair, but Rad51 appears to be the key regulator (Lazaro-Trueba et al., 2006; Chappell et al., 2015). Indeed, high levels of Rad51 can lead to genomic instability and contribute to cancer drug resistance (Wang et al., 2005; Hine et al., 2008). Conversely, downregulation of Rad51 by the cisplatin/Nutlin-3 combination in both sensitive and resistant cells is consistent with enhanced cytotoxicity in the present study and in accord with reports that targeting Rad51 directly overcomes drug resistance (Hine et al., 2008). Although downregulation of Rad51 with Nutlin-3 alone in our study has also been noted previously (Ireno et al., 2014), the underlying mechanism has remained unknown. We have now established that MDM2 upregulated in a p53-dependent manner can ubiquitinate Rad51 for its proteasomal degradation, which notably is accelerated with cisplatin/Nutlin-3 combination. This, however, is not surprising since MDM2 is a well-known E3 ligase that not only degrades p53 but also a number of other proteins (Toledo and Wahl, 2007; Nag et al., 2014; Urso et al., 2016). Interestingly, although cisplatin also induced MDM2, degradation of Rad51 was not observed. A possible explanation is a threshold effect, with low expression levels of MDM2 by cisplatin perhaps being inefficient at inducing degradation of Rad51, whereas enhanced upregulation of MDM2 with combination treatment results in facile degradation. Declines in general DNA repair after a robust increase in MDM2 by Nutlin-3 have been previously noted, but this was observed using conditions involving high nonphysiologic Nutlin-3 concentrations in p53-inactive cells (Carrillo et al., 2015). Although the mechanisms for these reported observations were not investigated, it is tempting to speculate that MDM2-dependent degradation of Rad51 may have been involved.
Downregulation of Rad51 by Nutlin-3 or cisplatin/Nutlin-3 combination was also observed at the transcriptional level in a p53-dependent manner, which is consistent with reports that Rad51 is a transrepression target of p53 (Lazaro-Trueba et al., 2006; Hine et al., 2008). In the present study, we also observed repression of other p53 targets: specifically, hypoxia-inducible factor 1α, Chk1, polo-like kinase 1, platelet-derived growth factor receptor β, and androgen receptor. Since cisplatin in fact modestly induced Rad51, it is likely that transrepression of Rad51 by p53 may also be threshold-dependent, and this is supported by reports of a switch in p53 function when its activation is substantially elevated (El Deiry, 2003; Meek, 2004; Helton and Chen, 2007; Luo et al., 2017). Based on these reports, it is possible to propose that a lower level or duration of p53 induction is likely to upregulate specific target genes that induce cell cycle arrest and repair, which, together with MDM2-dependent feedback inhibition of p53, attenuate cell killing. However, when this feedback inhibition of p53 by MDM2 is prevented by Nutlin-3, the resultant elevated and sustained increase in p53 then modulates additional genes to enhance cell killing.
Although in our study cisplatin did not induce degradation or transcriptional repression of Rad51, it was still able to reduce HR activity by about 50%. This strongly suggests that other mechanisms are likely involved. HR is composed of a number of proteins, and downregulation of any one of those, including Chk1, may inhibit HR (McCabe et al., 2006). Since Chk1 was downregulated by cisplatin in the present study, it is logical that the observed inhibition in HR by the platinum drug could be attributable in part to such an effect. Similarly, enhanced downregulation of Chk1 by the cisplatin/Nutlin-3 combination may also serve to augment HR inhibition.
In conclusion, we have demonstrated that Nutlin-3 can synergize cisplatin cytotoxicity in sensitive A2780 cells and resistant 2780CP/Cl-16 and A2780/Cl-24 tumor cells through inhibition of p53-MDM2 interaction. The mechanism is visualized schematically in Fig. 6D, in which cisplatin-induced DNA double-strand breaks (DSBs) are normally repaired by HR, and this leads to a moderate DNA damage effect, as seen from limited increases in γH2AX, p53 activation, transactivation of p21 and MDM2, G2/M arrest, and apoptosis. The combination of cisplatin with Nutlin-3, on the other hand, enhances levels of p53, which then inhibits HR through bimodal downregulation of Rad51 both directly (via transrepression) and indirectly (via MDM2-dependent proteasomal degradation). This then results in increased and/or sustained levels of unrepaired DSBs (that is, an apparent increase in DNA damage compared with controls), leading to further increases in γH2AX, p53 phosphorylation and stabilization, enhanced p53-dependent transactivation, robust G2/M arrest, and eventual increase in apoptosis. It is highly plausible that other MDM2 inhibitors undergoing clinical investigations may work similarly in concert with cisplatin to inhibit HR and provide greater benefit in the treatment of tumors that are proficient in this repair pathway.
Authorship Contributions
Participated in research design: Xie, He, Siddik.
Conducted experiments: Xie, He.
Performed data analysis: Xie, He, Siddik.
Wrote or contributed to the writing of the manuscript: Xie, He, Siddik.
Footnotes
- Received June 11, 2019.
- Accepted January 15, 2020.
The research support from the U.S. Public Health Service Grants CA211975 to Z.H.S. and Support Grant CA16672 to MD Anderson Cancer Center awarded by the National Cancer Institute, and in part from the Megan McBride Franz Endowed Research Fund, is gratefully acknowledged.
The authors declare no conflicts of interest.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- 53BP1
- p53-binding protein 1
- 53BP1trunc
- truncated form of p53-binding protein 1
- Chk1
- checkpoint kinase 1
- CI
- combination index
- DR
- damage recognition
- DSB
- double-strand break
- Fa
- fraction of cells affected by the drug treatment
- FACS
- fluorescence-activated cell sorting
- HR
- homologous recombination
- IPA
- Ingenuity Pathway Analysis
- MDM2
- mouse double minute 2 homolog
- MDM4
- mouse double minute 4 homolog
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- PCR
- polymerase chain reaction
- PI
- propidium iodide
- RO5963
- (Z)-2-(4-((6-Chloro-7-methyl-1H-indol-3-yl)methylene)-2,5-dioxoimidazolidin-1-yl)-2-(3,4-difluorophenyl)-N-(1,3-dihydroxypropan-2-yl)acetamide
- RITA
- reactivation of p53 and induction of tumor cell apoptosis
- RPPA
- reverse phase protein array
- siRNA
- small interfering RNA
- SJ-172550
- 2-[2-chloro-4-[(1,5-dihydro-3-methyl-5-oxo-1-phenyl-4H-pyrazol-4-ylidene)methyl]-6-ethoxyphenoxy]-acetic acid methyl ester
- TCPA
- The Cancer Proteome Atlas
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}