


Abstract
The major mechanism of agonist-induced internalization of G protein-coupled receptors (GPCRs) is β-arrestin- and dynamin-dependent endocytosis via clathrin-coated vesicles. However, recent reports have suggested that some GPCRs, exemplified by the AT1 angiotensin receptor expressed in human embryonic kidney (HEK) 293 cells, are internalized by a β-arrestin- and dynamin-independent mechanism, and possibly via a clathrin-independent pathway. In this study, agonist-induced endocytosis of the rat AT1A receptor expressed in Chinese hamster ovary (CHO) cells was abolished by clathrin depletion during treatment with hyperosmotic sucrose and was unaffected by inhibition of endocytosis via caveolae with filipin. In addition, internalized fluorescein-conjugated angiotensin II appeared in endosomes, as demonstrated by colocalization with transferrin. Overexpression of β-arrestin1(V53D) and β-arrestin1(1–349) exerted dominant negative inhibitory effects on the endocytosis of radioiodinated angiotensin II in CHO cells. GTPase-deficient (K44A) mutant forms of dynamin-1 and dynamin-2, and a pleckstrin homology domain-mutant (K535A) dynamin-2 with impaired phosphoinositide binding, also inhibited the endocytosis of AT1 receptors in CHO cells. Similar results were obtained in COS-7 and HEK 293 cells. Confocal microscopy using fluorescein-conjugated angiotensin II showed that overexpression of dynamin-1(K44A) and dynamin-2(K44A) isoforms likewise inhibited agonist-induced AT1 receptor endocytosis in CHO cells. Studies on the angiotensin II concentration-dependence of AT1 receptor endocytosis showed that at higher agonist concentrations its rate constant was reduced and the inhibitory effects of dominant negative dynamin constructs were abolished. These data demonstrate the importance of β-arrestin- and dynamin-dependent endocytosis of the AT1 receptor via clathrin-coated vesicles at physiological angiotensin II concentrations.
The pressor octapeptide hormone, angiotensin II (Ang II), exerts the majority of its physiological effects on cardiovascular regulation and salt-water balance by activating the Gq-coupled AT1 angiotensin receptor (De Gasparo et al., 2000). The AT1 receptor also activates intracellular signaling pathways that stimulate cell growth including activation of tyrosine kinases and small GTP-binding proteins (Berk, 1999; De Gasparo et al., 2000), and is rapidly internalized after Ang II binding (Thomas, 1999; Hunyady et al., 2000). Agonist-induced endocytosis of G protein-coupled receptors (GPCRs) initiates a process by which desensitized receptors are resensitized and recycled to the plasma membrane (Krupnick and Benovic, 1998). Sequestration of the β2-adrenergic receptor has been shown to require the binding of β-arrestin proteins to its cytoplasmic tail after agonist-induced activation and phosphorylation by G protein-coupled receptor kinases (Zhang et al., 1996; Ferguson et al., 1997; Krupnick and Benovic, 1998). β-arrestins direct the phosphorylated receptors to clathrin-coated pits and induce the formation of clathrin-coated vesicles (Goodman et al., 1996). The role of β-arrestins in receptor internalization has been demonstrated for several GPCRs (Bünemann et al., 1999). Although β-arrestins translocate to the plasma membrane upon agonist stimulation of many GPCRs (Zhang et al., 1999), it has been reported that the internalization of some of these GPCRs, including that of the AT1 receptor in HEK 293 cells, is independent of β-arrestin (Zhang et al., 1996; Bünemann et al., 1999).
The β-arrestin-dependent internalization of GPCRs also requires dynamins, GTPase proteins of ∼100 kDa that participate in the endocytosis of nutrient and growth factor receptors. Studies with GTPase-deficient dynamins [e.g., dynamin-1(K44A)] have suggested that internalization of GPCRs can occur via dynamin-dependent and -independent mechanisms (Zhang et al., 1996; Ferguson et al., 1997;Bünemann et al., 1999). Dominant negative dynamin-1, the neuronal isoform of dynamin, has been widely used to study the internalization of GPCRs. However, most Ang II target tissues contain the ubiquitous dynamin-2 isoform (Schmid et al., 1998). Although dynamin-2 has been shown to participate in clathrin-mediated endocytosis of the transferrin receptor (Altschuler et al., 1998; Kasai et al., 1999), dominant negative mutants of dynamin-2 have not been used to investigate the internalization of GPCRs. Recent studies have shown that pleckstrin homology (PH) domain mutants of dynamin-1 with impaired phosphoinositide binding (e.g., K535A) also act as dominant-negative inhibitors of transferrin receptor endocytosis (Achiriloaie et al., 1999; Lee et al., 1999; Vallis et al., 1999). However, the role of the PH domain of dynamins in GPCR internalization has not been studied extensively.
Most of the available data indicate that endocytosis of the AT1 receptor in Ang II target cells occurs via clathrin-coated vesicles (Thomas, 1999; Hunyady et al., 2000). Electron microscopic studies in rat aortic smooth muscle and adrenal glomerulosa cells have detected the internalized AT1 receptor in clathrin-coated pits and vesicles (Bianchi et al., 1986; Anderson et al., 1993). Also, inhibition of endocytosis via clathrin-coated vesicles by K+-depletion or phenylarsine oxide treatment prevents AT1 receptor internalization in several tissues, including smooth muscle, adrenal glomerulosa, and kidney epithelial cells (Hunyady et al., 2000). Recent studies in HEK 293 cells have shown that the internalized AT1receptor colocalizes with the transferrin receptor, which is known to be internalized by clathrin-mediated endocytosis (Hein et al., 1997). Also, in CHO-K1 cells stably transfected with the AT1A receptor, hyperosmotic sucrose treatment inhibited Ang II-mediated receptor endocytosis (Thomas et al., 1996). Endocytosis of the AT1 receptor in vascular smooth muscle cells has also been proposed to occur via caveolae (Ishizaka et al., 1998), but the relative contributions of coated pits and caveolae to AT1 receptor endocytosis have not been investigated.
Studies performed in HEK 293 cells have suggested that endocytosis of the AT1 receptor is the prototype for dynamin- and β-arrestin-independent internalization of GPCRs (Zhang et al., 1996). However, it has been suggested that the rapid agonist-induced phosphorylation of the AT1 receptor may cause its internalization in CHO and COS-7 cells (Smith et al., 1998; Thomas et al., 1998). Because phosphorylation of GPCRs promotes β-arrestin binding to the receptor, its proposed role in AT1receptor internalization is consistent with the possibility that β-arrestin-dependent mechanisms participate in AT1 receptor endocytosis in these cells.
The degree of internalization of GPCRs is determined by the balance between agonist-regulated endocytosis and constitutive recycling of the receptors to the cell surface (Koenig and Edwardson, 1997). In the present study, the mechanisms involved in endocytosis of the AT1 receptor were investigated in CHO, COS-7, and HEK 293 cells by inhibition of clathrin-mediated and caveolar endocytosis, and by the use of dominant negative mutants of β-arrestin1, dynamin-1, and dynamin-2.
Experimental Procedures
Materials.
The cDNA of the rat vascular smooth muscle AT1A receptor was obtained from Dr. K. E. Bernstein (Atlanta, GA). The cDNAs of the HA epitope-tagged wild-type and K44A mutant dynamin-1(aa) and 2(aa) sequences subcloned into pcDNA3 vector were kindly provided by Dr. K. Nakayama (Tsukuba Science City, Ibaraki, Japan). The cDNAs of wild-type and mutant β-arrestin1 constructs were generous gifts from Dr. M. G. Caron (Durham, NC) and Dr. S. S. G. Ferguson (London, Ontario, Canada). Anti-dynamin-1 and -2 antibodies and HRP-conjugated donkey anti-goat IgG antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-HA.11 monoclonal antibody was from Babco (Berkeley, CA) and HRP-conjugated goat anti-mouse antibody was from Pierce Chemical (Rockford, IL). Fluo-Ang II was obtained from NEN Life Science Products (Boston, MA), and Alexa Fluor 594 conjugate of transferrin was from Molecular Probes (Eugene, OR). All other chemicals and reagents unless otherwise stated were from SIGMA-Aldrich (St. Louis, MO).
Mutagenesis and Transfection.
Substitution of lysine535 of dynamin-2 with alanine [dynamin-2(K535A)] was created using the Muta-Gene kit (Bio-Rad, Hercules, CA). The sequence of the mutant was verified by dideoxy sequencing. CHO-K1, COS-7, and HEK 293 cells were transiently transfected in 24-well plates with plasmids containing AT1A receptor cDNAs, and wild-type or mutant dynamins or β-arrestins using 12 μg/ml Lipofectamine (Life Technologies, Gaithersburg, MD) as described previously (Hunyady et al., 1994). For confocal microscopy, cells were transfected on glass coverslips with the indicated constructs using 3 μl/ml FuGENE 6 (Roche Diagnostics, Nutley, NJ). CHO cells were maintained in NaHCO3-buffered Ham's F-12 medium containing 10% fetal bovine serum, 100 μg/ml streptomycin, and 100 IU/ml penicillin (Life Technologies). HEK 293 and COS-7 cells were maintained as described previously (Hunyady et al., 1994).
Receptor Endocytosis in Transiently Transfected CHO Cells.
To determine the internalization kinetics of the AT1A receptor, 125I-Ang II [2.5 kBq/ml (∼0.03 nM), or the indicated concentration] was added in 0.25 ml of HEPES-buffered Ham's F-12 or Dulbecco's modified Eagle's medium, and the cells were incubated at 37°C for the indicated times. Incubations were stopped by placing the cells on ice and rapidly washing them twice with ice-cold PBS. Acid-released and acid-resistant radioactivities were separated and measured by γ-spectrometry as described earlier (Hunyady et al., 1994). The percentage of internalized ligand at each time point was calculated from the ratio of the acid-resistant specific binding to the total (acid-resistant + acid-released) specific binding. The values for the endocytotic rate constant (k e) (defined as the probability of an occupied receptor being internalized within 1 min at 37°C) were calculated based on the data obtained 2, 3, and 5 min after addition of radiolabeled agonist, as described by Wiley and Cunningham (1982), to quantify endocytosis of the EGF receptor. The algorithms for this calculation were kindly provided by Dr. H. S. Wiley (Salt lake City, UT). Because agonist-induced endocytosis of the AT1 receptor behaved as a second-order process, similar to that of the EGF receptor (Lund et al., 1990),k e values were also determined at different Ang II concentrations.
Western Blot Analysis.
For immunodetection of expressed proteins 48 h after transfection, cells were scraped into 200 μl of Laemmli buffer containing protease inhibitors (10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml trypsin-chymotrypsin inhibitor, 10 μg/ml pepstatin A, 10 μg/ml benzamidine). After centrifugation, the supernatant proteins were analyzed on 8% denaturating polyacrylamide gels and transferred to nitrocellulose membranes. Blots were then probed with primary antibody and detected with HRP-conjugated secondary antibodies using the SuperSignal West Pico detection kit (Pierce Chemical).
Confocal Laser Scanning Microscopy.
For microscopy studies, CHO cells were grown on glass coverslips and transiently transfected as described above. The coverslips were mounted on the imaging chamber 48 h later, and the cells were maintained in HEPES-buffered Ham's F-12 medium at 37°C. Cells were incubated with 50 nM Fluo-Ang and, in some experiments, with 25 ng/μl Alexa Fluor 594 conjugate of transferrin in the imaging chamber for the indicated times at 37°C and images were detected with a Bio-Rad MRC-1024 Confocal Laser Scanning System with an Olympus BH2 microscope (Olympus, Tokyo, Japan). Fluorescein and Alexa Fluor 594 were excited with an argon/krypton laser and the emitted fluorescence was detected with 522/32 and 605/32 band filters, respectively.
Results
Inhibition of AT1A Receptor Endocytosis by Hyperosmotic Sucrose Treatment and Colocalization of the Receptor with Transferrin.
To study the role of clathrin-coated vesicles in AT1A receptor endocytosis, the effect of hyperosmotic sucrose on endocytosis of 125I-Ang II was measured in CHO cells transiently transfected with AT1A receptor cDNA. This treatment inhibits clathrin-mediated endocytosis by inducing abnormal clathrin polymerization into empty microcages on the membrane (Heuser and Anderson, 1989). Cells were preincubated for 30 min in HEPES-buffered Ham's F-12 medium containing 0.45 M sucrose, and internalization experiments were carried out in the same medium at 37°C. In these cells, the characteristically rapid agonist-induced endocytosis of the AT1A receptor was almost abolished by treatment with hyperosmotic sucrose (Fig. 1, upper part), and the k e value of the receptor was reduced from 0.44 ± 0.03 to 0.009 ± 0.003 per min (n = 3). In contrast, preincubation of the cells for 30 min with 5 μg/ml filipin, which inhibits the formation of caveolae (Anderson, 1998), had no detectable effect on the endocytosis of the AT1A receptor (Fig. 1, upper part). These findings suggest that the AT1A receptor expressed in CHO cells is predominantly internalized via clathrin-coated vesicles.

Effect of hyperosmotic sucrose treatment and filipin on the endocytosis of the rat AT1A receptor expressed in CHO cells (top) and colocalization of the internalized receptor with transferrin (bottom). Top, CHO cells were preincubated for 30 min at 37°C in HEPES-buffered Ham's F-12 medium in the absence (●) or presence of 0.45 M sucrose (■) or 5 μg/ml filipin (▵) before addition of 125I-Ang II (0.03 nM). Receptor endocytosis was measured at 37°C as described under Experimental Procedures. The percentage of internalized ligand at each time point was calculated from the ratio of the acid-resistant specific binding to the total (acid-resistant + acid-released) specific binding. Data are shown as means ± S.E.M. of three independent experiments each performed in duplicate. Bottom, confocal microscopy studies with Fluo-Ang II (A) and Alexa Fluo 594 transferrin (B) in CHO cells transiently transfected with the AT1A receptor. CHO cells were grown on glass coverslips and transfected 48 h before experiments. Cells were incubated at 37°C in HEPES-buffered Ham's F-12 medium containing 50 nM Fluo-Ang II and 25 ng/μl Alexa Fluor 594 transferrin for 35 min. Localization of the fluorescence of Fluo-Ang II and Alexa Fluor 594 conjugate of transferrin in the same cell is shown in A and B, respectively. Arrowheads indicate vesicles containing both Fluo-Ang II and Alexa Fluor 594 transferrin. Bar, 10 μm.
This conclusion was supported by the colocalization of internalized Fluo-Ang II with transferrin. Endocytosis of the AT1A and transferrin receptors was visualized by addition of 50 nM Fluo-Ang II and 25 ng/μl Alexa Fluor 594 conjugate of transferrin to transiently transfected CHO cells. After a 35-min incubation at 37°C, Fluo-Ang II showed cytoplasmic localization, and was colocalized with fluorescent Alexa Fluor 594-transferrin both under the plasma membrane and deep within the cell (Fig. 1, lower part). Most of the internalized Fluo-Ang II was observed in vesicles containing transferrin. Colocalization of the internalized Fluo-Ang II with transferrin, a well-established marker for endocytosis via clathrin-coated pits (Anderson, 1998), confirms that endocytosis of AT1A receptor occurs predominantly via the same mechanism.
Coexpression of mutant β-arrestins inhibits AT1Areceptor endocytosis.
The β-arrestin dependence of AT1A receptor endocytosis in CHO cells was investigated by overexpression of mutants of β-arrestin1. β-arrestin1(V53D) has reduced ability to bind to GPCRs (Ferguson et al., 1996; Krupnick et al., 1997), and has been widely used to investigate the role of β-arrestins in the internalization of GPCRs. The requirement for the clathrin or AP-2 adapter protein binding domain of β-arrestin1 was investigated by overexpression of β-arrestin1(1–349), in which the clathrin and the AP-2 binding domains are deleted (Krupnick et al., 1997; Laporte et al., 1999). 0.5 μg of wild-type or mutant β-arrestin1 were transiently coexpressed with the AT1A receptor in CHO cells, because no major effects on total 125I-Ang II binding or cell number were observed at this concentration of the constructs (data not shown). In transfected CHO cells, β-arrestin1(V53D) had a modest inhibitory effect on AT1A receptor endocytosis (Fig. 2), whereas wild-type β-arrestin1 had no effect on this process. Overexpression of β-arrestin1(1–349) caused more marked inhibition of endocytosis, and thek e value was reduced by almost 90% (Table1). These data suggest that β-arrestins participate in the endocytosis of the AT1Areceptors in CHO cells.

Effect of overexpression of dominant negative β-arrestins on AT1A receptor endocytosis in CHO cells. Cells were grown in 24-well plates and cotransfected with 0.5 μg of AT1A receptor cDNA and 0.5 μg of empty pcDNA3 vector (●), β-arrestin1(V53D) (■), or β-arrestin1(1–349) (▴). Endocytosis of 125I-Ang II was measured as described underExperimental Procedures. Data are shown as means ± S.E.M. of three independent experiments, each performed in duplicate.
Rate constants of AT1A receptor endocytosis. Thek e values (per min) were calculated based on results of endocytosis of 125I-Ang II in cells transfected with the rat AT1A receptor and cotransfected with empty pcDNA3 vector (Mock), or with the cDNA of the indicated wild-type or dominant negative mutant proteins. In each experiment, the data taken at 2, 3, and 5 min of incubation were used for calculation ofk e values, which are shown as means ± S.E.M. The number of independent experiments, performed in duplicate, is shown in parentheses.
Inhibition of AT1A Receptor Endocytosis by Coexpression of Mutant Dynamins.
GTPase-deficient mutant (K44A) and PH domain mutant (K535A) dynamins were used to study the role of dynamin in AT1A receptor endocytosis. Coexpression of 0.5 μg of the dynamin cDNA constructs with the AT1Areceptor in CHO cells had no major effect on total125I-Ang II binding or cell number (data not shown), and this amount of the wild-type or mutant cDNAs was used to study the endocytosis of the AT1 receptor. As shown in Fig. 3A, coexpression of dynamin-1(K44A), dynamin-2(K44A), and dynamin-2(K535A) inhibited AT1A receptor endocytosis with high to moderate efficacy. Under the same conditions, wild-type dynamin-1 and dynamin-2 exerted only minor inhibitory effects. The correspondingk e values are shown in Table 1. These data indicate that the GTPase activity and the phospholipid binding of dynamin are required for the endocytosis of the agonist-activated AT1A receptor. Western blot analysis of transfected cells from the same experiments with anti-HA epitope antibody showed that overexpression of dynamin-1(K44A) was slightly higher than that of dynamin-2(K44A) in CHO cells (data not shown), which may explain its greater inhibition of AT1receptor endocytosis. Immunoblot analysis with anti-dynamin-1 and anti-dynamin-2 antibodies also demonstrated the presence of the expressed constructs in the cells, and showed that dynamin-2 is the endogenously expressed isoform of dynamin in CHO and COS-7 cells (Fig.3B).

Effect of overexpression of wild-type and dominant negative dynamins on AT1A receptor endocytosis. CHO cells were grown in 24-well plates and cotransfected with 0.5 μg of AT1A receptor cDNA and 0.5 μg of empty pcDNA3 vector (●), dynamin-1 (■), dynamin-2 (▴), dynamin-1(K44A) (○), dynamin-2(K44A), (♦) or dynamin-2(K535A) (▵). A, effects of overexpression of wild-type and dominant negative dynamins on AT1A receptor endocytosis are shown. Endocytosis of125I-Ang II was measured as described underExperimental Procedures. Data are shown as means ± S.E.M. of three independent experiments, each performed in duplicate. B, detection of overexpressed dynamins in total cell lysates of CHO and COS-7 cells. CHO and COS-7 cells transiently transfected with 0.5 μg of AT1A receptor cDNA and 0.5 μg of empty pcDNA3 vector (first two lanes), dynamin-1(K44A) (lane 1), dynamin-1 (lane 2), dynamin-2(K44A) (lane 3), dynamin-2 (lane 4) or dynamin-2(K535A) (lane 5) were harvested 48 h after transfection. Equal amounts of cell lysates were analyzed by immunoblotting using anti-dynamin-1 (dyn1, top) or dynamin-2 (dyn2, bottom) antibodies as described underExperimental Procedures. These blots are representative of three independent experiments.
Confocal Microscopy Studies on AT1A Receptor Endocytosis.
Agonist-induced endocytosis of the AT1A receptor was visualized by the addition of 50 nM Fluo-Ang II to transiently transfected CHO cells. At 4°C, Fluo-Ang II was localized at the cell surface and no endocytosis was observed (Fig. 4A). During subsequent incubation at 37°C in the presence of 50 nM Fluo-Ang II, endocytosis of the ligand into vesicles was observed within 5 min (data not shown). After a 20-min incubation at 37°C, Fluo-Ang II showed cytoplasmic localization in 98% of 57 cells studied (Fig. 4B). The distribution of Fluo-Ang II was markedly altered by coexpression of dominant negative dynamin-1, with diminished uptake of the ligand after 20 min of incubation at 37°C in the cells expressing dynamin-1(K44A) (Fig. 4C). In 70% of the 48 cells studied, the ligand remained localized to the cell surface and no intracellular fluorescence was observed. Dynamin-2(K44A) had a similar inhibitory effect on AT1A receptor endocytosis in 68% of 119 cells studied (Fig. 4D). Wild-type dynamins had no detectable effect on the endocytosis of Fluo-Ang II (data not shown). These observations confirm that the endocytosis of the AT1A receptor in CHO cells is dynamin-dependent.

Visualization of the inhibitory effects of dominant negative dynamins on AT1A receptor endocytosis in CHO cells. Cells were grown on glass coverslips and transiently transfected 48 h before experiments. Fluo-Ang II was used for visualization of the expressed AT1A receptors. Localization of Fluo-Ang II is shown in: A, cells incubated in HEPES-buffered F-12 medium containing 50 nM Fluo-Ang II for 15 min at 4°C; B, cells incubated at 37°C for 20 min, which caused endocytosis of Fluo-Ang II into the cells; C, cells cotransfected with the AT1A receptor and dynamin-1(K44A), and incubated with 50 nM Fluo-Ang II at 37°C for 20 min; and D, cells expressing the AT1A receptor and dynamin-2(K44A), and incubated with 50 nM Fluo-Ang II at 37°C for 20 min. Bar, 10 μm.
Ang II Concentration-Dependence of AT1A Receptor Endocytosis.
In our previous kinetic experiments the concentration of radiolabeled Ang II was in the physiological range (0.03 nM). To determine whether the ligand concentration influences the kinetics of receptor endocytosis, endocytosis of the AT1Areceptor was analyzed at higher Ang II concentrations. The concentration of 125I-Ang II was increased to 0.2 nM, and the agonist concentration was further increased by adding unlabeled Ang II (up to 30 nM) to the medium, and endocytosis of the receptor was measured in CHO cells expressing the AT1A receptor with or without coexpression of dynamin-2(K44A). The endocytotic rate constant of the AT1A receptor progressively decreased at higher Ang II concentrations (Table 1). As shown in Fig.5, the inhibitory effect of dynamin-2(K44A) on AT1A receptor endocytosis was evident at 0.2 nM Ang II, but at 30 nM Ang II, its effects on the kinetics and the rate constant of AT1 receptor endocytosis were diminished (Fig. 5; Table 1). Similar results were obtained with dynamin-1(K44A) (data not shown). These data demonstrate that although endocytosis of the AT1A receptor is clearly dynamin-dependent at subnanomolar Ang II concentrations, the inhibitory effect of dominant negative dynamins is not demonstrable at high Ang II concentrations.
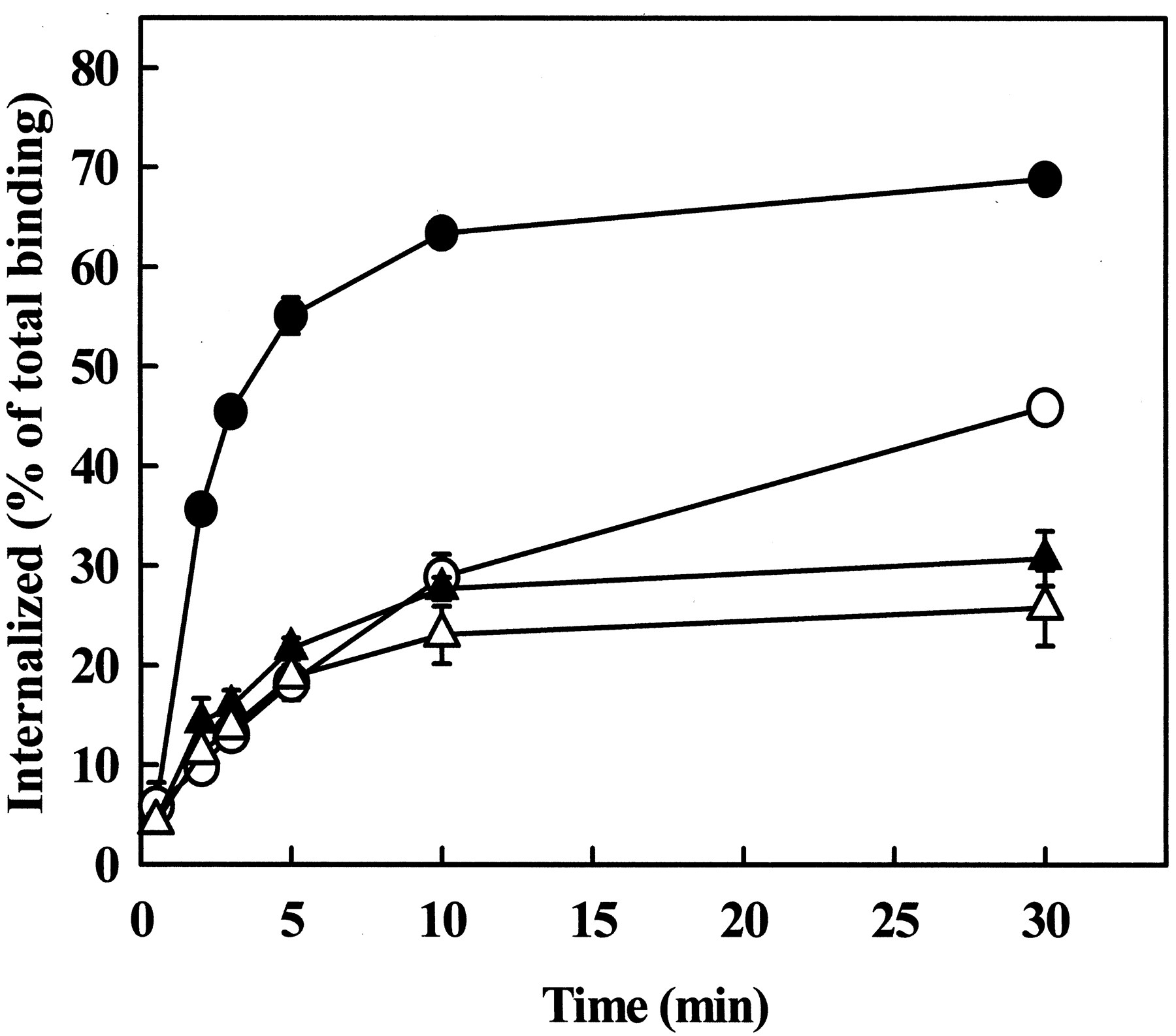
Effects of Ang II concentration on the kinetics of AT1A receptor endocytosis in CHO cells. CHO cells were grown in 24-well plates and transiently transfected with 0.5 μg of AT1A receptor only (solid symbols) or AT1Areceptor and 0.5 μg of dynamin-2(K44A) (open symbols). Endocytosis of125I-Ang II (0.2 nM) was measured in the absence (circles) or presence (triangles) of added Ang II to bring the agonist concentration to 30 nM. Data are shown as means ± S.E.M. of three independent experiments, each performed in duplicate.
Effects of coexpression of dominant negative dynamins and β-arrestins on AT1A receptor endocytosis in COS-7 cells.
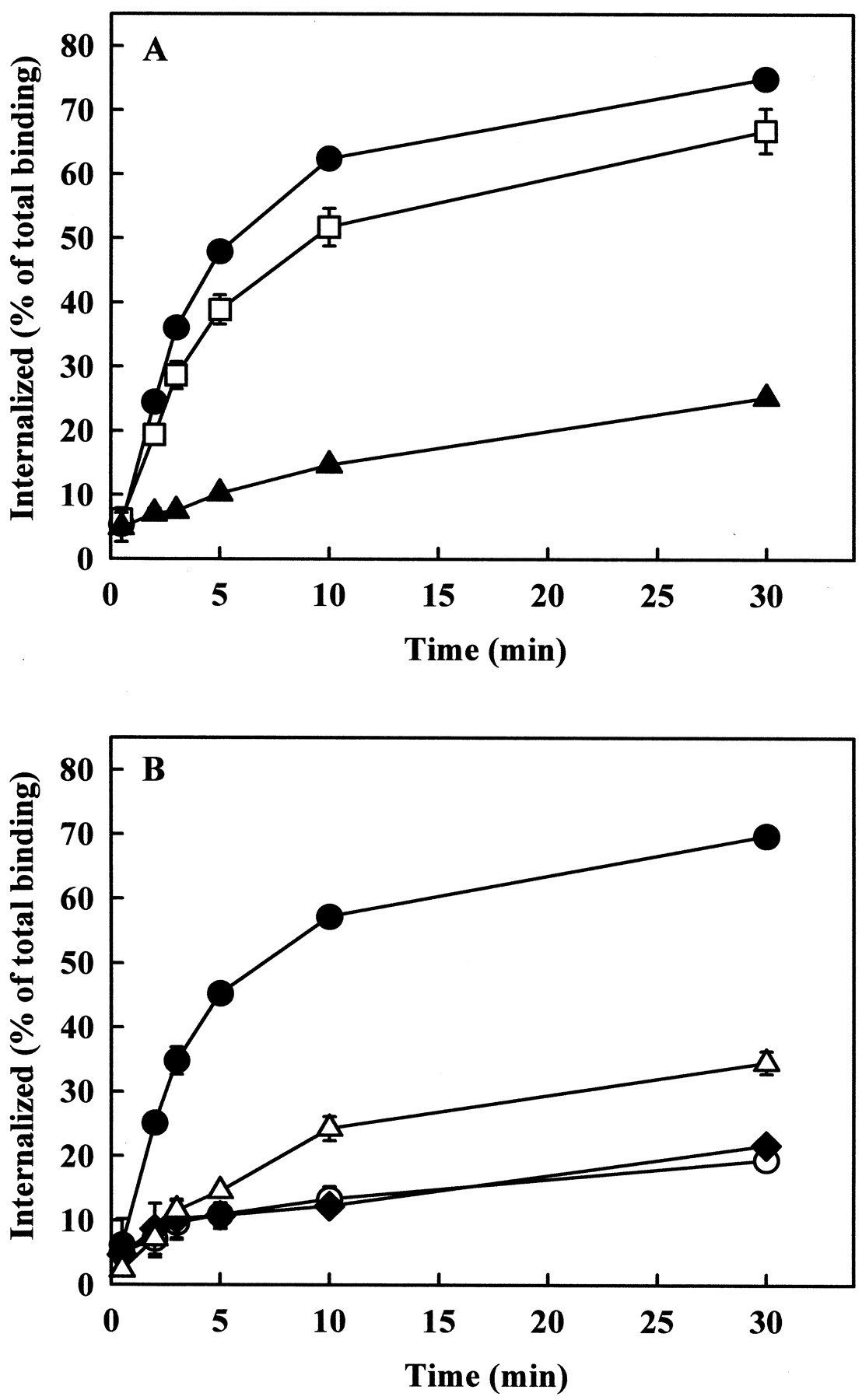
The dynamin- and β-arrestin dependence of AT1A receptor endocytosis in CHO cells prompted us to study the mechanism of AT1A receptor endocytosis in COS-7 cells, which have been reported to contain low levels of endogenous β-arrestins (Menard et al., 1997). The rate of endocytosis of the AT1A receptor was consistently lower in COS-7 cells than in CHO cells (Table 1). Hyperosmotic sucrose inhibited agonist-induced endocytosis of the AT1Areceptor, whereas filipin had no detectable effects on AT1A receptor endocytosis in these cells (data not shown). β-arrestin1(V53D) had only a partial inhibitory effect on AT1A receptor endocytosis, but overexpression of β-arrestin1(1–349) markedly reduced the endocytosis of the receptor, similar to its effect in CHO cells (Fig.6A). Overexpression of dynamin-1(K44A), dynamin-2(K44A), and dynamin-2(K535A) mutants caused similar or even greater inhibition of AT1A receptor endocytosis in COS-7 cells than that in CHO cells (Fig. 6B). The expression of dominant negative dynamin-1 and dynamin-2 constructs was detected with dynamin-1 and dynamin-2 antibodies, respectively. However, the endogenous dynamin of COS-7 cells was only detectable with dynamin-2 antibody (Fig. 3B), demonstrating that the endogenous dynamin in COS-7 cells, as in CHO cells, is dynamin-2. Wild-type dynamins and β-arrestin1 had no major effects on AT1Areceptor endocytosis (data not shown). Thek e values of the receptor in these experiments are shown in Table 1. These data provide evidence for the participation of dynamin and β-arrestin in endocytosis of the AT1A receptor in COS-7 cells.

Effects of overexpression of dominant negative β-arrestin1 constructs (A), and wild-type or dominant negative dynamins (B) on AT1A receptor endocytosis in COS-7 cells. COS-7 cells were grown in 24-well plates and cotransfected with 0.5 μg of AT1A receptor and 0.5 μg of empty pcDNA3 vector (●), β-arrestin1(V53D) (■), β-arrestin1(1–349) (▴), dynamin-1(K44A) (○), dynamin-2(K44A) (♦), or dynamin-2(K535A) (▵). Endocytosis of 125I-Ang II was measured as described under Experimental Procedures. Data are shown as means ± S.E.M. of three independent experiments, each performed in duplicate.
Effects of Coexpression of Dominant Negative Dynamins and β-Arrestins on AT1A Receptor Endocytosis in HEK 293 Cells.
The dynamin and β-arrestin dependence of AT1A receptor endocytosis was also studied in HEK 293 cells, in which the AT1A receptor has been reported to internalize in a dynamin- and β-arrestin-independent manner after stimulation with maximally effective concentrations of Ang II (Zhang et al., 1996). Endocytosis of the AT1Areceptor in HEK 293 cells was studied at low (0.03 nM) Ang II concentrations. Under these conditions, the rate of AT1A receptor endocytosis in HEK 293 cells was comparable with that observed in CHO cells (Table 1). Hyperosmotic sucrose inhibited the agonist-induced endocytosis of the AT1A receptor in HEK 293 cells, and thek e value of the receptor endocytosis was reduced to 0.04 ± 0.01 per min (n = 3). β-Arrestin1(V53D) had only a partial inhibitory effect on AT1A receptor endocytosis, but overexpression of β-arrestin1(1–349) markedly reduced receptor endocytosis, similar to its effect in the other two cell types (Fig.7A). Overexpression of dynamin-1(K44A) and dynamin-2(K44A) mutants caused inhibition of AT1A receptor endocytosis in HEK 293 cells similar to CHO and COS-7 cells (Fig. 7B). Wild-type dynamins and β-arrestin1 had no major effects on AT1Areceptor endocytosis (data not shown). These data suggest that the mechanism of AT1A receptor endocytosis in HEK 293 cells is similar to that in CHO and COS-7 cells.

Effects of overexpression of dominant negative β-arrestin1 constructs (A), and wild-type or dominant negative dynamins (B) on AT1A receptor endocytosis in HEK 293 cells. HEK 293 cells were grown in 24-well plates and cotransfected with 0.5 μg of AT1A receptor and 0.5 μg of empty pcDNA3 vector (●), β-arrestin1(V53D) (■), β-arrestin1(1–349) (▴), dynamin-1(K44A) (○), or dynamin-2(K44A) (♦). Endocytosis of125I-Ang II was measured as described underExperimental Procedures. Data are shown as means ± S.E.M. of three independent experiments, each performed in duplicate.
Discussion
This study demonstrates that treatment with hyperosmotic sucrose, which disrupts the clathrin coat at the plasma membrane (Heuser and Anderson, 1989), effectively inhibits endocytosis of the AT1A receptor. In contrast, inhibitors of caveolar endocytosis, such as filipin (Fig. 1) or nystatin (Z. Gáborik, L. Szidonya, A. J. L. Clark, and L. Hunyady, unpublished observation), had no major effect on the endocytosis of this receptor. These results suggest that, similar to the endocytosis of endogenous AT1 receptors, the major route of AT1A receptor endocytosis in transiently transfected cells is via clathrin-coated vesicles. This conclusion is in accordance with the observed β-arrestin and dynamin dependence of the AT1 receptor endocytosis pathway and is also supported by the colocalization of internalized Fluo-Ang II with transferrin. The latter is a well established marker for endocytosis via clathrin-coated pits, because only vesicles that are internalized by this mechanism, and not those taken up via caveolae, are known to fuse with endosomes (Anderson, 1998).
Recent studies have suggested that phosphorylation of serine/threonine residues in the cytoplasmic tail of the AT1receptor regulates its internalization in CHO and COS-7 cells (Smith et al., 1998; Thomas et al., 1998). Phosphorylation of GPCRs is believed to facilitate their binding of β-arrestins, which possess clathrin and AP-2 adaptor protein binding domains and have been proposed to target GPCRs to clathrin-coated pits (Goodman et al., 1996; Laporte et al., 1999). β-arrestin1(1–349), in which the clathrin and AP-2 binding domains are deleted (Krupnick et al., 1997; Laporte et al., 1999), strongly inhibited agonist-induced endocytosis of the AT1 receptor in CHO cells. The role of β-arrestin in AT1 receptor endocytosis pathway was not specific to CHO cells, because β-arrestin1(1–349) also strongly inhibited endocytosis of the AT1receptor in COS-7 and HEK 293 cells. Coexpression of β-arrestin1(V53D), a dominant negative mutant β-arrestin1 with impaired binding to GPCRs (Ferguson et al., 1996; Krupnick et al., 1997) partially inhibited the endocytosis of the AT1A receptor. Although previous studies using partially impaired β-arrestin1(V53D) and saturating concentrations of Ang II suggested that internalization of the AT1receptor is β-arrestin-independent (Zhang et al., 1996), Ang II has been shown to cause association of GFP-tagged β-arrestin2 with the AT1 receptor in HEK 293 cells (Zhang et al., 1999). Furthermore, the present data obtained with physiological concentrations of labeled Ang II demonstrate that AT1 receptor endocytosis is dependent on β-arrestin in CHO, COS-7, and HEK 293 cells.
Clathrin-mediated and β-arrestin-dependent internalization of GPCRs also requires the function of dynamin GTPases. Upon GTP binding, dynamin-1 undergoes self-assembly into helical collars that encircle the necks of deeply invaginated pits. Subsequent GTP hydrolysis by the assembled dynamin molecules is required for the fission of the coated vesicles from the plasma membrane (Schmid et al., 1998). Replacement of Lys44 by alanine in dynamin-1 prevents GTP binding to the molecule, and the mutant protein thus acts as a dominant negative inhibitor of the function of endogenously expressed dynamins during clathrin-mediated endocytosis (Herskovits et al., 1993; Schmid et al., 1998). After the first evidence for the role of dynamin in β2-adrenergic receptor internalization was uncovered, the endocytosis of several other GPCRs was found to be prevented by overexpression of dynamin-1(K44A) (Zhang et al., 1996;Bünemann et al., 1999). However, overexpression of this mutant dynamin-1 did not inhibit the internalization of AT1A angiotensin, secretin, D2 dopaminergic, and M2muscarinic receptors (Zhang et al., 1996; Vickery and von Zastrow, 1999; Vogler et al., 1999; Walker et al., 1999). Although endocytosis via caveolae was initially suggested to participate in the dynamin-independent internalization of GPCRs, it has been demonstrated recently that this process is also dynamin dependent (Henley et al., 1998).
The major isoform of dynamin detected by immunoblotting in CHO and COS-7 cells is dynamin-2. As noted above, previous studies on the role of dynamin in GPCR-mediated endocytosis have used dominant negative mutants of dynamin-1. The dynamin-1 and dynamin-2 isoforms are about 80% identical and contain the same structural domains, but exhibit differences in their cellular distribution (Cao et al., 1998). Because many GPCRs, including the AT1 receptor, are predominantly expressed in nonneural tissues, it is important to determine whether dominant negative mutants of the ubiquitous dynamin-2 isoform can also be used to analyze the roles of dynamins in the endocytosis of GPCRs. Although initial studies suggested that dynamin-2 participates in vesicle trafficking in the trans-Golgi network (Schmid et al., 1998), more recent observations have shown its role in dynamin-dependent endocytosis of the transferrin receptor via clathrin-coated vesicles (Altschuler et al., 1998; Kasai et al., 1999). Also, recent findings in cultured MDCK cells provided evidence for the differential efficacies of dominant negative dynamins. Although polarized MDCK cells express only the ubiquitous dynamin-2 isoform, overexpression of dynamin-1(K44A) inhibits receptor-mediated endocytosis specifically at the apical surface of the cell, whereas dynamin-2(K44A) has inhibitory effects on endocytosis at both the apical and basolateral membranes (Altschuler et al., 1998). Our evaluation of the actions of mutant dynamins demonstrated that not only dynamin-1(K44A) but also dynamin-2(K44A) and dynamin-2(K535A) inhibited AT1A receptor endocytosis in CHO, COS-7, and HEK 293 cells. These data indicate that these dynamin-2 constructs are applicable to studies on the mechanism of endocytosis of GPCRs. Furthermore, the previously reported dynamin-independence of the AT1 receptor endocytosis is not attributable to the use of dynamin-1, because dynamin-1 and dynamin-2 mutants had similar dominant negative inhibitory effects in the present study.
The participation of dynamins in agonist-induced endocytosis of the AT1A receptor was confirmed by morphological studies using confocal microscopy. Although Fluo-Ang II has lower affinity for the AT1A receptor than native Ang II, at 4°C it binds specifically to the surface of cells expressing the receptor. During incubation of CHO cells at 37°C in the presence of 50 nM Fluo-Ang II, fluorescence appeared in punctate intracellular structures within 5 min. Based on their size and time of appearance, these organelles were more likely to be early endosomes than clathrin-coated vesicles. Coexpression of dominant negative dynamin-1 and dynamin-2 markedly inhibited the endocytosis of Fluo-Ang II. These data demonstrate that internalization of Fluo-Ang II, which has at least 20-fold lower affinity to the AT1 receptor, occurs at 50 nM with the same dynamin-dependent mechanism as endocytosis of radiolabeled Ang II in lower concentrations.
Our findings on the mechanism of AT1 receptor endocytosis differ from those of earlier studies in which the internalization of the AT1A receptor in HEK 293 cells was found to be dynamin and β-arrestin independent (Zhang et al., 1996). This discrepancy does not reflect the operation of a unique receptor uptake mechanisms in these cells, because under our experimental conditions, the inhibitory effects of dominant negative dynamins and β-arrestins on AT1A receptor endocytosis are similar in HEK 293, CHO, and COS-7 cells. In the present study, endocytosis of the AT1 receptor was measured with tracer amounts of labeled Ang II that correspond to the physiological concentrations of the hormone, whereas previous studies on this topic have used saturating concentrations of Ang II (Zhang et al., 1996). When the importance of hormone concentration was addressed in the present study, the endocytotic rate constant of the AT1 receptor decreased with increasing ligand concentration, and at higher Ang II concentration, the inhibitory effect of dominant negative dynamin-2 on AT1receptor endocytosis was diminished.
These results demonstrate that although endocytosis of the AT1 receptor is clearly dynamin-dependent at physiological Ang II concentrations, inhibitory effects of dominant negative dynamins cannot be detected at higher Ang II concentrations. One possible explanation for this finding is that high levels of occupied surface receptors in overexpression systems saturate the endocytotic apparatus, as observed previously for the EGF receptor (Lund et al., 1990). Under these conditions, the inhibitory effects of partially impaired molecules, such as β-arrestin1(V53D) and dynamin-1(K44A), may not be detected because an intrinsic component of the endocytosis machinery (e.g., an adaptor protein or clathrin) becomes the rate-limiting step. This concept is supported by a very recent report that N-terminal deletion of dynamin-1, which completely eliminates the GTPase activity of the molecule, or K535M mutation of its PH domain, produces dominant negative mutants that reveal a role of dynamin during the internalization of the AT1 and M2 muscarinic receptors when steady-state levels of receptor internalization are measured at saturating Ang II concentrations in HEK 293 cells (Werbonat et al., 2000).
In summary, the present data demonstrate that clathrin-mediated endocytosis of the agonist-activated AT1 receptor in at least three cell types requires β-arrestins, as well as the GTPase activity of dynamin and the intact lipid binding region of its PH domain. Based on these findings, it would be interesting to reevaluate the importance of these mechanisms for other GPCRs reported to show dynamin- and β-arrestin-independent internalization in previous studies.
Acknowledgments
The excellent technical assistance of Judit BakacsinéRácz, Istvánné Sneider and Katinka Süpeki is greatly appreciated. We thank Drs. K. E. Bernstein, M. G. Caron, S. S. G. Ferguson, K. Nakayama, and T. C. Südhof for providing plasmid DNA constructs, and Dr. H. S. Wiley for algorithms to calculate the endocytotic rate constants.
Footnotes
-
Send reprint requests to: Dr. László Hunyady, Department of Physiology, Semmelweis University Medical School, H-1444 Budapest, P. O. Box 259, Hungary. E-mail:hunyady{at}puskin.sote.hu
-
This work was supported in part by a Collaborative Research Initiative grant from the Wellcome Trust (051804/Z/97/Z), an International Research Scholar's award from the Howard Hughes Medical Institute (HHMI 75195–541702) and by grants from the Hungarian Ministry of Culture and Education (FKFP-0318/1999), the Hungarian Science Foundation (OTKA T-032179) and the Semmelweis University.
-
Preliminary data of this work were presented at the American Society for Cell Biology Annual Meeting, Washington, DC, December 1999 [Hunyady L, Gáborik Z, Mihalik B, Clark AJL, Catt KJ (1999) Dynamin-dependent mechanism of internalization of the AT1Aangiotensin receptor (Abstract). Mol Biol Cell 10:316a].
Abbreviations
- Ang II
- Angiotensin II
- GPCR
- G protein-coupled receptor
- HEK
- human embryonic kidney
- PH
- pleckstrin homology
- CHO
- Chinese hamster ovary
- HA
- influenza hemagglutinin epitope
- HRP
- horseradish peroxidase
- ke
- endocytotic rate constant
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}