


Abstract
G protein–coupled receptors (GPCRs) are the largest class of transmembrane receptors and serve as signal mediators to transduce information from extracellular signals such as neurotransmitters, hormones, or drugs to cellular responses. They are exposed to the strong electrical field of the plasma membrane. In the last decade voltage modulation of ligand-induced GPCR activity has been reported for several GPCRs. Using Foerster resonance energy transfer–based biosensors in patch clamp experiments, we discovered a robust voltage dependence of the thromboxane receptor (TP receptor) on the receptor level as well as on downstream signaling. TP receptor activity doubled upon depolarization from −90 to +60 mV in the presence of U46619, a stable analog of prostaglandin H2. Half-maximal effective potential (V0.5) determined for TP receptor was −46 mV, which is within the physiologic range. We identified that depolarization affected the agonist affinity for the TP receptor. Depolarization enhanced responses of several structural analogs of U46619 with modifications to a similar extent all around the molecule, indicating that voltage modulates the general conformation of TP receptor. By means of site direct mutagenesis, we identified TP receptor R2957.40, which showed alteration of voltage sensitivity of TP receptor upon mutation. Voltage sensitivity was not limited to TP receptor because prostaglandin F receptor activated with U46619 and prostaglandin E2 receptor subtype 3 activated with iloprost showed a similar reaction to depolarization as TP receptor. However, prostacyclin receptor activated with iloprost showed no detectable voltage dependence.
SIGNIFICANCE STATEMENT Prostanoids mediate many of their physiological effects via transmembrane receptors expressed in the plasma membrane of excitable cells. We found that agonist-mediated activation of prostaglandin F receptors and prostaglandin E2 receptors as well as thromboxane receptors are activated upon depolarization, whereas prostacyclin receptors are not. The voltage-induced modulation of thromboxane receptor activity was observed on the level of receptor conformation and downstream signaling. The range of voltage dependence was restricted by R2957.40 in the agonist-binding pocket.
Introduction
G protein–coupled receptors (GPCRs) constitute the largest family of integral membrane proteins with over 800 members encoded in the human genome. They are targets for many approved drugs as well as for new drugs under development (Hauser et al., 2017; Sriram and Insel, 2018). All membrane proteins are located in a strong electrical field (Yang and Brackenbury, 2013). Therefore, it was a striking finding when first published that the M2-muscarinic receptor, a class A GPCR, showed voltage dependence (Ben-Chaim et al., 2003). Since this first discovery at the M2-receptor, a number of GPCRs have been investigated under this aspect (reviewed in: Vickery et al., 2016). The majority of investigated GPCRs showed voltage dependence and the effect that voltage had on GPCR activity was ligand-specific (Navarro-Polanco et al., 2011; Sahlholm et al., 2011; Rinne et al., 2013, 2015; Birk et al., 2015; Moreno-Galindo et al., 2016). Voltage was able to alter either affinity, efficacy, or both (Rinne et al., 2013; Birk et al., 2015). In contrast, in the absence of ligands, no modulation of GPCR activity was observed. Point mutation studies supported this observation as the mutation of amino acids, which are involved in the ligand binding process, could alter the voltage dependence of GPCRs (Rinne et al., 2015; Barchad-Avitzur et al., 2016; Hoppe et al., 2018). Taken together, this indicates that observed voltage effects act on receptor-ligand interaction. Not much is known about the mechanism behind this voltage sensing process, and a voltage sensing domain similar to voltage-gated ion channels has not yet been found. For the M2-receptor, a gating charge was directly observed (Ben-Chaim et al., 2006). With around 0.8 elementary charges, this charge movement was rather small compared with voltage-gated ion channels. Nevertheless, modulation of ligand-induced GPCR activity might have relevance in the understanding of physiology, pathophysiology, and potential use for pharmacology. Voltage dependence of GPCRs can have a potential role in excitable cells, which undergo fast changes in the electrical field during action potentials, e.g., smooth muscle, cardiac, and neuronal cells. This role might not be limited to excitable cells because studies have shown changes in membrane potentials (VM) over time in cells during the cell cycle and in cancer cells and different VM of various differentiated cells (Arcangeli et al., 1995; Yang and Brackenbury, 2013). One group of GPCRs with almost ubiquitous expression are prostanoid receptors. They belong to the lipid-receptor group of class A GPCRs and are comprised of nine members: prostacyclin receptor (IP receptor), prostaglandin F receptor (FP receptor), prostaglandin D2 receptor subtypes 1 and 2, prostaglandin E2 (PGE2) receptor subtypes 1–4 (EP1, EP2, EP3, EP4 receptors) and the thromboxane receptor (TP receptor). The prostanoids are locally generated by cyclooxygenases from arachidonic acid and have a very limited lifetime. Besides the prostaglandins, there are so-called isoprostanes, which are produced by nonenzymatic free radical-catalyzed peroxidation of arachidonic acid under conditions of oxidative stress. Isoprostanes are also able to activate certain prostanoid receptors and have an increased lifetime (Milne et al., 2014). Prostanoid receptors fulfill a variety of physiologic and pathophysiological functions. These widespread functions are also reflected in diseases in which they are targeted. TP receptor is inhibited indirectly by aspirin, e.g., for secondary prevention of heart attacks and directly by Seratrodast, which is used to treat asthma. In pulmonary hypertension, IP receptor is targeted by different IP receptor agonists such as iloprost. FP receptor is targeted by Latanoprost in glaucoma therapy and by Cloprostenol for luteolysis (Coleman et al., 1994). Recently, prostanoid receptors received extensive attention in the field of cancer research (reviewed in: (Pannunzio and Coluccia, 2018; Wang and DuBois, 2018; Zmigrodzka et al., 2018; Hashemi Goradel et al., 2019; Karpisheh et al., 2019). In a previous study, Ca2+-levels in megakaryocytes induced with U46619, a stable analog of prostaglandin H2, have been measured at different holding potentials, suggesting activation of endogenous TP receptors upon depolarization (Martinez-Pinna et al., 2005). Because prostanoid receptors are also important pain sensitizers and reside on excitable cells such as neurons, we decided to set out and investigate this important receptor group under this aspect using combined FRET and patch clamp techniques. This combination of methods enabled us to perform direct and indirect measurements of receptor activity at controlled VM, which provided us with insight into voltage effects. We started out to investigate the voltage dependence of prostanoid receptors, focusing on the thromboxane receptor.
Materials and Methods
Plasmids and Agonist
cDNAs for human TP receptor wt (henceforth referring to the α-isoform), mouse Gα13-mTurq2 (Bodmann et al., 2017); human Gβ1 wt, bovine Gγ2 wt (Bünemann et al., 2003); human Gβ1-Cer (Frank et al., 2005); mouse Gαq wt, mouse Gαq-YFP receptor (Hughes et al., 2001); exchange protein directly activated by cAMP (Epac)1-camps (cAMP sensor) (Nikolaev et al., 2004); human GRK2 wt (Winstel et al., 1996); and human GRK2-mTurq2 (Wolters et al., 2015) have been described previously. We used mouse Gα13 (NM_010303.3, cDNA from N. Wettschureck, Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany), Human FP receptor (prostaglandin F receptor, AY337000, cat. no. #PTGFR00000) and IP receptor (prostaglandin I2 [prostacyclin, AY242134, cat. no. #PTGIR00000] receptor). cDNA was obtained from the Missouri S&T cDNA Resource Center (http://www.cdna.org). Mutations were introduced into TP receptor wt by site directed mutagenesis and were verified by sequencing.
The following mutagenesis primers were used: TP receptor R295A 5′-aggagctgctcatct-acttggctgtggccacctggaaccagat-3′; TP receptor R295K 5′-gctgctcatctacttgaaagtggccacctggaacc-3′; TP receptor D193E forward: 5′-ggcgccgagtccggggaagtggccttcgggctg-3′, reverse: 5′- cagcccgaaggccacttccccggactc-ggcgcc -3′; TP receptor S201T 5′- ttcgggctgctcttcaccatgctgggcggcctc -3′; TP receptor S255T 5′-gatcat-ggtggtggccaccgtgtgttggctgccc -3′; TP receptor W299L 5′- ttgcgcgtggccaccttgaaccagatcctggac -3′. eYFP-p115 was cloned by inserting p115, which was a kind gift from Thomas Worzfeld (Pharmacological Institute, University of Marburg, Germany), into pcDNA3 backbone vector expressing an N-terminal eYFP, using restriction sites AgeI and NotI. TP receptor-mVenus was cloned from TP receptor (Bodmann et al., 2017) by inserting TP receptor into pcDNA3-mVenus backbone vector using HindIII and BamHI restriction sites. TP receptor-eYFP was cloned by replacing mVenus of TP receptor-mVenus with eYFP from Gα13-eYFP (Bodmann et al., 2017) using BamHI and XhoI restriction sites. A FRET-based TP receptor-sensor was generated by exchanging the C-terminal eYFP of TP receptor-eYFP with mTurq2 of Gα13-mTurq2 (Bodmann et al., 2017), using the restriction sites BamHI and XhoI. Next, the BamHI restriction site was eliminated by PCR using a mutagenesis primer: 5′-ctccgggctgcaggggtccatggtgagcaag-3′. Subsequently, we inserted a BamHI restriction site, a spacer consisting of the nucleotides GGGGGG and a NheI restriction site between A232 and Q233. This was done by PCR using a mutagenesis primer 5′-gggcaggaggcg-gccggatccgggggggctagccagcagcgtccccgg-3′. Finally, we inserted eYFP, which was amplified from eYFP-p115 with flanking BamHI and NheI sites, between A232 and Q233 using BamHI and NheI restriction sites. mCherry-IP receptor was cloned by inserting mCherry N-terminally into IP receptor-pcDNA3-backbone vector using BamHI and EcoRI restrictions sites. The gene for isoform 1 of EP3 receptor (prostaglandin E2 receptor subtype 3, henceforth referring to isoform 1, Genbank accession: L27490.1) optimized for Homo sapiens codon usage table, was synthesized by Eurofins genomics (Eurofins, Genomics Germany GmbH, Ebersberg). EP3 receptor arrived in pEX-A128 vector and was cloned into pcDNA3 using HindIII and XbaI restriction sites. Mutation D124R was introduced into EP3 receptor wt analogously to the procedure used for TP receptor. The mutagenesis primer used had the sequence 5′-agatgggagcacatccgccccagcggcagactg-3′.
In this study we used U46619 (16450), 8-iso prostaglandin E1 (13360), 8-iso prostaglandin E2 (14350), I-BOP (19600), 15-keto prostaglandin E2 (14720), 15(S)-15-methyl prostaglandin E2 (14730), prostaglandin E2 ethanolamide (14012), prostaglandin E2 methyl ester (14011), and iloprost (18215). The manufacturer was Cayman Chemical, Ann Arbor, MI. The preparation and solution steps were carried out analogously as described before (Bodmann et al., 2017).
The Gαq inhibitor FR900359 was a kind gift from Dr. Evi Kostenis, University of Bonn, Germany.
Cell Culture and Transfections
All experiments in this study were carried out with transiently or stably transfected HEK293 cells. Cells were cultured using standard conditions (Rinne et al., 2015). To investigate TP receptor-sensor, a stable cell line was generated by transfecting HEK293 with 1 µg of TP receptor-sensor plasmid cDNA (in a dish with 6 cm ∅) and subsequently culturing the cells under selection with G418. Generation of a stable cell line expressing TP receptor-eYFP construct was done analogously. Cells were transfected with Effectene reagent (Qiagen) according to the manufacturer’s instructions using the following cDNAs (per dish with 6 cm ∅).
Transfections into HEK293T Cells
TP receptor induced Gα13-p115-interaction assay: TP receptor wt or mutant receptor (0.5 µg), Gα13-mTurq2 (0.8 µg), Gβ1 wt (0.5 µg), Gγ2 wt (0.2 µg), YFP-p115 (1 µg); FP receptor induced G protein activation: FP receptor (0.5 µg), Gαq-YFP (1.5 µg), Gβ1-Cer (0.5 µg), Gγ2 wt (0.2 µg), as negative control FP receptor was replaced by the same amount of empty pcDNA3; TP receptor induced G protein activation: TP receptor (0.5 µg), Gαq-YFP (1.5 µg), Gβ1-Cer (0.5 µg), Gγ2 wt (0.2 µg), GRK2 (0.5 µg) was added to enhance the signal; IP receptor cAMP measurements: mCherryIP receptor (0.5 µg), Epac1-camps (1 µg); β2-AR cAMP measurements and IP receptor negative control: Epac1-camps (1 µg); TP receptor cAMP measurements: TP receptor wt (0.5 µg), Epac1-camps (1 µg) EP3 receptor GIRK measurements: EP3 receptor (0.3 µg) or EP3 receptor-D124R, pcDNA3-CFP receptor (0.2 µg), and a bicistronic plasmid expressing GIRK1 and GIRK4 subunits (0.5 µg).
Transfections into the Stable Cell Line Carrying TP Receptor-eYFP
TP receptor-Gα13-interaction assay: Gα13-mTurq2 (0.8 µg), Gβ1 wt (0.5 µg), Gγ2 wt (0.2 µg); TP receptor-GRK2-interaction assay: Gαq wt (0.8 µg), Gβ1 wt (0.5 µg), Gγ2 wt (0.2 µg), GRK2-mTurq2 (0.5 µg).
Fluorescence measurements were performed 48 hours after transfections. Transfected HEK293 cells were split on sterile, poly-L-lysine–coated glass coverslips and measured the next day.
FRET Measurements and Electrophysiology
FRET measurements with simultaneous control of the VM: FRET signals between CFP or variants and YFP or variants were recorded from selected single cells using an inverted microscope (Axiovert 135), as described previously (Birk et al., 2015). In brief, CFP was excited with short light flashes and the emitted donor fluorescence (F480) and acceptor fluorescence (F535) were recorded with photodiodes (TILL Photonics Dual Emission System) (see schematic representation in Fig. 1A) with a sampling frequency of 1-, 2.5-, or 5-Hz. The ratio of F535:F480 (termed emission ratio) was calculated and plotted using Patchmaster software (version 2.65; HEKA). Correction for photobleaching (Supplemental Fig. 1) was performed with Origin Pro 2016 for assays shown in: Fig. 1, Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Supplemental Fig. 1, Supplemental Fig. 2, Supplemental Fig. 3, Supplemental Fig. 4, Supplemental Fig. 5 and Supplemental Fig. 6. Data acquired from FRET-based cAMP measurements was processed by subtracting the emission ratio before agonist application (Fig. 7; Supplemental Fig. 7). Simultaneously to FRET measurements, the cells were patched in whole-cell configuration and the membrane potential was set to the desired values with an EPC-10 amplifier (HEKA). Patch pipette resistances ranged from 3 to 7 MΩ and the patch-pipettes were filled with an internal buffer solution (in millimolars: 105 K+-aspartate, 40 KCl, 5 NaCl, 7 MgCl2, 20 HEPES, 10 EGTA, 0.025 GTP, 5 Na+-ATP, pH 7.2). During the measurements, the patched cells were continuously superfused with extracellular buffer (in millimolars: 137 NaCl, 5.4 KCl, 1 MgCl2, 10 HEPES, pH 7.3) or agonist-containing solution.

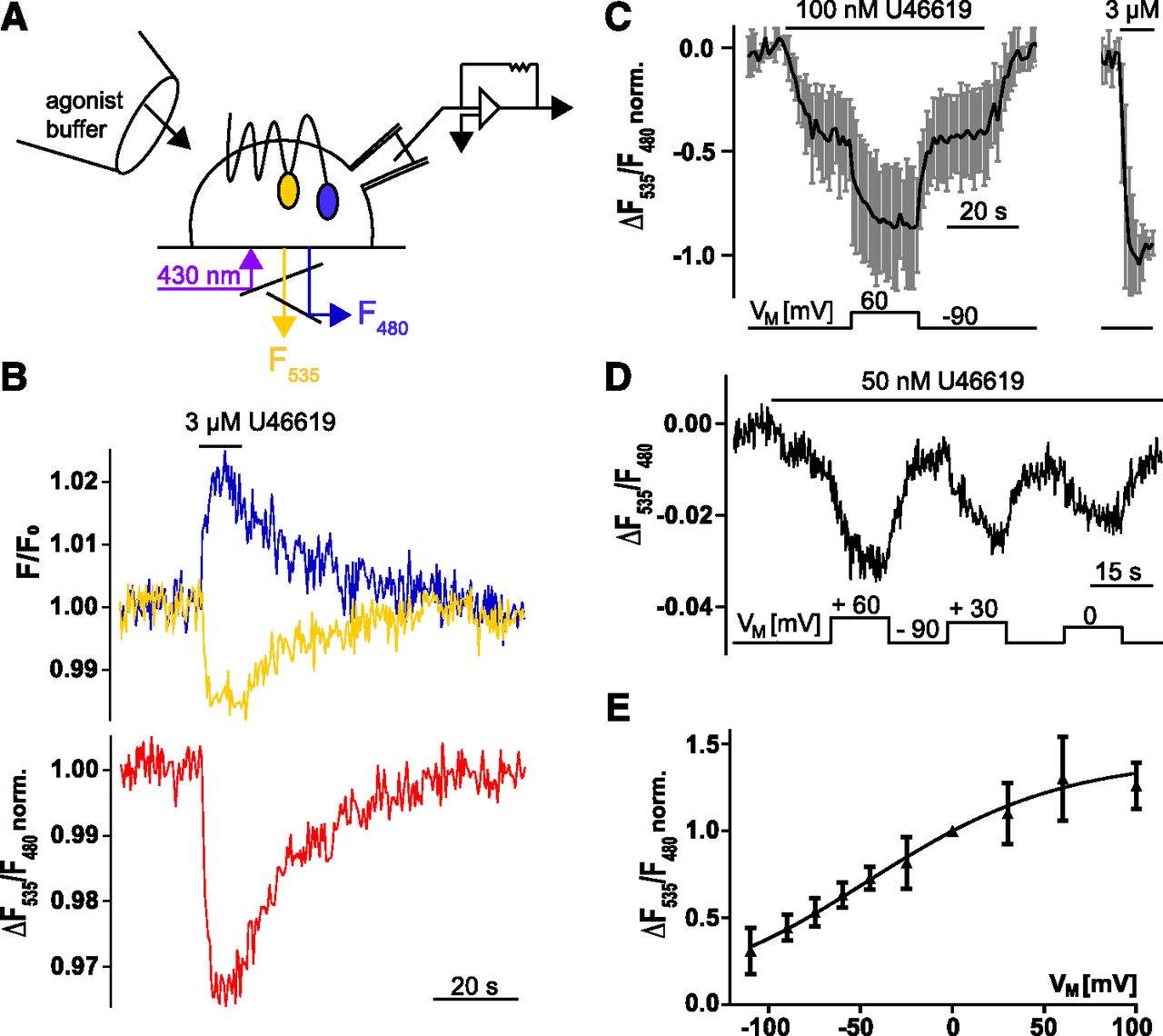
Depolarization of the membrane potentiates TP receptor activation. (A) The scheme illustrates the experimental setup used for FRET measurements under voltage clamp conditions in HEK293 cells expressing the TP receptor-sensor. Cells were continuously superfused with buffer or agonist-containing buffer. (B) The emission of CFP and YFP in a single cell stably expressing TP receptor-sensor superfused with 3 µM U46619 was recorded over time, corrected for photobleaching, and plotted as indicated by the colors as shown in the representative trace (out of n = 6). The corresponding emission ratio YFP:CFP is shown in red (normalized to initial values). (C) In experiments similarly described in (B), cells were exposed to 100 nM U46619 at −90 mV. Subsequently, cells were depolarized to +60 mV for 20 seconds as indicated; after wash-out, a high concentration of 3 µM U46619 was applied (obtained emission ratio was normalized to the negative amplitude evoked by 3 µM U46619) n = 5; means ± S.D.). (D) Representative recording and (E) show the voltage-dependence of the TP receptor-sensor for indicated voltages. Zero and −90 mV were always included as reference potentials. (E) Summarized data (n = 5–15 cells per data point; means ± S.D.) of the voltage-induced alterations in the emission ratio after normalization to values obtained at 0 mV. These data were fitted to a Boltzmann function giving rise to value for V0.5 = −46 mV.

Propagation of voltage effect on TP receptor downstream signals. Cells transfected with indicated FRET-based biosensor for TP receptor-Gα13 (representative recording n = 3) (A), TP receptor induced Gα13-p115 RhoGEF (representative recording n = 7) interactions (B), and TP receptor induced Gαq activation (representative recording n = 5) (C) were subjected to single-cell FRET recording under voltage clamp conditions using the indicated voltage and superfusion protocol. (D) Values in the presence of agonist of ΔF535/F480 at +60 mV were divided by values of ΔF535/F480 at −90 mV; the values for the effect voltage had on TP receptor-sensor activity, TP receptor-Gα13 interaction, TP receptor induced Gα13-p115 RhoGEF interaction, and TP receptor induced Gαq activation were compared: no significant difference could be detected (analyzed by Kruskal-Wallis-Test, P value = 0.10; means ± S.D., TP receptor-sensor = 2.21 ± 0.85, n = 29; TP receptor-Gα13 = 2.23 ± 0.24, n = 3; TP receptor induced Gα13-p115 RhoGEF interaction = 2.53 ± 0.69, n = 15, and TP receptor induced Gαq activation = 1.72 ± 0.42, n = 5).

Voltage effect on U46619 mediated TP receptor-sensor activation. (A) Means ± S.D. concentration-response curves of TP receptor-sensor at −90 (EC50 93 nM) and at +60 mV (EC50 21 nM,) respectively (n = 7–15 per data point), were recorded. All amplitudes of agonist-evoked declines in FRET were normalized to the amplitude of a reference concentration (3 µM U46619 at −90 mV). (B) Voltage effect on TP receptor-sensor activity in presence of a saturating agonist concentration (n = 6). (C) Means ± S.D. of the time course of TP receptor-sensor activation induced upon depolarization from −90 to +60 mV in the presence of U46619 (50 nM [blue]: n = 7, 200 nM [red] n = 15). Left: Averaged data were fitted to a monoexponential function. Right: Half times determined by fitting of individual experiments are illustrated as means ± S.D. (t1/2 (50 nM) = 2.2 ± 0.33 seconds, t1/2 (200 nM) = 1.2 ±0.4 seconds; ***P = 0.0001, Mann Whitney test).

Voltage effect on TP receptor activated with U46619 analogs. (A) Structures of tested U46619 analogs (https://www.caymanchem.com/; From Cayman Chemicals). Left, from top to bottom: U46619, I-BOP, 8-iso PGE2, 8-iso PGE1. Right, from top to bottom: PGE2 ethanolamide, PGE2 methyl ester, 15-keto PGE2, 15(S)-15-methyl PGE2. Ligands were tested in similar experiments as shown in (B). (B) PGE2 Ethanolamide tested in TP receptor induced Gα13-p115 RhoGEF interaction assay under voltage clamp conditions (representative recording n = 3). (C) Summary of ligands tested in terms of voltage dependence. “+” indicates an increase of at least 50% in ΔF535/ΔF480 between −90 and +60 mV in presence of a nonsaturating agonist concentration.

TP receptor R295 is involved in the voltage sensing process. (A) Left: Snakeplot created with GPCRdb.org of TP receptor indicating positions of mutated amino acids either in blue or in green. Right: Crystal structure of TP receptor cocrystallized with ramatroban (Protein Data Bank: 6IIU). The black numbers indicate minimum heavy atom distances (in angstrom) between the carboxyl group of ramatroban and R7.40. (B) Summary of mutants tested in terms of voltage dependence. “+” indicates an increase of at least 50% in ΔF535/ΔF480 between −90 and +60 mV in presence of a nonsaturating agonist concentration. (C–E) HEK293T cells transiently transfected with Gα13-p115 RhoGEF interaction assay and as indicated with either TP receptor wt or TP receptor R295K or R295A. (C) Left: Means ± S.D. cells were depolarized from −90 to +60 mV in the presence of U46619 (TP receptor wt [blue]: n = 6, TP receptor R295K [green] n = 5). Right: Cells were selected for a similar response to U46619 at −90 mV (included: ΔF535/F480 in presence of U46619 at −90 mV: 0.02 ≤ ΔF535/ΔF480 ≤ 0.08). The responses of the selected groups to +60 mV were then compared showing a significantly stronger activation upon depolarization for TP receptor R295K compared with wt (TP receptor wt = 3.13 ± 0.30, n = 7; TP receptor R295K = 5.31 ± 1.85, n = 6; Mann Whitney test **P = 0.0012). (D) Means ± S.D. TP receptor R295A induced Gα13-p115-interaction (n = 6). (E) Summarized data (n = 3–14 cells per data point) of the voltage-induced alterations in the emission ratio after subtraction of the −90 mV response for TP receptor wt, TP receptor R295K, and R295A recorded in the TP receptor induced Gα13-p115-interaction in presence of U46619. For −45 mV values TP receptor R295K and TP receptor R295A showed significant difference against wt (means ± S.D., wt n = 7, ΔF535/F480: 0.07 ± 0.01; R295K n = 5, ΔF535/F480: 0.02 ± 0.01; R295A n = 5, ΔF535/F480: 0.04 ± 0.03; Mann Whitney test R295K vs. wt: *P = 0.03, R295A vs. wt **P = 0.003); 60 mV (means ± S.D., wt n = 6, ΔF535/F480: 0.09 ± 0.03; R295K n = 7, ΔF535/F480: 0.17 ± 0.04; R295A n = 7, ΔF535/F480: 0.16 ± 0.06; Mann Whitney test R295K vs. wt: *P = 0.04, R295A vs. wt **P = 0.001); 100 mV (means ± S.D., wt n = 5, ΔF535/F480: 0.11 ± 0.04; R295K n = 6, ΔF535/F480: 0.21 ± 0.05; Mann Whitney test R295K vs. wt: **P = 0.009).

Voltage dependence of FP receptor means ± S.D. of HEK293T cells transfected with FP receptor and FRET Gαq–activation assay, depicted in the scheme on top. The single traces were normalized to the negative response on stimulation with 10 µM U46619 and averaged (n = 6). Cells were activated with 333 nM U46619. Agonist-activated cells were clamped from −90 to +60 mV and vice versa.

Voltage dependence of IP receptor. (A) Left: Means ± S.D. of HEK293T cells transfected with mcherry-IP receptor and FRET based cAMP-Sensor Epac1-camps. The single traces were normalized to the response on stimulation with 0.1 µM iloprost and averaged (0 mV: n = 5; −90 mV n = 4). A nonsaturating agonist concentration of iloprost was applied to cells that were held at −90 mV, and after 2 minutes the mean cAMP production was measured (T1). Subsequently, cells were either clamped to 0 mV (blue) or kept at −90 mV (black traces) for 2 minutes. The mean cAMP production was measured for both groups (T2). Right: T1 blue: 30.5% ± 14.1%; black: 26.2% ± 5.8%; T2 blue 49.1% ± 12.1%; black 48.6% ± 8.6%. No significant difference between the two conditions in the production of cAMP was observed (n.s., not significant). (B) Measurements of HEK293T cells transfected with TP receptor wt and Epac1-camps. Cells were stimulated with U46619 and clamped to the indicated potentials; traces were normalized on response to 3 µM U46619. To the nonsaturating U46619 concentration the Gαq inhibitor FR900359 with a final concentration of 1 µM has been added (means ± S.D.; n = 5). (C) Measurement of β2-AR voltage dependence in cAMP assay. Cells were stimulated with isoproterenol (Iso) and clamped to the indicated potentials; traces were normalized on response to 0.1 µM isoproterenol (means ± S.D.; n = 5).
FRET measurements without simultaneous control of the membrane potential: performed as described above, except the measurement of concentration-response curve for TP receptor wt induced interaction of Gα13 with p115 (Supplemental Fig. 5E), for which a previously described microscope setup was used (Bodmann et al., 2017).
Measurements of GIRK currents: GIRK currents were measured in the whole-cell configuration, analog to the measurements of FRET with simultaneous control of the membrane potential in 1 kHz sampling-intervals. The applied extracellular solution contained a high concentration of potassium (in millimolars: 137 KCl, 5.4 NaCl, 10 HEPES, 1 MgCl2, pH 7.3). Therefore, the recorded GIRK currents were inward currents (holding potential: −90 or −30 mV, as indicated).
Measurements in HEK293 Cells Were Performed at Room Temperature
Confocal Microscopy.
Confocal images of stable cell lines expressing either TP receptor-sensor or TP receptor-eYFP were taken with an inverted fluorescence microscope (TCS SP5; Leica, Wetzlar, Germany) equipped with a Lambda Blue 363/1.4 NA oil objective (Leica) analogously as described in Bodmann et al. (2017). Images were taken in a 1024x1024 pixel format.
Data Analysis and Statistics.
All data represent individual observations or an average of individual recordings and are presented as mean ± S.D. of n individual cells. The data were analyzed with Origin Pro 2016 or GraphPad Prism 7 (GraphPad Software) or Excel 2016. Data sets were tested for normal distribution with D’Agostino-Pearson normality test. For statistical analysis of data sets that did not pass testing for normality, nonparametric tests were used. Statistical analysis was performed with either Kruskal-Wallis test, Mann Whitney test, or Wilcoxon matched-pairs signed rank test. Differences were considered statistically significant for P ≤ 0.05.
For the IP receptor voltage dependence experiment (Fig. 7A), only cells with a higher response than 15% of max agonist concentration after 2 minutes of agonist application were included (at T1).
For the comparison of the voltage effect on TP receptor and TP receptor R295K (Fig. 5C), cells were selected for a similar response to U46619 at −90 mV (included: ΔF535/F480 in presence of U46619 at −90 mV: 0.02 ≤ ΔF535/F480 ≤ 0.08).
Analysis of charge movements, deactivation kinetics, concentration–response relationships.
Normalized values for the degree of receptor activation (R), measured with TP receptor-sensor were fitted to a single Boltzmann function (Fig. 1E) to analyze the voltage-dependence of gating charge movement and to obtain V0.5. Analysis was performed using GraphPad Prism 7 (GraphPad Software). The equation used for fitting: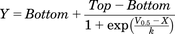 Where bottom and top are the minimal and maximal plateau, X is the respective membrane potential, V0.5 the voltage for half-maximal effect on the observed interaction, and k the slope factor.
Where bottom and top are the minimal and maximal plateau, X is the respective membrane potential, V0.5 the voltage for half-maximal effect on the observed interaction, and k the slope factor.
Values obtained were normalized to the calculated top of the Boltzmann function, which set Top as a constant equal to 1.0, so Y can be viewed as the fraction of receptors that are activated. For the calculation of the z value, a new Boltzmann function was fitted with the retrieved values.
Determination of kinetics of receptor activation upon agonist application was performed by fitting the FRET response of TP receptor-sensor (indicated in Fig. 3C) to monoexponential function, with constrained plateau. Curve-fitting and calculation of respective half time values were performed using GraphPad Prism 7 (GraphPad Software). Determination of kinetics of receptor deactivation upon agonist withdrawal or repolarization were performed analog without constraint (Supplemental Fig. 3D).
Concentration–response relationships were evaluated without voltage control for TP receptor-sensor (Fig. 3A) and without voltage control for TP receptor-sensor (Supplemental Fig. 3A) and for TP receptor or TP receptor R295K or TP receptor R295A induced Gα13-p115 RhoGEF interaction (Supplemental Fig. 5E). Single cells were superfused with test concentrations, followed by a reference concentration, and ΔF535/F480 of the tested concentrations were evaluated relative to ΔF535/F480 at the reference concentration. Concentration–response curves shown in Fig. 3A were fitted with GraphPad Prism 7 (GraphPad Software) with variable top, Hill slope, as well as EC50 and bottom. Concentration–response curves shown in Supplemental Figs. 3A and 5E were fitted with constrained top and bottom and variable EC50. Hill slope was variable except for TP receptor R295K and R295A curves (Supplemental Fig. 5E) with a hill slope set to the value obtained by TP receptor wt curve (0.85).
Results
To investigate voltage effects on TP receptor (henceforth referring to the α-isoform) activity, HEK293 cells, expressing proteins of interest, were subjected to whole-cell voltage-clamp conditions to control VM while simultaneously measuring FRET as a readout for receptor activity (Fig. 1A). We constructed a FRET-based TP receptor conformation sensor in which mTurq2 was cloned to the C-terminus and eYFP into intracellular loop three. This construct is referred to as TP receptor-sensor. A stable cell line of HEK293 expressing this construct was established and confocal microscopy showed that the TP receptor-sensor was well expressed and localized at the cell membrane (Supplemental Fig. 1A). For FRET measurements, cells stably expressing the construct were superfused with buffer and excited with light at 430 nm. Emission was recorded at 480 nm (F480, for mTurq2 fluorescence) and 535 nm (F535, for eYFP fluorescence). Upon stimulation with a saturating concentration of TP receptor agonist U46619 YFP emission decreased, while CFP emission simultaneously increased, indicating the occurrence of FRET (Fig. 1B, top). The ratio F535/F480 was calculated and plotted, referred to as emission ratio, showing a decrease upon agonist stimulation (Fig. 1B, bottom). Changes in emission ratio were reversible after agonist removal. The observed decrease in emission ratio was in line with previously published GPCR-FRET-conformation sensors, which were labeled in intracellular loop three and at the C-terminus (Kauk and Hoffmann, 2018). The traces shown in Fig. 1A and all traces in which the TP receptor-sensor is shown in this study are corrected for photobleaching by subtraction of a monoexponential function (Supplemental Fig. 1B).
Ligand-Induced Activation of TP Receptor Is Potentiated upon Depolarization within the Physiologic Range of VM.
To study the effect of voltage on the receptor activity, FRET was measured in HEK293 cells stably expressing the TP receptor-sensor under voltage clamp conditions. TP receptor was activated with 100 nM U46619, a nonsaturating agonist concentration, at a holding potential of −90 mV. An agonist concentration was considered henceforth nonsaturating if the agonist-evoked response at −90 mV was less than 70% of the response to subsequently applied high concentration of a full agonist. In steady-state conditions, cells were clamped to +60 mV. This depolarization led to a robust decrease in the emission ratio reflecting receptor activation upon depolarization. This effect was reversible upon repolarization to −90 mV (Fig. 1C). In contrast, in the absence of agonist, no change of TP receptor-sensor emission ratio was observed (Supplemental Fig. 1C). To find out whether voltage dependence of TP receptor occurred in a physiologic range of VM, we measured the relation of receptor activity and VM with a nonsaturating agonist concentration. Values were normalized to values obtained at 0 mV and fitted to a Boltzmann function resulting in V0.5 = −46 mV (Fig. 1, D and E). The z value was calculated to be 0.5 elementary charges.
Voltage Effect on TP Receptor Is Transduced to Downstream Effectors.
Next, we wanted to know whether this observed voltage effect at the TP receptor-sensor propagates to downstream signaling. TP receptor couples primarily to Gαq and Gα13. Therefore, voltage dependence of TP receptor-Gα13-interaction, TP receptor induced interaction of Gα13 with p115, a RhoGEF family member, and TP induced Gαq activation was measured. We observed activation upon depolarization consistent with that seen for TP receptor-sensor (Fig. 2, A–C vs. Fig. 1C). Note that TP receptor in the Gα13-p115-interaction and Gαq activation assay is the wt receptor and not labeled with a fluorophore or modified in any other form. Activation upon depolarization could not be observed in the absence of agonist (Supplemental Fig. 2, A and B). Voltage effect on four different FRET assays turned out to exhibit similar magnitudes (Fig. 2D).
Voltage Effect on TP Receptor’s Affinity.
The observed increase of TP receptor activity upon depolarization could, in theory, be due to a voltage effect on efficacy or affinity of the agonist or a mixture of both. To investigate this, we measured concentration-response curves for the TP receptor-sensor at −90 and at +60 mV. The EC50 was left-shifted approximately 4.5-fold upon depolarization (Fig. 3A). We further applied 3 µM U46619 to saturate the receptor (see Supplemental Fig. 3A), no voltage induced changes of FRET of the TP receptor-sensor were detected (Fig. 3B). Similarly, on the level of the TP receptor-Gα13-interaction (Supplemental Fig. 3B) and at TP receptor-GRK2-interaction (Supplemental Fig. 3C) we also failed to see additional stimulation. This is in line with the hypothesis that voltage primarily regulates agonist affinity for TP receptor. The third aspect that we considered in this context was the kinetics of the voltage induced receptor activation and deactivation. If voltage changes lead to a difference in affinity, this process would require agonist binding; therefore, the kinetics of this effect would be dependent on the agonist concentration. We compared the kinetics of receptor activation upon depolarization in presence of either 50 or 200 nM U46619 and observed significantly faster on-set kinetics for the higher agonist concentration (Fig. 3C). If voltage modulated TP receptor affinity for U46619, one would expect that deactivation upon hyperpolarization from +60 to −90 mV in presence of agonist should have the same kinetics as wash-out of the same concentration at −90 mV because in both processes agonist leaves the receptor in case of an affinity change. To our surprise, the voltage induced deactivation kinetics were approximately three times faster than those for washout (Supplemental Fig. 3D), possibly reflecting rebinding of the lipophilic ligand in the plasma membrane.
TP Receptor’s Voltage Dependence Is Not Dependent on Specific Moieties of the Agonist.
Prostanoids have a very conserved structure and we hypothesized that systematically testing ligands substituted in one or more functional group(s) could provide information on which contact of U46619 is important for the observed voltage dependence. As TP receptor activated by U46619 showed activation upon depolarization, we searched for TP receptor ligands that differ in their structure from U46619. U46619 as a prostanoid-like ligand comprises of an α-chain and ω-chain connected to a central ring. Substituted ligands were tested in terms of voltage dependence either with the TP receptor-sensor assay (activation is reflected by a decrease in FRET) or TP receptor induced Gα13-p115-interaction (reflected by an increase in FRET), as this assay provides a very robust signal, which also allows us to test agonists with a small efficacy. Structures of the tested ligands are shown in Fig. 4A. The following ligands were tested: I-BOP (difference in ring structure and more lipophilic ω-chain) (Supplemental Fig. 4A), 15-keto-PGE2 (Supplemental Fig. 4B), and 15(S)-15-methyl PGE2 (Supplemental Fig. 4C) (prostaglandin E2 [PGE2]-like ring structure and substituted at the hydroxyl group at position 15). Furthermore, PGE2 ethanolamide (Fig. 4B) and PGE2 methyl ester (Supplemental Fig. 4D) (PGE2-like ring structure and substituted C1 position at the carboxylic acid taking away the charge), 8-iso PGE2 (Supplemental Fig. 4E) (PGE2-like ring structure and chiral inversion at C8) and 8-iso PGE1 (Supplemental Fig. 4F) (like 8-iso PGE2 but lacked the double bond between position 5 and 6) have been investigated. TP receptor activated with either ligand showed robust activation upon depolarization as seen before when activated with U46619, summarized in Fig. 4C (for detailed data see Supplemental Fig. 4G).
Mutation of R295 Alters the Voltage Effect on TP Receptor.
Because we could not identify important contact(s) between ligand and receptor by testing different agonists, we mutated amino acids known to be involved in the ligand binding process of TP receptor (Funk et al., 1993; Chiang et al., 1996; D’Angelo et al., 1996; Khasawneh et al., 2006) (Fig. 5A, left) with the goal to change the ligand binding mode. Studies have found that the agonist-binding mode can be crucial for the effect voltage has on GPCR activity (Rinne et al., 2015). Based on the recently published crystal structure of antagonist-bound TP receptor (Fan et al., 2019), we mutated R295, which interacts with the carboxyl group of the ligand (Fig. 5A, right) either to alanine or to lysine (TP receptor R7.40295A or TP receptor R7.40295K). In addition, we constructed the following receptor mutants: TP receptor S5.44201T (superscript indicates residue numbering using the Ballesteros–Weinstein nomenclature [Ballesteros and Weinstein, 1995]), TP receptor S6.45255T, TP receptor W7.44299L, and TP receptor D5.36193E. TP receptor S201T, TP receptor S255T, TP receptor W299L, and TP receptor D193E showed wild-type–like behavior in terms of voltage dependence (Supplemental Fig. 5, A–D), summarized in Fig. 5B (for detailed data see Supplemental Fig. 5E). TP receptor R295K showed ligand-induced receptor activation (consistent with previous studies [Tai et al., 1997]) and had an EC50 right shifted by about two orders of magnitude (Supplemental Fig. 5F). Therefore, U46619 concentration was adjusted from 1 to 2 nM for wt to 200 nM for the mutant receptor. TP receptor R295K showed significantly stronger activation upon depolarization compared with wt measured in Gα13-p115-interaction (Fig. 5C). As R7.40295 is a charged residue under physiologic pH, it could potentially serve as part of a voltage sensor for TP receptor. Removing the charge in this position by mutating arginine to alanine should then remove or reduce voltage sensitivity. According to Stitham et al. (2003), mutation of R7.40 to alanine abolished the ligand binding for IP receptor. We observed agonist-induced receptor activation for TP receptor R295A in the Gα13-p115-interaction assay, with an EC50 value shifted further right in comparison with TP receptor R295K (Supplemental Fig. 5F). TP receptor R295A showed robust activation upon depolarization (Fig. 5D), indicating that the positive charge was not required for voltage dependence. To study the role of R295 on voltage dependence in more detail, we measured activation voltage relation for wt (with measurements similar as in Supplemental Fig. 5G) and for R295K and R295A (with measurements similar as shown in Supplemental Fig. 5H) in Gα13-p115-interaction assay, when TP receptor was activated with U46619. The retrieved values were subtracted by the agonist-induced response to −90 mV of the same cell. We measured membrane potentials between −110 and +100 mV (Fig. 5E). The curve for the mutants was shallow compared with wt and did not reach a plateau. Because no plateau was reached, no Boltzmann fit could be performed, and neither V0.5 nor z-value could be calculated for the mutants. At −45 and +60 mV values for TP receptor R295K and TP receptor R295A showed significant difference against wt (n: wt = 7; R295K,A = 5; Mann Whitney test R295K: P = 0.03, R295A P = 0.003); 60 mV (n: wt = 6; K,A = 7; Mann Whitney R295K: P = 0.04, R295A P = 0.01); At +100 mV values for TP receptor R295K showed significant difference against wt (n: wt = 5; K = 6; Mann Whitney test R295K: P = 0.01) (Fig. 5E). These results indicate that even though R295 is not part of the voltage sensor, its side chain, but not the positive charge, is important for the range of voltage sensitivity.
Voltage Dependence of FP Receptor, IP Receptor, and EP3 Receptor.
The thromboxane receptor belongs to the prostanoid receptor group. Endogenous ligands that can activate other receptors of this family are also cyclooxygenase products of arachidonic acid. We decided to investigate which voltage behavior other members of this receptor family show, if activated by prostanoid-like ligands. FP receptor affinity to U46619 is only 10 times lower than for TP receptor (Abramovitz et al., 2000), which allowed us to use U46619 in our voltage dependence measurements at FP receptor. We measured Gαq activation with FRET while we simultaneously controlled VM with patch clamp. Here a decrease in emission ratio reflects receptor activation (Fig. 6). We transfected HEK293T cells with FP receptor and plasmids necessary for Gαq activation. Our results show a strong decrease in emission ratio upon depolarization in the presence of U46619, indicating receptor activation upon depolarization (Fig. 6). In HEK293T cells without transfected FP receptor, no Gαq activation could be observed (Supplemental Fig. 6). Because there was no obvious difference in voltage dependence between TP receptor and FP receptor, we wanted to test a receptor that has a lower affinity for U44619 and therefore likely a binding pocket with less similarity than those of TP receptor’s and FP receptor’s. Therefore, we characterized IP receptor, the physiologic counterpart of TP receptor in terms of voltage dependence. Due to a lack of robust FRET-based assays on receptor or G protein level, we measured cAMP production with the FRET-based Epac sensor Epac1-camps (Nikolaev et al., 2004) as a readout for the activity of Gαs-coupled IP receptor at −90 or 0 mV. HEK293T cells without additionally transfected IP receptor did not show cAMP production upon stimulation with iloprost, which is a stable analog of prostacyclin and is known to activate IP receptor (Supplemental Fig. 7). Due to difficulties reaching stable steady-state signals, we compared responses to a nonsaturating concentration of iloprost in cells held at −90 mV during the whole measurement with those of cells that were held at −90 mV for 2 minutes and clamped to 0 mV for another 2 minutes (Fig. 7A, left). The FRET-based Epac1-camps signal was compared after 2 and 4 minutes in both groups. Surprisingly, no significant difference could be observed, suggesting no detectable voltage dependence of IP receptor activated with iloprost in our system (Fig. 7A, right). To test whether this system is sufficiently sensitive to pick up changes in receptor activity mediated by voltage changes, we measured TP receptor in this assay. TP receptor showed robust activation upon depolarization, as observed before (Fig. 7B). As a further positive control measurement of Gαs-coupled β2-AR showed a decrease in cAMP production upon depolarization, in line with the previously reported moderate voltage dependence of this receptor (Birk et al., 2015), (Fig. 7C), indicating that voltage dependence even of GPCRs poorly sensitive to regulation by membrane potential, with an opposing polarity of voltage dependence compared with TP receptor, can be detected in the applied assay. Next, we chose to measure the Gαi-coupled EP3 receptor (prostaglandin E2 receptor subtype 3, henceforth referring to isoform 1, Genbank accession: L27490.1). As a sensitive readout, we used GIRK channels as a reporter system. Therefore, we transiently transfected HEK293T cells with EP3 receptor and GIRK1/4. Responses to a submaximal concentration of 10 nM iloprost were compared with maximal currents evoked by a saturating agonist concentration of 1 µM iloprost at −90 and at −30 mV in the same recording (Fig. 8, left). Measured currents could be blocked with barium, and I/V curve showed inward rectification (Supplemental Fig. 8A), supporting that the measured currents were, in fact, GIRK currents. The fraction of GIRK channels activated by 10 nM iloprost relative to those activated by 1 μM iloprost (ratio response to 10 nM/response to 1000 nM) was much larger at −30 mV than at −90 mV (Fig. 8, right), suggesting EP3 receptor activation upon depolarization in the presence of iloprost. Attempts to test EP1 receptor for voltage dependence failed as none of our assays showed a specific signal with this receptor.

Voltage dependence of EP3 receptor. Left: Representative inward K+ current of HEK293T cells expressing wild-type EP3 receptor and GIRK channels evoked by 10 nM or 1 µM iloprost, measured at −90 or −30 mV (n = 6). Right: The GIRK current ratio (10 nM:1 µM) indicates that the response to 10 nM iloprost is potentiated at −30 mV (current ratio [10 nM/1 µM] at −90 mV = 0.13 ± 0.10; at −30 mV = 0.44 ± 0.20; *P = 0.016. Wilcoxon matched-pairs signed rank test).
We then performed a structure-based alignment with GPCRdb.org between the four prostanoid receptors characterized in terms of voltage dependence in this study. In particular, we screened for differences in charged residues between the voltage-activated prostanoid receptors TP receptor, FP receptor, EP3 receptor, and IP receptor, which did not show a detectable activation upon depolarization. Position 3.19 was the only position, which fulfilled the criteria: TP receptor, FP receptor, and EP3 receptor contained a negatively charged aspartate in this position, whereas IP contained a positively charged arginine. We wondered whether a mutation in this position could lead to a change in voltage dependence, which was not the case because EP3 receptor D124R showed EP3 receptor wt-like activation upon depolarization (Supplemental Fig. 8B), suggesting a more complex mechanism underlying the differences in voltage sensitivity.
Discussion
In the present study, we discovered that the majority of the tested prostanoid receptors exhibit robust voltage dependence. Depolarization of the electrical membrane potential enhanced receptor signaling via FP receptor, EP3 receptor, and TP receptor, whereas IP receptor signaling was not affected. The findings for TP receptor are in line with previous findings, which suggested an activation upon depolarization (Martinez-Pinna et al., 2005). Interestingly, for another member of the lipid-receptor group, the lysophosphatidic acid G protein–coupled receptors, activation upon depolarization has also been observed (Martinez-Pinna et al., 2010). Using TP receptor as a model system, we found the highest sensitivity toward voltage in the physiologic range of VM. Depolarization-induced potentiation of TP receptor signaling was already detectable on the level of TP receptor conformations. To study voltage dependence of prostanoid receptors single cell–based assay systems were needed. FRET-based assays as readout systems to measure receptor conformation, receptor interactions with downstream signaling molecules such as G proteins and G protein–coupled receptor kinases as well as G protein activity or G protein effector interaction as used here have the advantage that they do not rely on a detection unit with intrinsic voltage dependence such as ion channels. Using these assays for most of the receptors of the prostanoid family, we found that depolarization enhanced prostanoid signaling except for IP receptor (Fig. 7A). For Gαs-coupled receptors, we used the Epac1-camps biosensor assay due to its great sensitivity and signal to noise ratio. In this assay, we succeeded in detecting the modest voltage sensitivity of β2-AR (Fig. 7C) and observed a large enhancement of TP receptor stimulated raises in cAMP in response to depolarization (Fig. 7B). This result is in line with those observed with all the other FRET-based applied to TP receptor assays. For the IP receptor, application of agonist led to cAMP production; however, these rises in cAMP were insensitive toward changes of VM. We tested in all experiments for signal saturation. IP receptor induced cAMP elevations showed in both conditions a signal to 1 to 2 nM iloprost below 50% of the signal observed in response to 3 µM iloprost (Fig. 7A), indicating that the absence of voltage dependence can’t be attributed to a saturation of the signal at −90 mV. Because all three receptors (TP receptor, IP receptor, and β2-AR) exhibited similar global rises in cAMP upon agonist application, the effect of voltage was specific to the receptor expressed: β2-AR reacted with a decrease in cAMP levels upon depolarization. IP receptor induced responses were not altered by voltage changes to a detectable degree, whereas TP receptor-mediated elevations in cAMP were strongly potentiated upon depolarization. Cytosolic cAMP levels are not only influenced by receptor-mediated stimulation of adenylyl cyclases but also by phosphodiesterases, which can be quite receptor specific. It seems very unlikely that phosphodiesterase activity is voltage dependent based on their intracellular localization. Although no evidence for a voltage dependence of IP receptors could be found, we cannot rule out that some voltage dependence exists below our detection limit or requiring different agonists.
The finding that depolarization enhances prostanoid receptor signaling is of special interest because prostanoid receptors are widely expressed in the human body and the potential impact might not be limited to excitable cells. Studies show changes in VM over time in cells during cell cycle and in cancer cells and different membrane potentials of various differentiated cells (Arcangeli et al., 1995; Yang and Brackenbury, 2013). Furthermore, it has been shown for platelets, which express different voltage-gated ion channels (Mahaut-Smith, 2012), that physiologic changes of membrane potential occur, e.g., endothelial cells release epoxyeicosatrienoic acids, which hyperpolarize platelets in turn impairing GPCR-mediated signaling (Krotz et al., 2004).
For the TP receptor that we studied in depth, ligand-induced TP receptor activity for nonsaturating concentrations doubled if the membrane potential was depolarized from −90 to +60 mV both at TP receptor-sensor level and for TP receptor-Gα13 interaction (Fig. 1C; Fig. 2, A and C). With a V0.5 = −46 mV, this voltage modulation resided within the physiologic range of membrane potentials (Fig. 1, D and E). The strength of the effect was remarkable as there was no signal amplification in these assays—allowing us to see exactly the fraction of receptors that were modulated by voltage. At very low agonist concentrations leading to only a small but detectable induction of the amplified Gα13/p115-RhoGEF interaction showed a threefold effect by depolarization from – 90 to +60 mV (Fig. 5C). This is a remarkably strong effect of voltage in comparison with other receptors published (Navarro-Polanco et al., 2011; Sahlholm et al., 2011; Birk et al., 2015).
For other GPCRs described to be voltage dependent, voltage induced alterations of agonist affinity or efficacy have been described. We could show that the voltage effect on TP receptor activity was clearly due to a change in affinity because: 1) the EC50 was right shifted approximately 4.5 times at −90 mV compared with +60 mV (Fig. 3A). 2) At saturating agonist concentrations, the voltage effect was missing even at the level of the receptor-sensor (Fig. 3B). 3) On-kinetics of voltage-induced receptor activation were dependent on agonist concentration (Fig. 3C). The kinetics of re- or hyperpolarization-induced deactivation of receptor responses should ideally be similar to those measured in response to agonist withdrawal if voltage regulates affinity of the agonist toward the receptor. In case of TP receptor voltage-induced deactivation, kinetics were significantly faster than washout kinetics. One reason for this could be the slow washout of the agonist due to its lipophilic property. Nevertheless, our results strongly suggest voltage-induced modulation of TP receptor’s affinity toward the agonist.
To date, the molecular correlate for a general voltage sensor of GPCRs remains unknown, even though the tyrosine lid above the agonist-binding pocket on muscarinic receptors have been proposed to serve as such a sensor (Barchad-Avitzur et al., 2016). However, if this tyrosine lid indeed represents a voltage sensor, which is still under debate (Hoppe et al., 2018), it is not found in all other voltage-sensitive GPCRs, including prostanoid receptors. We therefore attempted to at least gain some molecular information about voltage sensitive structures on TP receptor. We screened different agonists that carried chemical modification on distinct sites of the molecule as depicted in Fig. 4A in respect to voltage sensitivity of their TP receptor activation, all agonists reacted similarly (summarized in Fig. 4C; for detailed data see Supplemental Fig. 4G). This suggests that depolarization leads to a global alteration of the TP receptor conformation in a way that it enhances binding to all agonists in a similar way, for example by increasing the probability of TP receptor to reach active conformations, or by inducing a receptor conformation, that alters the access for agonists.
In line with this, our results on the voltage sensitivity of TP receptor mutants, previously reported to be important for ligand binding, yielded no hint for a specific modulation of voltage dependence. The only exception are results obtained with mutation of arginine 295, a site proposed and confirmed to be the interaction partner for the C1 carboxylic group of prostanoids (Sugimoto et al., 1992; Audet et al., 2019; Fan et al., 2019; Toyoda et al., 2019). Upon mutation of 295 to either lysine or alanine agonist, potency was not only reduced but also enhanced the effect of voltage, extending the range of effective voltages to more positive potentials (Fig. 5, C and D). Based on these results, we can conclude that the carboxylic acid, and its interaction site is not required for voltage sensitivity of TP receptor. In our opinion, the extended site chain of R295 but not the charge might cause a restriction of the voltage range possibly by reducing the flexibility of the receptor. Taken together, our results suggest that the voltage sensitivity of TP receptor leads to an enhanced probability of the receptor to bind ligand and become activated, indicating a general influence of voltage on receptor conformation, instead of specific switches in certain sites of the receptor.
Acknowledgments
We thank Dr. Cornelius Krasel for providing reagents for the GRK2-assays. Furthermore, we would like to thank Dr. Evi Kostenis for providing the Gαq inhibitor FR900359.
Authorship Contributions
Participated in research design: Kurz, Bünemann.
Conducted experiments: Kurz, Krett.
Performed data analysis: Kurz, Krett.
Wrote or contributed to the writing of the manuscript: Kurz, Bünemann.
Footnotes
- Received September 25, 2019.
- Accepted January 25, 2020.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CFP
- cyan fluorescent protein
- EP3 receptor
- prostaglandin E2 receptor subtype 3
- Epac
- exchange protein directly activated by cAMP
- eYFP
- enhanced yellow fluorescent protein
- FP receptor
- prostaglandin F receptor
- FRET
- Foerster resonance energy transfer
- GIRK
- G protein–activated, inwardly rectifying K+
- GPCR
- G protein–coupled receptor
- GRK
- G protein–coupled receptor kinase
- HEK
- human embryonic kidney
- I-BOP
- [1S-[1α,2α(Z),3β(1E,3S*),4α]]-7-[3-[3-hydroxy-4-(4-iodophenoxy)-1-butenyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid
- IP receptor
- prostacyclin receptor
- iso
- isoproterenol
- mTurq
- monomeric turquoise fluorescent protein
- pcDNA3
- Mammalian Expression Vector
- TP receptor
- thromboxane receptor
- U46619
- 9,11-dideoxy-9α,11α-methanoepoxy-prosta-5Z,13E-dien-1-oic acid
- V0.5
- half maximal effective potential
- VM
- membrane potential
- wt
- wild type
- YFP
- yellow fluorescent protein
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}