


Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that mediates the biological responses to environmental contaminants such as 2,3,7,8-tetrachlorodibenzo-p-dioxin. Embryonic fibroblast (EF) isolated from AHR-null mice exhibited slow cell growth compared with wild-type EF. Reintroduction of AHR into AHR-null EF increased cell growth, suggesting that AHR is involved in cell cycle control. The role of the AHR in cell cycle control was examined using the adenovirus oncoprotein E1A. EF, derived from wild-type and AHR-null mice, were transfected with two mutant E1A expression plasmids that inactivate either p300/CBP or retinoblastoma protein (pRb). Although DNA synthesis of wild-type EF was induced by both E1A mutants, DNA synthesis in the AHR-null EF was induced only by the mutant that binds pRb, not by the mutant to p300/CBP. These data show that both pRb and p300/CBP were the target of E1A-induced DNA synthesis in wild-type EF. In AHR-null mice, however, only pRb was the target of E1A-induced DNA synthesis and p300/CBP cannot be inactivated by E1A in the absence of AHR. Immunoprecipitation revealed that AHR directly bound to p300, thus suggesting the intriguing possibility that AHR is involved in control of the cell cycle via interaction with p300.
The aryl hydrocarbon receptor (AHR) is an intracellular protein that binds exogenous ligands such as halogenated aromatic hydrocarbon, 2,3,7,8-tetracholorodibenzo-p-dioxin (TCDD), and mediates their biological actions including their toxic effects (Poland, 1982;Safe, 1986; Swanson and Bradfield, 1993; Okey et al., 1994). The AHR is a basic helix-loop-helix protein that dimerizes with another basic helix-loop-helix protein, the AHR nuclear translocator (ARNT) to form a heterodimeric transcription factor (Hoffman et al., 1991; Burbach et al., 1992; Ema et al., 1992; Reyes et al., 1992; Hankinson, 1995). The liganded AHR/ARNT heterodimer binds to an enhancer sequence called a xenobiotic response element (XRE, also called DRE and AhRE) located upstream of the AHR target genes, such as CYP1A1, CYP1A2, CYP1B1, CYP2A8, UDPGT*6, and NQO1 (Fujii-Kuriyama et al., 1990; Wu and Whitlock, 1992; Whitlock et al., 1997; Kurose et al., 1999). These genes encode enzymes that metabolize foreign compounds including drugs and carcinogens.
The AHR has been implicated in cell cycle control. AHR-deficient mouse hepatoma cells (Hepa1c1c7) exhibited a prolonged doubling time compared with wild-type cells (Ma and Whitlock, 1996). Flow cytometric and biochemical analyses indicated that the slow growth rate of AHR-defective cells reflects the prolonged G1 phase of the cell cycle. Embryonic fibroblast (EF) from AHR-null mice showed slower cell growth rate compared with the wild-type cells by increasing TGFβ production and down-regulation of mitotic kinases such as Cdc2 and polo kinase (Elizondo et al., 2000). Other studies have shown that stimulation of AHR affected the cell cycle. For example, TCDD induced G1 arrest in rat hepatoma 5L cells (Gottlicher et al., 1990) and benzo[a]pyrene, another AHR ligand, induced an AHR-dependent G1 arrest in murine Swiss 3T3 cells (Vaziri and Faller, 1997). These results suggested that the AHR/ARNT heterodimer might be involved in the cell cycle control in addition to the stimulation of target gene transcriptions. Two factors, p300/cAMP response element-binding protein (CBP) and retinoblastoma protein (pRb), have been suggested to be involved in AHR/ARNT mediated biological effects (Kobayashi et al., 1997; Ge and Elferink, 1998). CBP was originally found to be a coactivator for cAMP-responsive element binding protein (Chrivia et al., 1993) and is now known to be a common coactivator for numerous DNA-binding transcription factors, including activator protein 1 (Arias et al., 1994; Bannister and Kouzarides, 1995), steroid hormone receptors (Chakravarti et al., 1996; Kamei et al., 1996), MyoD (Yuan et al., 1996), and Myb (Dai et al., 1996;Oelgeschlager et al., 1996). p300 closely resembles CBP, both structurally and functionally (Arany et al., 1994, 1995; Lundblad et al., 1995). Yeast two hybrid experiments revealed that p300/CBP binds to the ARNT protein in the AHR/ARNT heterodimer complex (Kobayashi et al., 1997). pRb, which is believed to regulate the progression through G1 phase of the cell cycle (Weinberg, 1995;Sherr, 1996; Lundberg and Weinberg, 1998), also binds to AHR in the AHR/ARNT heterodimeric complex (Ge and Elferink, 1998).
Cell cycle control can be investigated using the E1A proteins. The E1A gene of human adenovirus type 2 and adenovirus type 5 generates two major species of mRNAs with sizes of 13 S and 12 S that encode proteins of 289 and 243 amino acid residues, respectively (Bellett et al., 1985). The E1A 12 S has the ability to immortalize primary rodent cells (Houweling et al., 1980; Cone et al., 1988) and to induce DNA synthesis when introduced into quiescent cells (Stabel et al., 1985; Kaczmarek et al., 1986; Nakajima et al., 1987). This function of E1A 12 S on the cell cycle in primary cells was found to correlate with its ability to bind with cellular proteins, including pRb (Whyte et al., 1988, 1989; Egan et al., 1989) and p300/CBP (Arany et al., 1994; Arany et al., 1995; Lundblad et al., 1995). The relationship between the structure and the activity of E1A was studied using E1A mutants (Bayley and Mymryk, 1994). The conserved region 1 (CR1; amino acid residues 40 to 80) and the conserved region 2 (CR2; amino acid residues 120 to 149) of E1A 12 S binds to pRb, resulting in release of the E2F family of transcription factors from pRb (Bagchi et al., 1990; Bayley and Mymryk, 1994). This permits unscheduled DNA synthesis (Bagchi et al., 1990; Raychaudhuri et al., 1991; Helin et al., 1992; Kaelin et al., 1992). The N terminus and CR1 of E1A 12 S binds to p300/CBP resulting in complete inactivation of this factor (Bayley and Mymryk, 1994). Although the role of the p300/CBP-binding region of E1A in either cell cycle deregulation or cellular transformation is much less clear, p300/CBP is believed to suppress the cell cycle because inactivation of p300/CBP with E1A 12 S leads to stimulation of cell growth (Howe et al., 1990; Eckner et al., 1996; Liu and Kitsis, 1996; Wang et al., 1998). These observations suggested that E1A stimulates DNA synthesis by influencing two functionally distinct pathways involving pRb and p300/CBP. These results further suggested that pRb and p300/CBP act on the cell cycle machinery by distinct mechanisms, because either the pRb- or the p300/CBP-interacting domain of E1A is sufficient for the induction of DNA synthesis (Howe et al., 1990; Wang et al., 1991, 1993; Liu and Kitsis, 1996).
In the present study, the roles of pRb and p300/CBP in cell cycle control was investigated using AHR-null EF derived from the AHR-null mouse (Fernandez-Salguero et al., 1995) and two E1A deletion mutants (Nakajima et al., 1992). The results indicate that both pRb and p300/CBP were the target of E1A-induced DNA synthesis in wild-type EF. However, in AHR-null EF, pRb was the target of E1A-induced DNA synthesis, whereas p300/CBP was refractory to E1A treatment. These data suggest that AHR plays an important role in regulating p300/CBP activity. Immunoprecipitation revealed that AHR binds to p300, suggesting that AHR is involved in control of the cell cycle via interaction with p300.
Experimental Procedures
Materials.
To prepare the AHR expression plasmid, a full-length mouse AHR cDNA was introduced into the mammalian expression vector, pCI vector (Promega, Madison, WI), in which a cDNA insert was expressed under the control of the CMV promoter. The resulting construct was designated pCI/AHR. pHβAPrE1A, NTdl598, and dl922/947, in which transcription of the wild-type and mutant E1A 12 S genes are driven by the human β-actin promoter, were provided by Dr. Kinichiro Oda. An XRE-containing luciferase reporter gene (pGL-2A8) was prepared as described previously (Kurose et al., 1999). Monoclonal antibodies against p300 and pRb were purchased from Upstate Biotechnology Inc. (Lake Placid, NY), monoclonal and polyclonal antibodies against AHR and ARNT, respectively, were obtained from AffnityBioreagent (Golden, CO). LipofectAMINE reagent was purchased from Gibco-BRL (Bethesda, MD).
Culture of the Mouse EF and the [3H]Thymidine Incorporation Assay.
EFs were prepared from AHR-null and wild-type mouse embryos of embryonic day 14.5 as described previously (Fernandez-Salguero et al., 1995; Elizondo et al., 2000). After enzymatic dissociation, the cells from three embryos were plated into one gelatin-coated 175-cm2 flask and cultured in media consisting of Dulbecco's modified Eagle's medium (DMEM), 10% (v/v) fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2atmosphere. After 2 days in culture, the cells were divided into four 175-cm2 flasks and cultured for an additional 2 days. Then the cells were transferred to a 12-well plate (2 × 105 cells/well) for the [3H]thymidine incorporation experiment or to 100-mm plates (2 × 106 cells/plate) for preparation of cell nuclear extracts. To synchronize the cell cycle, cells were cultured in DMEM containing 0.1% (v/v) FBS (starvation media) for an additional 24 h after 24 h cultivation in 10% FBS containing DMEM. Under starvation media, these cells did not exhibit significant levels of thymidine incorporation. For transfection experiments, the E1A and AHR expression plasmids were introduced into cells using the LipofectAMINE reagent during the synchronization period in starvation media. After transfection, cells were washed with PBS, then cultured in 1, 5, or 10% FBS-containing media for 6 h. The cells were then pulsed with [3H]thymidine (1 μCi/ml) for 4 h and washed twice with ice-cold PBS. Macromolecules were precipitated with the addition of an equal volume of 10% trichloroacetic acid. Cells were lysed on ice for 10 min using 0.5 ml of 0.5 N NaOH, then 0.25 ml of 1 N HCl and 40% trichloroacetic acid were added to the cell lysates. Insoluble materials were removed using Whatman GF/C filters. The filter paper was washed with ethanol three times, dried, and analyzed for radioactivity by liquid scintillation. Activity of the pGL-2A8 luciferase reporter gene was determined using the Dual-Luciferase Reporter Assay System (Promega). To normalize transfection efficiencies, the pRL-CMV vector (Renilla reniformis luciferase expression vector, Promega E2261) was cotransfected.
Western Blot and Immunoprecipitation.
For detection of pRb and p300 in the nuclear fractions of wild-type and AHR-null EFs, nuclear extracts were prepared from subconfluent EFs in 100-mm plates as described previously (Kurose et al., 1999). The extracted nuclear proteins were fractionated by 7.5% or 10% SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h with gentle shaking, then incubated with an anti-pRb or anti-p300 antibody for an additional 1 h. A peroxidase-conjugated goat anti-mouse IgG antibody and the ECL-PLUS detection kit (Amersham Pharmacia Biotech, Piscataway, NJ) were used for visualization of immune complexes. To detect p300-associated proteins, subconfluent EF cultures in 100-mm plates were treated with 5 nM TCDD or 0.05% (v/v) dimethyl sulfoxide for 30 min. The cells were washed with ice-cold PBS, then lysed on ice for 1 h by addition of 1 ml/plate of lysis buffer (50 mM Tris·HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 1 mM dithiothreitol, 0.1% Tween 20, 10% glycerol, 10 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM NaF, 1 μM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A). The cell debris was removed by centrifugation at 12,000g for 20 min at 4°C. Immunoprecipitations were performed with total cell lysates prepared from a 100-mm plate using an anti-p300 monoclonal antibody (4 μg/ml) followed by adsorption to protein G-Sepharose for 1 h at 4°C. Protein G-Sepharose complexes were washed five times with lysis buffer and the bound proteins were subjected to Western blot analysis. The membranes were incubated with antibodies against p300, AHR, or ARNT for 1 h at room temperature before washing and development.
Results
Lower Rates of DNA Synthesis in AHR-Null EF.
To compare DNA synthesis between AHR-null and wild-type EF, fibroblasts were prepared from the AHR-null and wild-type mouse embryos on gestational day 14.5. When placed into primary cultures in DMEM containing 10% FBS, AHR-null EF stopped proliferation at around passage 5, whereas wild-type EF still had high proliferating activity (data not shown). Therefore, passage 3 or 4 fibroblasts were used for further studies. For both EFs derived from wild-type and AHR-null, DNA synthesis rates were dependent on the cell density and serum concentration in the media (Fig.1). The DNA synthesis rate in AHR-null EF is slower than that of wild-type EF at all conditions tested. To confirm whether the slower DNA synthesis rate observed with AHR-null EF is caused by the lack of AHR function, the AHR cDNA was introduced into the AHR-null EF and DNA synthesis was measured. To assess the function of introduced exogenous AHR in recipient cells, a luciferase reporter gene (pGL-2A8) containing an XRE was cotransfected and luciferase activity was measured in the presence of TCDD. Introduced exogenous AHR functioned as a TCDD-dependent transcription factor as evident by the elevated luciferase activity (Fig. 2A). In addition, [3H]thymidine incorporation was increased by the transfection of AHR expression plasmid in a dose-dependent manner (Fig. 2B), indicating that the slow DNA synthesis rate observed with AHR-null EF is the result of loss of AHR function.

Effects of cell density and serum concentration in the culture medium on DNA synthesis in wild-type (▪) and AHR-null (■) EFs. Embryonic fibroblasts from gestational day 14.5 embryos were prepared from wild-type and AHR-null mice. The cells were plated in 12-well dishes at three different cell densities and synchronized in starvation medium. The cells were then cultured in the different concentrations of serum and pulse-labeled with 1 μCi/well of [3H]thymidine for 4 h as described. Data represent means and S.D. from three or four samples for each group.
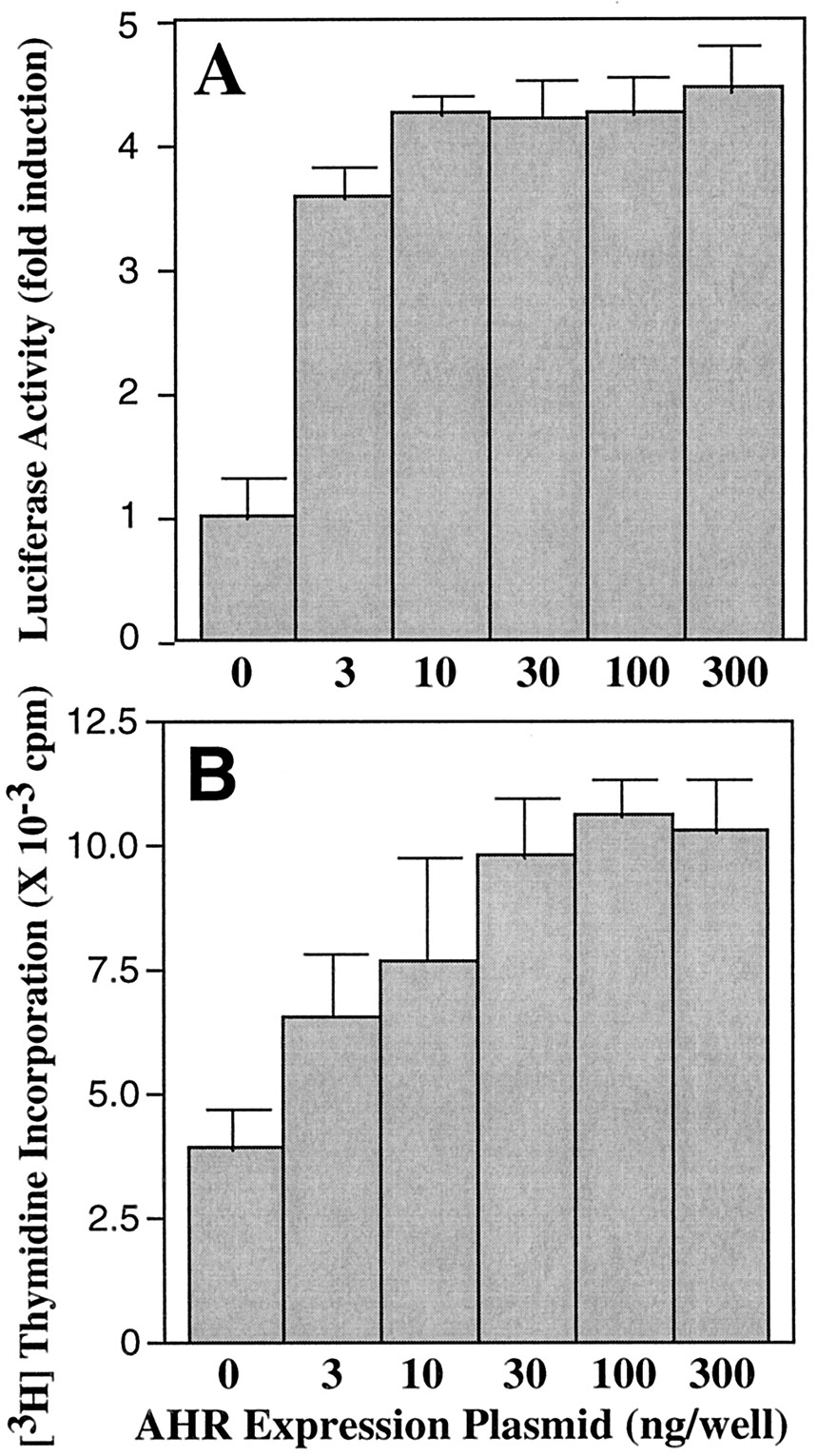
Effect of exogenously introduced AHR on XRE-mediated transcription activity and DNA synthesis in AHR-null EF. AHR-null EF, cultured in starvation medium for 24 h in 12-well dishes, were transfected with different amounts of the AHR expression plasmid (pCI/AHR) and 1 μg/well of XRE reporter gene plasmid (pGL-2A8). The medium was changed to 1% FCS-containing and cells were treated with 10 nM TCDD for 24 h before measuring luciferase activities (A). The fold induction refers to the ratio of luciferase activity of AHR-reintroduced EF to AHR-null EF. Other plates of cells were pulse-labeled with 1 μCi/well of [3H]thymidine for 4 h (B). Data represent means and S.D. from three or four samples for each group.
E1A Stimulates DNA Synthesis in AHR-Null and Wild-Type EFs.
Because it was established that adenovirus oncoprotein E1A enhances DNA synthesis by binding to Rb and p300/CBP, the effect of E1A was examined using AHR-null and wild-type EFs. An E1A expression plasmid pHβAPrE1A, in which transcription of the Ad2 E1A gene is driven by the human β-actin promoter, was transfected into EF cultured in starvation media before being pulse-labeled with [3H]thymidine. The maximal effect of E1A on DNA synthesis was observed at a cell density of 2 × 105 cells/well (data not shown). In addition, the FBS concentration during labeling with [3H]thymidine was found to alter the E1A effect; FBS concentration at 1% in the medium showed the maximal effect of E1A on the DNA synthesis. Therefore, these conditions were adopted to examine the effect of E1A on DNA synthesis. E1A stimulated DNA synthesis when introduced into wild-type EF in a dose-dependent manner. A maximal stimulation of approximately 5-fold-induction relative to the basal level of wild-type EF was achieved with 100 ng/well concentration. (Fig. 3). E1A stimulation of DNA synthesis in AHR null-EF cells was also observed, but the maximal induction, observed at the concentration of 1 μg/well, was lower than that of wild-type EF (3.5-fold of the basal level of AHR-null EF). These results indicated that AHR-null EF also responded to E1A-induced DNA synthesis, but were less sensitive than wild-type EF.
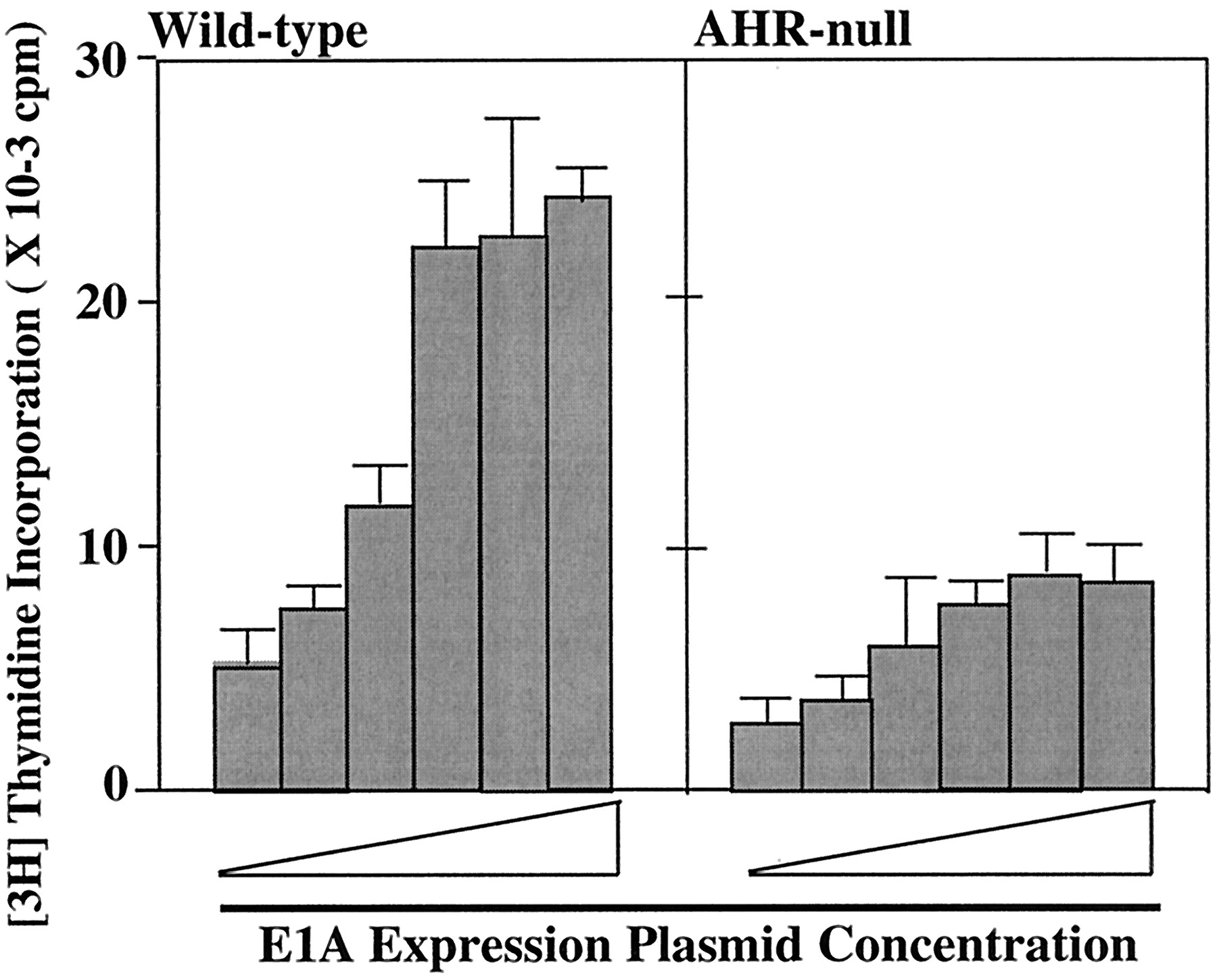
E1A-induced DNA synthesis in wild-type and AHR-null EFs. Wild-type and AHR-null EFs were cultured in 12-well dishes, and transfected with different amounts of the E1A expression plasmid (pHβAPrE1A) in starvation medium for 24 h. The DNA concentrations were at 0, 0.001, 0.01, 0.1, 1.0, or 10 μg/well. Cells were then cultured in 1% serum containing medium, and pulse-labeled with 1 μCi/well of [3H]thymidine for 4 h. Data represent means and S.D. from three or four samples for each group.
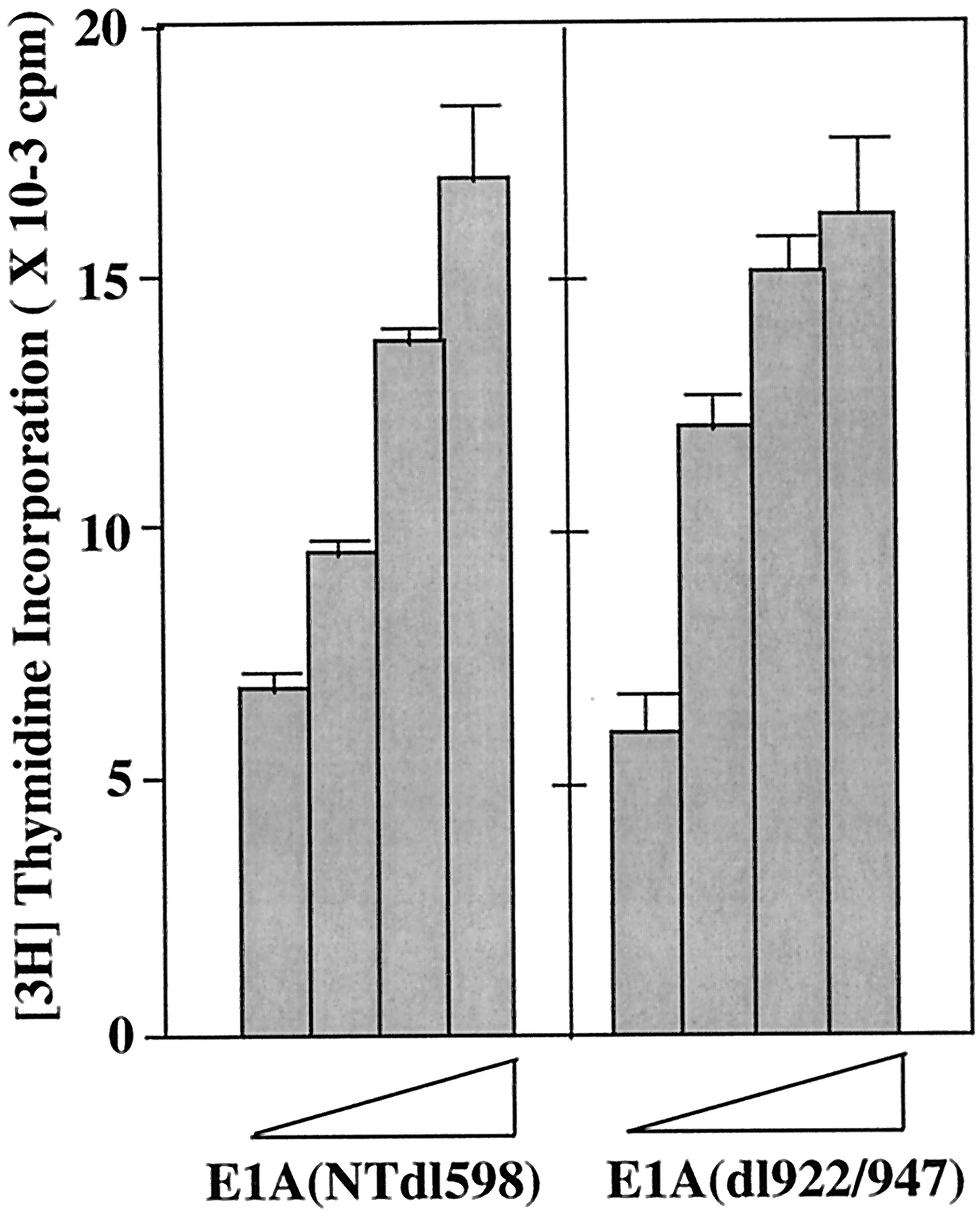
E1A is thought to bind to pRb and p300/CBP and inhibit their function (Trouche and Kouzarides, 1996). To investigate whether the slow DNA synthesis observed with AHR-null is caused by the loss of pRb or p300/CBP functions in the absence of AHR, two E1A mutants that have been shown to be selectively defective in binding to one of these proteins were tested. An E1A mutant, NTdl598, that lacks amino acid residues from 1 to 13 at the N terminus, was shown to be defective in p300/CBP binding but was able to bind pRb, p107, and p130 (Nakajima et al., 1992). Expression of NTdl598 in EF stimulated DNA synthesis in both wild-type and AHR-null EF to the same extent (Fig.4) suggesting that AHR is not required for Rb-dependent stimulation of DNA synthesis. On the other hand, the other E1A mutant, dl922/947 (lacks amino acid residues from 122 to 129), which retains the p300/CBP binding capacity but does not bind to pRb, p107, and p130 (Nakajima et al., 1992), stimulated DNA synthesis in the wild-type EF but not in AHR-null EF (Fig. 4). These data suggest that p300/CBP-dependent stimulation of DNA synthesis failed to occur in the absence of AHR. Taken together, these results suggest that both pRb and p300/CBP are involved in E1A-stimulated DNA synthesis in wild-type EF, and that AHR seems to be required for the function of E1A in p300/CBP-mediated cell cycle control.

Ability of E1A deletion mutants to stimulate DNA synthesis in wild-type and AHR-null EFs. Wild-type and AHR-null EFs were cultured in 12-well dishes and transfected with different amounts of the E1A deletion mutant expression plasmids (NTdl598 or dl922/947) in starvation medium for 24 h. The DNA concentrations were 0, 0.001, 0.01, 0.1, 1.0, 10, or 30 μg/well. Cells were then treated with 1% serum containing medium and pulse-labeled with 1 μCi/well of [3H]thymidine for 4 h. Data represent means and S.D. from three or four samples for each group.
To ensure that failure to induce DNA synthesis in the dl922/947-treated AHR-null EF is due to the loss of AHR function, the AHR-null EF expressing recombinant AHR were examined. In these cells, the extent of stimulation of DNA synthesis achieved by dl922/947 was similar to that of NTdl598 (Fig. 5). This result demonstrates that the lower rates of DNA synthesis observed with dl922/947-treated AHR-null EF are most likely caused by the absence of the AHR.

E1A mutants stimulate DNA synthesis in the AHR-reintroduced EF. AHR-null EF were cultured in 12-well dishes, and transfected with different amounts of the E1A deletion mutant expression plasmids (NTdl598 or dl922/947), and the AHR expression plasmid (pCI/AHR) in starvation medium for 24 h. The DNA concentrations were at 0, 1, 10, or 100 ng/well (NTdl598 or dl922/947) and 10 ng/well (pCI/AHR). Cells were then cultured in 1% serum containing medium, and pulse-labeled with 1 μCi/well of [3H]thymidine for 4 h. Data represent means and S.D. from three samples for each group.
pRb and p300 Are Expressed at Similar Levels in AHR-Null and Wild-Type EFs.
To determine whether the decreased amount of DNA synthesis in dl922/947-treated AHR-null cells is not the result of altered levels of p300 expression, the levels of p300 expression were examined. Nuclear extracts from wild-type and AHR-null EF were prepared and subjected to Western blot analyses using an anti-p300 antibody. For comparison, the amount of pRb was also determined. A representative blot from several experiments shows that the relative levels of p300 and pRb were similar in wild-type EF and AHR-null EF nuclear extracts (Fig. 6). The assignment of the pRb band was based solely on molecular weight estimates. The identity of the lower mobility protein in the wild-type and AHR-null EF is not known.

Western blot analysis of pRb and p300 in the nuclear extracts from wild-type and AHR-null EFs. Cell nuclear extracts (20 μg of protein per lane) from wild-type and AHR-null EFs were subjected to electrophoresis on SDS-polyacrylamide gel electrophoresis (10% for pRb and 7.5% for p300), transferred to a nitrocellulose membrane, and immunoblotted with an anti-pRb and an anti-p300 antibodies. Nuclear extract from NIH 3T3 cell was loaded as a positive control. Bars show the position of pRb (A) and p300 (B), respectively. Molecular mass standards are indicated in kilodaltons.
Interaction of AHR-ARNT Complex with p300.
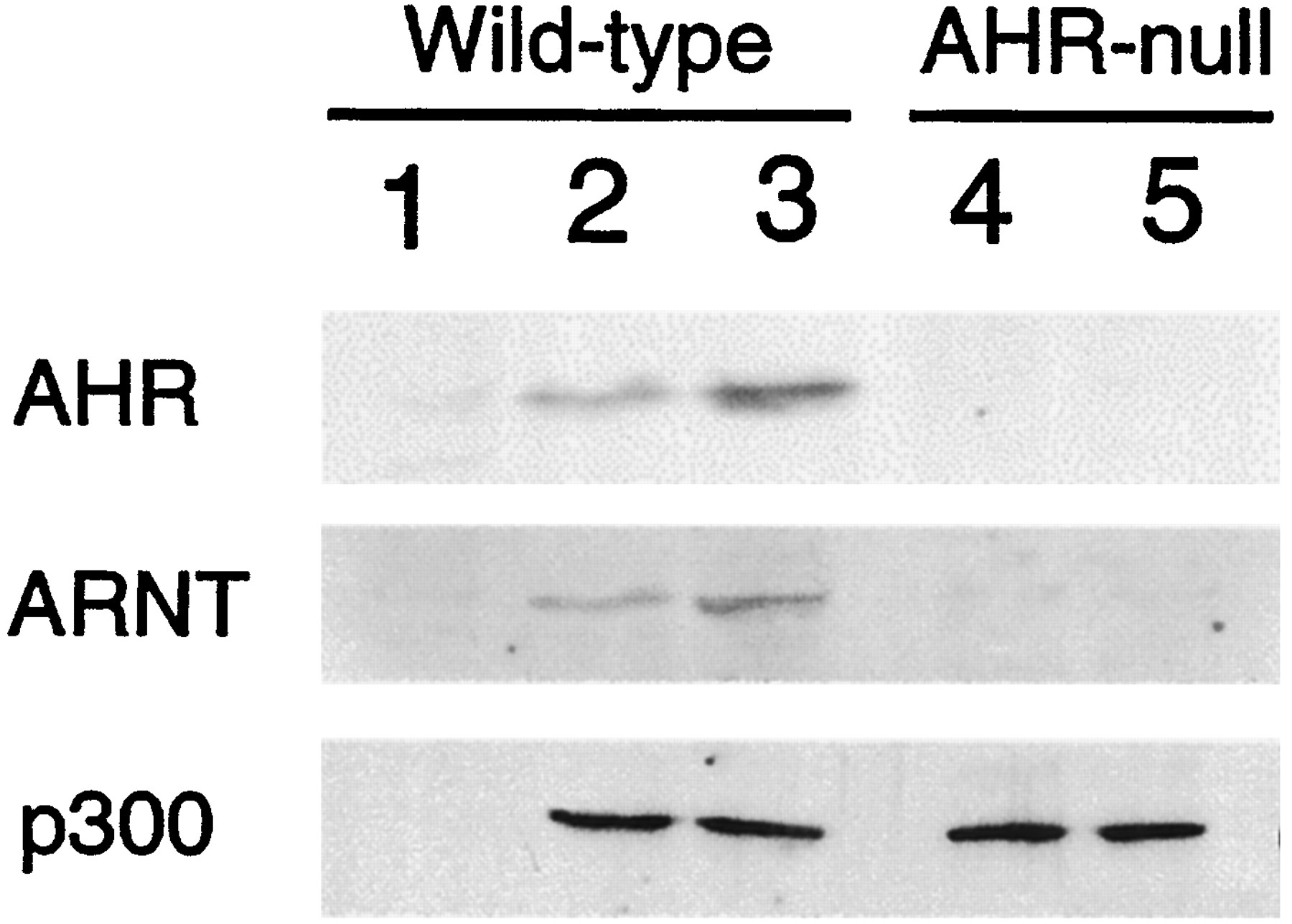
To determine whether there is a physical interaction between p300 and AHR/ARNT heterodimer in EF cells, immunoprecipitation was carried out using an anti-p300 antibody. To precipitate p300 and its associated proteins, cell lysates were prepared from control or TCDD-treated wild-type and AHR-null EFs, and incubated with an anti-p300 antibody. Both ARNT and AHR were coprecipitated with p300 in wild-type EF total cell extracts (Fig. 7, lanes 2 and 3). Their association with p300 was significantly enhanced when wild-type cells were treated with TCDD (Fig. 7, lane 3). Both AHR and ARNT were absent in the AHR-null EF (Fig. 7, lanes 4 and 5). To verify that the presence of AHR and ARNT in the precipitate were not caused merely by nonspecific absorption on the resin, a control immunoprecipitation experiment using the wild-type cell lysate and preimmune serum instead of anti-p300 antibody was carried out. AHR, ARNT, or p300 were not detected in the immunoprecipitated proteins (Fig. 7, lane 1). These results suggest that p300 associated with the AHR/ARNT heterodimer.

Coimmunoprecipitation of p300 and AHR-ARNT complex. Wild-type and AHR-null EFs were treated with dimethyl sulfoxide vehicle (lanes 1, 2, and 4) or with 10 nM TCDD for 1 h (lanes 3 and 5). Cell lysates were prepared from each group and incubated with an anti-p300 antibody. The immunoprecipitates of p300 and its associated proteins were subjected to SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and incubated with anti-AHR, anti-ARNT, or anti-p300 antibodies. Control immunoprecipitation experiment was performed using the wild-type cell lysate and preimmune serum instead of anti-p300 antibody (lane 1).
Discussion
The results in this study showed that DNA synthesis rates of AHR-null EF are slower than that of wild-type EF. Reintroduction of AHR into the AHR-null restored thymidine incorporation to levels comparable with wild-type cells. However, dose response for restoration of cell division differed from the transactivation through the XRE. This suggests that the mechanism for control of cell division by AHR is distinct from transactivation through the XRE. Perhaps higher levels of AHR are required for sequestering other nuclear factors that affect cell division. The present finding of a role for AHR in control of cell division is consistent with the previous observation with mouse hepatoma Hepa 1c1c7, in which cell doubling time is dependent on AHR content (Ma and Whitlock, 1996). It was also suggested that a finite level of AHR function is required for Hepa 1c1c7 cells to survive in culture (Ma and Whitlock, 1996). Interestingly, cells used in this study are completely viable, although the AHR expression is completely absent. This observation indicates that although AHR affects DNA synthesis, it is not an essential factor in cell cycle progression. In this regard, it is noteworthy that the AHR-null mice are viable (Fernandez-Salguero et al., 1995) whereas mice lacking expression of pRb (Sherr, 1996) or p300 (Yao et al., 1998) are reported to be embryonic lethal.
DNA synthesis in AHR-null EF stimulated by E1A was slower than that of wild-type EF, suggesting AHR plays a role in cell cycle control. Among factors that are known to be targets of E1A are pRb and p300/CBP (Trouche and Kouzarides, 1996). Because these factors are reported to interact with the AHR/ARNT heterodimer (Kobayashi et al., 1997; Ge and Elferink, 1998), they were examined using two E1A mutants. NTdl598, which can bind to pRb but not p300/CBP (Nakajima et al., 1992), stimulated DNA synthesis both in AHR-null and wild-type EFs. In contrast, dl922/947, which binds to p300/CBP but not pRb (Nakajima et al., 1992), stimulated DNA synthesis only in wild-type EF. This does not seem to be attributable to the differences in p300 levels between AHR-null and wild-type Efs (Fig. 6). Because p300 associates with the AHR/ARNT complex, the role of AHR in modulation of p300 function is likely to be direct. The immunoprecipitation data suggest that formation of the AHR/ARNT heterodimeric complex may be a prerequisite for p300 binding, because ARNT did not bind to p300 in AHR-null EF. Kobayashi et al. showed the interaction between p300 and ARNT protein from the results of two-hybrid assay and pull-down assay using human embryonic kidney cell line (Kobayashi et al., 1997). It should be noted that there remains a possibility that endogenous AHR is present in the human embryonic kidney cell line used in their study; therefore, AHR might be involved in the interaction of p300 and ARNT in their system. Thus, their observation may not differ from ours.
These data suggest that the AHR in some way modulates the function of p300 in cell cycle control. However, the mechanism by which AHR affects the function of p300 in the cell cycle is not clear. The p300 is a coactivator required for p53 induction of the p21WAF1/CIP1 gene resulting in inhibition of the cell division (Avantaggiati et al., 1997; Gu et al., 1997; Lill et al., 1997; Somasundaram and El-Deiry, 1997). Because E1A inhibits the interaction of p53 and p300 (Somasundaram and El-Deiry, 1997), there is a possibility that the AHR/ARNT complex could modulate or influence the interaction between these two proteins either directly or indirectly.
Post-translational modifications, such as phosphorylation, may modulate the function of p300. In fact, phosphorylated p300 appears between M phase and S phase in the cell cycle (Banerjee et al., 1994; Kitabayashi et al., 1995). Therefore, the possibility exists that post-translational modification of p300 differs between wild-type and AHR-null EFs and that modifications of the protein might alter the responsiveness to E1A-induced DNA synthesis. It was recently revealed that AHR-null EFs are enriched in the G2/M phase of the cell cycle with lower Cdc2 and Plk protein contents (Elizondo et al., 2000). Thus, it cannot be ruled out that low activity of mitotic kinases may affect the phosphorylation status of p300 to modulate its p300 function in AHR-null EF.
In the cells used in this study and the Hepa1c1c7 cells used previously (Ma and Whitlock, 1996), stimulation of DNA synthesis is dependent on AHR, but independent of AHR activation by exogenous ligands such as TCDD. However, the AHR ligand benzo[a]pyrene was reported to suppress the cell cycle in murine Swiss 3T3 cells (Vaziri and Faller, 1997), and TCDD arrests rat 5L hepatoma cells in the G1 phase of the cell cycle (Gottlicher et al., 1990; Ge and Elferink, 1998). In addition, it was suggested that AHR-mediated G1 arrest is achieved by the interaction of AHR with pRb (Ge and Elferink, 1998). Thus, different effects of AHR on the cell cycle may be caused in part by the balance between AHR/ARNT-pRb complex-mediated pathway that is influenced by exogenous ligands and the AHR/ARNT-p300 complex-mediated pathway that may be ligand-independent or dependent on an endogenous ligand. Further studies will be required to determine the role of AHR in mediating p300-dependent cell cycle regulation.
Acknowledgments
We thank Kinichiro Oda, Tokyo Science University (Noda, Japan) for providing the E1A expression plasmids. We are deeply indebted to Kyung Lee, Shioko Kimura, and Albert Fornace for helpful suggestions and review of the manuscript.
Footnotes
- Received February 22, 2000.
- Accepted June 23, 2000.
-
Send reprint requests to: Dr. Frank J. Gonzalez, Laboratory of Molecular Carcinogenesis, National Cancer Institute, 9000 Rockville Pike, Bldg. 37, Room 3E24, Bethesda, MD 20892-0001.
-
M.T. was supported by a grant from the Japan Health Science Foundation.
Abbreviations
- AHR
- aryl hydrocarbon receptor
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- ARNT
- AHR nuclear translocator
- XRE
- xenobiotic response element
- EF
- embryonic fibroblast
- CBP
- cAMP response element-binding protein
- pRb
- retinoblastoma protein
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}