


Abstract
The anti-inflammatory drugs indomethacin and ramatroban, the latter showing clinical efficacy in treating allergic asthma, have been shown to act as a classic agonist and antagonist, respectively, of the G protein-coupled chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2 receptor). Here, we report the identification of two indole derivatives 1-(4-ethoxyphenyl)-5-methoxy-2-methylindole-3-carboxylic acid and Nα-tosyltryptophan (hereafter referred to as 1 and 2, respectively), which are structurally related to indomethacin and ramatroban but which selectively interfere with a specific G protein-independent signaling pathway of CRTH2. In whole-cell saturation-binding assays, 1 and 2 both increase the number of [3H]prostaglandin D2 (PGD2)-recognizing CRTH2 sites and the affinity of PGD2 for CRTH2. Enzyme-linked immunosorbent assays show that they do not alter the total number of CRTH2 receptors on the cell surface. Analysis of their binding mode indicates that unlike indomethacin or ramatroban, 1 and 2 can occupy CRTH2 simultaneously with PGD2. On a functional level, however, 1 and 2 do not interfere with PGD2-mediated activation of heterotrimeric G proteins by CRTH2. In contrast, both compounds inhibit PGD2-mediated arrestin translocation via a G protein-independent mechanism. In human eosinophils endogenously expressing CRTH2, 1 selectively decreases the efficacy but not the potency of PGD2-induced shape change, unlike ramatroban, which displays competitive antagonistic behavior. These data show for the first time that “antagonists” can cause markedly dissimilar degrees of inhibition for different effector pathways and suggest that it may be possible to develop novel classes of specific signal-inhibiting drugs distinct from conventional antagonists.
The proximal event mediating cellular signaling by a seven-transmembrane (7TM) receptor is the binding of ligand, which causes the receptor to change its behavior toward the cell. In the past, ligands targeting 7TM receptors have been believed to cause a single type of functional response for all effectors linked to a given receptor; thus, compounds were classified as agonists or antagonists/inverse agonists, respectively, according to their intrinsic efficacy. Drugs with positive intrinsic efficacy stabilize the active receptor conformation and elicit a signaling response (agonists), whereas drugs with negative intrinsic efficacy preferentially stabilize the inactive receptor conformation and shut down a signaling response (antagonists/inverse agonists). In addition, this traditional view of either turning on or off receptor responses has been linked exclusively to signaling pathways involving the activation of heterotrimeric G proteins. However, the fact that ligands acting at a single 7TM receptor can cause markedly dissimilar—even opposing—effects on different intracellular signaling pathways challenges the traditional concept of ligand classification. For example, Azzi et al. (2003) have shown that β2 adrenoceptor ligands such as propranolol and ICI118551, which are inverse agonists for Gαs-mediated stimulation of adenylyl cyclase, act as partial agonists for the MAP kinase pathway. Unlike conventional agonists such as isoproterenol, MAP kinase activation occurred in a G protein-independent manner but required β-arrestin for intracellular signal propagation. Likewise SR121463B, an inverse agonist on V2 vasopressin receptor-stimulated adenylyl cyclase, simultaneously acts as an agonist of the MAP kinase pathway in an arrestin-dependent, G protein-independent manner (Azzi et al., 2003). This phenomenon ascribed to as “trafficking of receptor stimuli” (Kukkonen et al., 2001; Kenakin 2003, 2004) has important consequences for the classification of ligands modulating 7TM receptor function: 1) the term “efficacy” not only should be confined to cellular responses involving signaling through heterotrimeric G proteins but should rather be expanded to the complete range of 7TM receptor behaviors such as phosphorylation, desensitization, arrestin recruitment, internalization, dimerization, and interaction with scaffolding proteins; and 2) ligands may not simply be categorized into agonists and antagonists/inverse agonists without additional specifications of the particular signaling pathway/“receptor behavior” examined. To date, a range of different studies exist which describe differential effects of agonists inducing dissimilar degrees of activation for different effector pathways through a single 7TM receptor (Kenakin, 2003; Gay et al., 2004). However, no reports are available so far showing that antagonists are endowed with the ability to selectively suppress certain 7TM receptor signaling pathways.

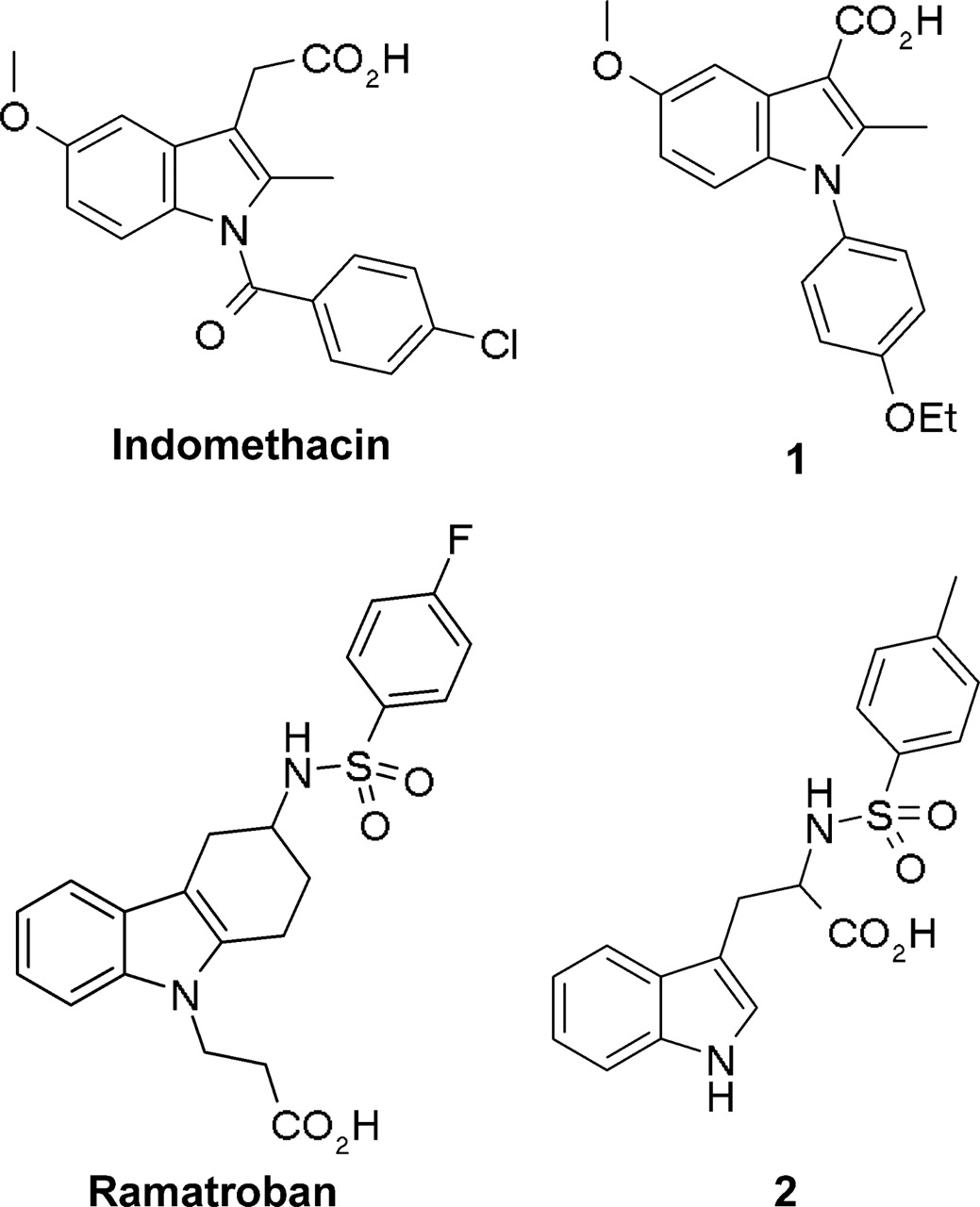
Structures of the indole derivatives indomethacin, ramatroban, and compounds 1 and 2.
Chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) has recently been discovered as the second high-affinity receptor for prostaglandin D2 (PGD2) (Hirai et al., 2001). It induces intracellular calcium mobilization and chemotaxis in Th2 cells in a Gαi-dependent manner (Hirai et al., 2001). CRTH2 has also been shown to mediate PGD2-induced shape change of eosinophils, an effect that could be attenuated with the phospholipase Cβ inhibitor U73122 (Bohm et al., 2004). Functional experiments performed in HEK293 cells stably expressing CRTH2 have shown that it negatively regulates adenylyl cyclase via Gαi/o proteins (Sawyer et al., 2002). Upon coexpression with the promiscuous G protein Gα15, CRTH2 effector specificity can be switched to stimulation of phospholipase Cβ, generation of inositol phosphates, and subsequent calcium mobilization (Sawyer et al., 2002). So far, the cellular responses elicited by CRTH2 involved signaling through heterotrimeric G proteins and could be abrogated with the small-molecule antagonist ramatroban, which has been developed for clinical use and is currently marketed in Japan for the treatment of allergic rhinitis. In an effort to identify small molecules competing with PGD2 for CRTH2 binding and activation, we identified two indole derivatives with a fairly unconventional mechanism of action. Both compounds, 1-(4-ethoxyphenyl)-5-methoxy-2-methylindole-3-carboxylic acid and Nα-tosyltryptophan (Fig. 1; referred to as 1 and 2 in this study, respectively), were found to occupy CRTH2 simultaneously with PGD2 by binding to a site that is topologically distinct from the orthosteric site used by the endogenous agonist PGD2. Occupation by compounds 1 and 2 of the CRTH2 receptor did not prevent PGD2 from activating cellular responses, which required heterotrimeric G proteins for signal propagation, but exclusively interfered with the ability of PGD2 to recruit β-arrestin in a G protein-independent fashion. A thorough study of the functional and binding profile of these novel “biased antagonists” will be presented and compared with the known conventional receptor ligands indomethacin and ramatroban, which represent a classic agonist and antagonist, respectively.
Materials and Methods
Materials. White 96-well Optiplates and DeepBlueC were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Tissue culture media and reagents were purchased from Invitrogen (Breda, Netherlands). PGD2 was from Cayman Chemical (Ann Arbor, MI), and [3H]PGD2 was from PerkinElmer. Compound 1 was obtained from ChemDiv (San Diego, CA). Compound 2 was obtained from Chembridge Research Laboratories (San Diego, CA). Eotaxin was from Preprotech EC (London, UK). CellFix and FACSFlow were from BD Biosciences Immunocytometry Systems (Vienna, Austria). Fixative solution was prepared by diluting CellFix 1:10 in distilled water and 1:4 in FACSFlow. All other laboratory reagents were from Sigma-Aldrich (St. Louis, MO) unless explicitly specified.
Generation/Origin of the cDNA Constructs. The coding sequence of human CRTH2 (Genbank accession number NM_004778) was amplified by PCR from a human hippocampus cDNA library and was inserted into the pcDNA3.1(+) expression vector (Invitrogen) via 5′ HindIII and 3′ EcoRI. To generate a CRTH2-Renilla reniformis luciferase (CRTH2-Rluc) fusion protein, the CRTH2 coding sequence without a STOP codon and Rluc were amplified, fused in frame by PCR, and subcloned into the pcDNA3.1(+)Zeo expression vector. For ELISA experiments, a 78-base pair sequence containing a signal peptide and the M1 FLAG-epitope tag was introduced by PCR at the extreme N terminus, and the resulting construct was inserted via 5′ NheI and 3′ EcoRI into pcDNA3.1(+). The thromboxane A2 (TXA2) receptor (Genbank accession number BC074749) was cloned from a leukocyte cDNA library and was inserted via 5′ HindIII and 3′ BamHI into pcDNA3.1(+). A TXA2-receptor-Rluc fusion protein was generated by PCR and was inserted via 5′ HindIII and 3′ XbaI into pcDNA3.1(+). Human β-arrestin2 (β-arr2) N-terminally tagged with GFP2 (βarr2-GFP2) and R. reniformis luciferase were purchased from BioSignal Packard Inc. (Montreal, QC, Canada). The sequence identity of the constructs was verified by restriction endonuclease digests and sequencing in both directions on a Prism sequence detection system (Applied Biosystems, Foster City, CA).
Cell Culture and Transfection. COS-7 cells were grown in Dulbecco's modified Eagle's medium 1885 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 1 mg/ml streptomycin and kept at 37°C in a 10% CO2 atmosphere. CHO cells were maintained in Ham's F-12 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 1 mg/ml streptomycin. HEK293 cells were maintained in minimum essential medium (MEM) supplemented with 10% (v/v) heat-inactivated fetal calf serum, 2 mM Glutamax-I, 1% nonessential amino acids, 1% sodium pyruvate, and 10 μg/ml gentamicin. CHO and HEK cells were kept at 37°C in a 5% CO2 atmosphere. For binding experiments, COS-7 cells were transiently transfected with CRTH2 using a calcium phosphate/DNA coprecipitation method with the addition of chloroquine as described by Kostenis et al. (2005). CHO cells were transiently transfected with CRTH2 using Lipofectamine (Invitrogen) according to the manufacturer's instructions. For functional inositol phosphate assays, COS-7 cells were transiently cotransfected with CRTH2 and a promiscuous Gα protein facilitating inositol phosphate production by a Gi-selective CRTH2 receptor (Kostenis, 2001). To perform the functional bioluminescence resonance energy transfer (BRET) assays, an HEK293 cell clone stably expressing βarr2-GFP2 and CRTH2-Rluc was generated (CRTH2-HEK293 cells). The functional and binding properties of the CRTH2-Rluc fusion protein were not changed compared with the wild-type CRTH2 receptor (data not shown). The BRET assay on the TXA2 receptor was performed upon transient transfection of the TXA2-Rluc fusion protein into an HEK293 cell clone stably expressing βarr2-GFP2.
Binding Experiments
Whole-Cell Binding. Twenty-four hours after transfection, COS-7 cells were seeded into 96-well plates at a density of 30,000 cells/well. Competition binding experiments on whole cells were then performed approximately 18 to 24 h later using 1.2 nM [3H]PGD2 (PerkinElmer; 170 Ci/mmol) in a binding buffer consisting of HBSS (Invitrogen) and 10 mM HEPES, pH 7.5. Competing ligands were diluted in DMSO, which was kept constant at 1% (v/v) of the final incubation volume. Total and nonspecific binding were determined in the absence and presence of 10 μM PGD2, respectively. Binding reactions were routinely conducted for 3 h at 4°C and were terminated by two washes (100 μl each) with ice-cold binding buffer. Radioactivity was determined by liquid scintillation counting in a TopCount counter (PerkinElmer) after overnight incubation in MicroScint 20. The binding assays for CHO cells transiently expressing CRTH2 or HEK293 cells stably transfected with CRTH2 were performed essentially as described above for COS-7 cells. For saturation binding experiments, CRTH2-HEK293 cells were incubated with 1.5 to 48 nM [3H]PGD2 for 3 h, and nonspecific binding was determined in the presence of 10 μM ramatroban. The exact concentration of [3H]PGD2 used was determined from experiment to experiment. Determinations were made in duplicates.
Membrane Binding. Scintillation proximity assay (SPA) binding was carried out in a total volume of 150 μl containing cell membranes (4 μg of protein) from stably transfected CHO-K1 cells ([3H]PGD2: Kd = 12.1 nM, Bmax = 10.24 pmol/mg protein) (Euroscreen, Brussels, Belgium), [3H]PGD2 (1.2 nM) and varying concentrations of PGD2 or competitor compounds, and 0.4 mg/well wheat germ agglutinin-coupled yttrium silicate SPA beads (Amersham Biosciences Inc., Piscataway, NJ). Competitor drug dilutions, radioligand, and cell membranes were all prepared in assay buffer [50 mM HEPES, pH 7.5, 5 mM MgCl2, 100 mM NaCl, 10 μg/ml saponin, and 0.1% (w/v) BSA (protease-free)]. Each data point was performed in duplicate, and reactions were incubated under continuous shaking at room temperature for 1 h to allow equilibration. Radioactivity was then counted with TopCount (PerkinElmer).
Dissociation Kinetics. Whole hCRTH2-HEK293 cells (250,000 cells/ml) were incubated at 4°C with 3 nM [3H]PGD2 in binding buffer (HBSS + 10 mM HEPES, pH 7.5) for 60 min to reach equilibrium. Dissociation was initiated by adding 10 μM ramatroban alone or in combination with 10 μM concentrations of modulator 1 or 2. After various time intervals, 200-μl aliquot samples were taken, and the reaction was terminated by sample filtration on a Millipore vacuum manifold (Millipore Corporation, Billerica, MA), using Whatman GF/F filters (presoaked in 0.5% BSA for at least 1 h; Whatman, Clifton, NJ). The filters were washed rapidly three times with 3 ml of ice-cold binding buffer, and radioactivity was determined in a β counter (PerkinElmer).
Two-Point Kinetic Experiments to Estimate the Affinity of 1 and 2 for the [3H]PGD2-Occupied CRTH2 Receptor. hCRTH2-HEK293 cells (250,000 cells/ml) were incubated with 3 nM [3H]PGD2 at 4°C for 60 min. Thereafter, 1-ml aliquots were distributed to tubes that contained either DMSO or a final concentration of 10 μM ramatroban alone or combined with a number of different concentrations of 1 or 2. Seven minutes later (corresponding to ∼2 dissociation half-lives), three 200-μl aliquots were taken and filtered as described above. Nonspecific binding was determined after preincubation with 10 μM ramatroban for 60 min in separate tubes. All incubations were performed at 4°C. The data were transformed to dissociation rate constants k-1 and expressed as a percentage of the [3H]PGD2 dissociation rate constant in the absence of modulator as described in detail by Kostenis and Mohr (1996). Normalized k-1 values were then fitted to a logistic function using nonlinear regression analysis with the inflection point of the curve corresponding to the Kd value of the modulator for the [3H]PGD2-occupied CRTH2 receptor.
BRET Assay. Functional BRET assays were performed on HEK293 cells stably expressing human CRTH2-Rluc and GFP2-β-arr2 or on HEK293 cells stably expressing GFP2-β-arr2 and transiently expressing the TXA2-Rluc or PGD2 D prostanoid receptor-Rluc fusion proteins. Before their use in the BRET assay, cells were detached and resuspended in Dulbecco's PBS with 1000 mg/l l-glucose at a density of 2 × 106 cells/ml. DeepBlueC was diluted to 50 μM in Dulbecco's PBS with 1000 mg/l l-glucose (light-sensitive). A 100-μl sample of cell suspension was transferred to wells in a 96-well microplate (white OptiPlate) and placed in the Mithras LB 940 instrument (Berthold Technologies, Bad Wildbad, Germany). Agonist (12 μl/well) was then injected by injector 1, and 10 μl/well DeepBlueC was injected simultaneously by injector 2. Five seconds after the injections, the light output from the well was measured sequentially at 400 and 515 nm, and the BRET signal (mBRET ratio) was calculated as the ratio of the fluorescence emitted by GFP2-β-arr2 (515 nm) over the light emitted by the receptor-Rluc (400 nm). Antagonists were added before placing the microplates into the Mithras LB 940 and were allowed to incubate for 15 min before the addition of agonist and DeepBlueC. Compounds were dissolved in DMSO, and the final DMSO concentration was kept constant at 1% in the assay.
[35S]GTPγS Binding Assays. SPA [35S]GTPγS binding was performed on membranes from CHO-K1 cells stably expressing CRTH2. Membrane protein (4 μg) was incubated in GTPγS binding buffer (50 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.1% BSA, and 10 μg/ml saponin) with 50 nCi [35S]GTPγS, 1 μM GDP, and 0.4 mg of wheat germ agglutinin-coupled SPA beads (Amersham Biosciences) with or without increasing concentrations of PGD2 in the absence or presence of compounds 1 or 2. Parallel assays containing 100 μM nonradioactive GTPγS defined nonspecific binding. Samples were incubated for 30 min at ambient temperature on a plate shaker and centrifuged for 5 min, and radioactivity was counted in a TopCount (PerkinElmer).
Inositol Phosphate Accumulation Assays. Twenty-four hours after transfection, cells were seeded in 24-well tissue-culture plates and loaded with 5 μCi of [myo-2-3H]inositol (Amersham Biosciences). The next day, cells were washed twice in HBSS buffer (including CaCl2 and MgCl2; Invitrogen) and stimulated with the respective agonists in HBSS buffer supplemented with 5 mM LiCl for 45 min at 37°C. The reactions were terminated by aspiration and addition of 10 mM concentrations of ice-cold formic acid, and they were incubated for 30 min on ice. The lysate was applied to AG 1-X8 anion exchange resin (Bio-Rad, Hercules, CA) and washed twice with buffer containing 60 mM sodium formate and 5 mM borax. The [3H]inositol phosphate fraction was then eluted by adding 1 M ammonium formate and 100 mM formic acid solution and counted after the addition of HiSafe3 scintillation fluid (PerkinElmer).
ELISA. Determination of cell-surface expression levels of CRTH2 was performed using an N-terminally FLAG-tagged CRTH2 receptor in an ELISA assay. Twenty-four hours after transfection, cells were seeded in poly-d-lysine-coated 48-well tissue-culture plates at a density of 100,000 cells/well. Approximately 48 h after transfection, cells were washed once in minimum essential medium + 0.1% BSA and stimulated with the indicated compounds diluted in the same buffer for 30 min at 37°C. Cells were then fixed with 4% paraformaldehyde, washed three times with washing buffer (150 mM NaCl, 25 mM Tris base, 2.7 mM KCl, and 1 mM CaCl2 · 2H2O, pH 7.4), and blocked with blocking buffer (3% dry milk, 1 mM CaCl2, and 50 mM Tris-HCl, pH 7.5). Cells were then incubated with the primary monoclonal mouse anti-FLAG M1 antibody (Sigma-Aldrich) diluted in blocking buffer at 1:500 for 1 h at room temperature followed by three washes and a 1-h incubation with secondary antibody (1:2500, goat anti-mouse conjugated to horseradish peroxidase; Bio-Rad) in blocking buffer. After three final washes, the secondary antibody was detected and quantified after adding the colorimetric horseradish peroxidase substrate 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich). When adequate color change was reached, the reaction was terminated by the addition of 0.5 M H2SO4. Samples were then transferred to a 96-well plate, and colorimetric readings were obtained at optical density of 450 nM on a Tecan Sunrise absorbance reader (Tecan, Maennedorf, Switzerland). All experiments were performed in triplicate.
Human Eosinophil Shape-Change Assay. Blood samples were obtained from healthy volunteers according to a protocol approved by the Ethics Committee of the University of Graz and were processed as described previously (Bohm et al., 2004). Preparations of polymorphonuclear leukocytes (containing eosinophils and neutrophils) were prepared by dextran sedimentation of citrated whole blood and Histopaque gradients. The resulting cells were washed and resuspended in assay buffer (comprising PBS with Ca2+/Mg2+ supplemented with 0.1% BSA, 10 mM HEPES, and 10 mM glucose, pH 7.4) at 5 × 106 cells/ml. Cells were incubated with the antagonists or vehicle (PBS or DMSO) for 10 min at 37°C and then stimulated with various concentration of the agonists (PGD2 or eotaxin) for 4 min at 37°C. To stop the reaction, samples were transferred to ice and fixed with 250 μl of fixative solution. Samples were immediately analyzed on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and eosinophils were identified according to their autofluorescence in the FL-1 and FL-2 channels. Shape-change responses were quantified as a percentage of the maximal response to PGD2 or eotaxin in the absence of an antagonist.
Calculations and Data Analysis. IC50 and EC50 values were determined by nonlinear regression using the Prism 3.0 software (GraphPad Software Inc., San Diego, CA). Kd and Ki values were estimated from competition binding experiments using the equations Kd = IC50 - L (homologous competition experiments) and Ki = IC50/(1 + L/Kd) (heterologous competition experiments), where L is the concentration of radioactive ligand and Kd is its dissociation constant. Data sets of saturation binding isotherms were analyzed via nonlinear regression according to a hyperbolic, one-site binding model, and individual estimates for total receptor number (Bmax) and radioligand dissociation constant (Kd) were subsequently determined. Dissociation kinetic experiments were fitted to a model of one-phase exponential decay by Prism 3.0 to calculate the dissociation rate constants (k-1). Data from two-point kinetic experiments were transformed to off-rate constants k-1 applying the formula k-1 = ln(B0/Bt)/t, where B0 is initially bound radioligand at t = 0, and Bt corresponds to specifically bound radioligand after t min of dissociation. These k-1 values were then expressed as a percentage inhibition of the [3H]PGD2 k-1 value in the absence of modulating agent 1 or 2.
Results
Effect of Indole Derivatives on [3H]PGD2 Binding at Human CRTH2 Stably Expressed in HEK293 Cells. First, ligand binding of the human CRTH2 receptor was characterized in whole cells using the agonist [3H]PGD2 as a radiotracer. [3H]PGD2 saturation isotherms revealed the presence of a single population of specific binding sites with an equilibrium dissociation constant log Kd of 12.9 ± 2.1 nM and a Bmax of 57.5 ± 3.5 fmol/30,000 cells (mean ± S.E., n = 3). Our binding assay confirmed that the affinity for CRTH2 of PGD2 is similar to the affinity reported by Gervais et al. (2005). Four different indole derivatives (Fig. 1) were then tested for their ability to displace [3H]PGD2-specific binding in hCRTH2-HEK293 cells: indomethacin, a nonsteroidal anti-inflammatory drug, recently reported to bind to and activate CRTH2 (Hirai et al., 2002; Sawyer et al., 2002; Hata et al., 2005); ramatroban, an orally active small-molecule CRTH2 antagonist originally developed to block the thromboxane A2 receptor (Shichijo et al., 2003; Sugimoto et al., 2003); and two compounds structurally closely related to indomethacin and ramatroban, respectively (Fig. 1, compounds 1 and 2). In agreement with published data (Hirai et al., 2002; Sugimoto et al., 2003; Hata et al., 2005), indomethacin and ramatroban competed with [3H]PGD2 for CRTH2-specific binding with log Ki values of -5.81 ± 0.059 and -8.53 ± 0.091 (mean ± S.E., n = 4-8) (Fig. 2A). On the contrary, 1 and 2 were found to concentration-dependently increase the number of CRTH2 receptors accessible to [3H]PGD2, with log EC50 values of -6.46 ± 0.036 and -5.29 ± 0.059, respectively (mean ± S.E., n = 3-6) (Fig. 2A). [3H]PGD2 saturation isotherms in the presence of 10 μM concentrations of 1 and 2 revealed that the modulators significantly increased the affinity of PGD2 for the CRTH2 receptor from a log Kd of 12.9 ± 2.1 nM without modulator to 2.8 ± 0.3 (p < 0.01) and 5.9 ± 1.1 nM (p < 0.05) in the presence of 1 and 2, respectively (one-way analysis of variance followed by Dunnett's multiple comparison test). Furthermore, the total number of CRTH2 agonist binding sites was increased from a Bmax of 57.5 ± 3.5 without modulator to 97.5 ± 5.9 (p < 0.05) and 60.9 ± 5.4 fmol/30,000 cells (not significant) in the presence of 1 and 2, respectively (Fig. 2, B and C). These findings are consistent with a direct allosteric effect of the modulators on PGD2 affinity and with the ability of modulator 1 to induce or expose additional PGD2 binding sites within the CRTH2 receptor. Enhancement of [3H]PGD2 binding was also observed in COS-7 and CHO whole cells transiently transfected with the hCRTH2 receptor, indicating that the observed phenomenon is cell-type-independent (data not shown). It is noteworthy that the enhancement of [3H]PGD2 equilibrium binding was detectable when compounds were preincubated for 30 min either at 37 or 4°C, ruling out molecular chaperone action or inhibition of receptor endocytosis as a mechanistic principle. Quantification of receptor density with agonist radioligands ([3H]PGD2 was the only CRTH2 radiotracer available to us) does not allow for discrimination between a difference in total receptor number as opposed to an increase in high-affinity ternary complexes, because agonists are known to recognize only a fraction of the complete receptor population, particularly in recombinant receptor systems, in which the amount of G protein available for the formation of such complexes is limited. To unequivocally distinguish whether the observed increase by compound 1 and 2 of [3H]PGD2 binding sites was caused by conversion of cell-surface receptors from a non-agonist-recognizing to an agonist-recognizing conformation as opposed to an increase of the total receptor number at the cell surface, ELISA assays were conducted in HEK293 cells transiently transfected with an N-terminally FLAG-tagged CRTH2 receptor (Fig. 2D). The introduction of the FLAG tag did not alter ligand binding or signaling properties of CRTH2 (data not shown). HEK293 cells expressing FLAG-tagged CRTH2 did not display any appreciable difference in cell-surface receptor number upon treatment with both 1 and 2. Hence, both compounds act to selectively increase the fraction of CRTH2 receptors capable of recognizing PGD2 but do not alter the total cell-surface receptor number. We then investigated whether an analogous increase in [3H]PGD2 equilibrium binding is also detectable in membrane preparations from CRTH2-expressing cells. It is interesting that both modulators lost their ability to increase [3H]PGD2 equilibrium binding in membranes from CHO cells stably expressing CRTH2 (Fig. 2E). Therefore, enhancement by 1 and 2 of [3H]PGD2 binding to the CRTH2 receptor seems to require intact cells.
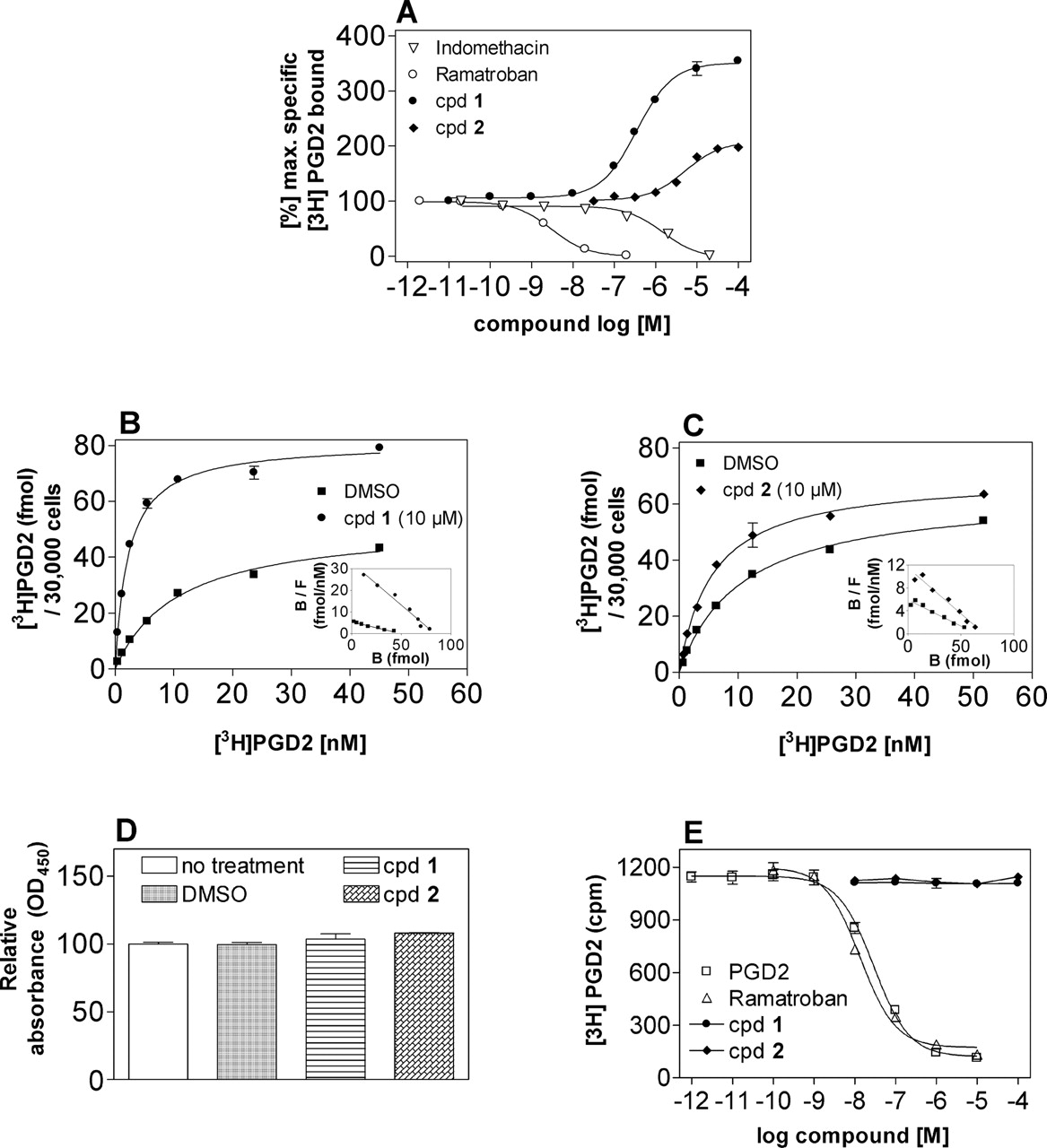
Effects of CRTH2 ligands on [3H]PGD2 binding and CRTH2 surface expression in mammalian whole cells and cell-membrane preparations expressing the human CRTH2 receptor. A, equilibrium competition binding analysis of indomethacin, ramatroban, and the structurally related indole derivatives 1 and 2. hCRTH2-HEK293 whole cells were incubated with 1 nM [3H]PGD2 (3 h, 4°C) in the presence of increasing concentrations of the indicated compounds. B and C, representative saturation analysis and Scatchard plots (insets) of [3H]PGD2 binding to CRTH2 receptors in whole hCRTH2-HEK293 cells in the absence and presence of 10 μM 1 (B) or 2 (C). In three such assays, 1 and 2 caused a significant decrease in log Kd from 12.9 ± 2.1 to 2.8 ± 0.3 (1) and 5.9 ± 1.1 nM (2) and an increase in Bmax (1, 161 ± 8%; 2, 106 ± 4%) (refer to text for statistical analysis). D, ELISA assay on HEK293 cells transiently transfected with an N-terminally FLAG-tagged CRTH2 receptor in the absence (substituted with DMSO) and presence of 10 μM 1 and 2. The compounds do not alter cell-surface CRTH2 receptor expression. E, competition binding analysis of the indicated CRTH2 ligands in membranes from stably transfected CHO cells. PGD2 and ramatroban but not compounds 1 and 2 show effects on [3H]PGD2 equilibrium binding. All data points are shown as mean values ± S.E. of three to eight independent experiments (A) or individual experiments (B-E) each representative of at least three such experiments. Where not shown, error bars lie within the dimensions of the symbols.
Enhancement by 1 and 2 of [3H]PGD2 binding is only possible if both PGD2 and the modulators occupy mutually exclusive binding sites. This is an intriguing observation given the similarity of 1 and 2 to indomethacin and ramatroban, respectively, compounds that both act competitively in [3H]PGD2 equilibrium binding assays (Fig. 2, A and E) (Hirai et al., 2002; Sawyer et al., 2002; Sugimoto et al., 2003; Hata et al., 2005). To further probe for an allosteric interaction and test whether the two binding sites were conformationally linked such that binding to one site influences the nature and the extent of binding to the other site, [3H]PGD2 dissociation kinetic experiments were undertaken. The clearest indication that a compound is acting allosterically is its ability to inhibit the dissociation of an orthosteric ligand (Kostenis and Mohr, 1996; Christopoulos and Kenakin, 2002). Thus, we determined whether 1 and 2 had this property. After a 60-min radioligand/receptor equilibration period at 4°C, [3H]PGD2 dissociation was initiated by the addition of 10 μM ramatroban alone or in combination with a high concentration (10 μM) of 1 and 2 (Fig. 3A). Complete dissociation of [3H]PGD2 was observed within 20 min and yielded a k-1 value of 0.193 ± 0.01 min-1 (mean ± S.E., n = 3) corresponding to a dissociation half-life of 3.6 min. Both 1 and 2 significantly retarded the dissociation rate of [3H]PGD2 with the k-1 values and half-lives being 0.049 ± 0.002 min-1 (t1/2 = 14 min) and 0.138 ± 0.03 min-1 (t1/2 = 5.0 min), respectively (p < 0.01, one-way analysis of variance followed by Dunnett's multiple comparison test).
To further quantify the effect of 1 and 2 on [3H]PGD2 dissociation from the CRTH2 receptor, dissociation rate constants were measured at a single time point in two-point kinetic experiments in the absence and presence of increasing concentrations of modulators as described in detail under Materials and Methods. Both 1 and 2 concentration-dependently retarded dissociation induced by 10 μM ramatroban and essentially completely prevented [3H]PGD2 dissociation at high concentrations (Fig. 3B), with log IC50 values of -6.14 ± 0.11 and -4.85 ± 0.10, respectively (mean ± S.E., n = 4). The IC50 value of the curves corresponds to the Kd of the modulators for the [3H]PGD2-occupied CRTH2 receptor and mirrors the occupancy by the modulators of the PGD2-occupied form of the CRTH2 receptor regardless of the exact mechanism of action (Kostenis and Mohr, 1996). Together, these data represent unequivocal evidence that 1 and 2 bind to a domain on CRTH2 that is distinct from the orthosteric site accommodating PGD2.

Effects of 1 and 2 on the dissociation kinetics of [3H]PGD2 in hCRTH2-HEK293 cells. A, whole cells were incubated with 3 nM [3H]PGD2 at 4°C for 1 h before dissociation was visualized by the addition of 10 μM ramatroban alone (□) or in combination with 10 μM 1 (•) or 2 (♦). The incubations were terminated by filtration through Whatman GF/F filters on a Millipore vacuum manifold after the indicated time intervals. Data are from one experiment representative of three such experiments. B, concentration-effect relationship of 1 and 2 on the dissociation rate of [3H]PGD2 from CRTH2 receptors induced by 10 μM ramatroban. Dissociation rate constants were determined at a single time point (approximately after ∼2 dissociation half-lives) and are expressed as a percentage of control as described under Materials and Methods. The log IC50 values of 1 (-6.14 ± 0.11) and 2 (-4.85 ± 0.10) were obtained using nonlinear regression analysis of the curves to a logistic function and reflect the affinity of the compounds to the [3H]PGD2-occupied CRTH2 receptor. Each data point is given as mean ± S.E. of four independent experiments performed in triplicate.
Effect of Indole Derivatives on PGD2-Mediated CRTH2-Dependent Cellular Signaling Responses. We next wanted to explore the functional consequences of simultaneous occupation of CRTH2 by 1 or 2 and PGD2 and tested the effects of both compounds on PGD2-mediated CRTH2 activation in a set of functional assays. 1 and 2 completely lacked both agonistic and antagonistic activity and did not show any signs of enhancement of the PGD2 dose-response curve in assays measuring agonist-mediated stimulation of [35S]GTPγS binding in hCRTH2 receptor expressing CHO membranes (Fig. 4, A and B) or inositol phosphate accumulation in hCRTH2 expressing COS-7 whole cells (Fig. 4C, data not shown for compound 2). For control purposes, ramatroban was included in the functional experiments and displayed the expected antagonistic behavior in agreement with literature data (Fig. 4D) (Shichijo et al., 2003; Sugimoto et al., 2003; Ulven and Kostenis, 2005). It is interesting that both compounds acted as inhibitors of PGD2-mediated CRTH2-dependent arrestin translocation, a cellular event known to occur distal to (Gurevich and Gurevich, 2004) or independent of (Azzi et al., 2003; Wei et al., 2003) heterotrimeric G protein activation (Fig. 4E). PGD2-mediated arrestin translocation was also inhibited by ramatroban in agreement with its role as a functional CRTH2 receptor antagonist (Fig. 4E). Thus, ramatroban represents a classic antagonist inhibiting functional responses for all effector pathways studied, whereas 1 and 2 display markedly dissimilar degrees of inhibition of different effector pathways and represent antagonists with functional selectivity. It is important to note that neither compound interfered with arrestin recruitment stimulated upon agonist treatment of the thromboxane A2 (Fig. 4F), adrenergic β2, or neurokinin NK1 receptors (data not shown), ruling out nonspecific effects by both indoles on cellular arrestin translocation. Because 1 and 2 neither compete for PGD2 binding nor inhibit PGD2-mediated CRTH2-dependent G protein signaling, the antagonistic efficacy in BRET assays cannot be caused by antagonizing PGD2-induced arrestin translocation occurring distal to G protein activation. Hence, both compounds ought to inhibit arrestin translocation independent of G protein activation. To corroborate this hypothesis, we tested whether arrestin recruitment occurs in the absence of G protein activation and whether perturbation of G protein function is without consequences on the modulator-receptor-arrestin interaction. Toward this end, hCRTH2-HEK293 cells were incubated overnight in the presence of pertussis toxin (PTX) to disrupt CRTH2-Gαi/o interaction. CRTH2 is exclusively linked to Gαi/o-dependent signaling pathways in HEK293 cells because inhibition of cAMP formation as well as mobilization of inositol phosphates or intracellular calcium is completely sensitive to the pretreatment of cells with PTX (Sawyer et al., 2002). PGD2 induced the translocation of arrestin irrespective of whether cells were pretreated with PTX (Fig. 4G) or not (Fig. 4H). It is important to note that inhibition by compounds 1 and 2 of PGD2-mediated arrestin translocation was virtually indistinguishable when comparing PTX-treated cells with untreated cells. Together, the data suggest that PGD2-mediated CRTH2-dependent arrestin translocation mainly occurs independent of G protein activation and that both modulators interfere with PGD2-mediated arrestin recruitment in a G protein-independent fashion.
The identification of molecules that exert antagonistic efficacy on a specific signaling pathway of CRTH2 could be valuable in dissecting 7TM receptor signaling events in native cells (ex vivo) or in vivo. Thereafter, the functional selectivity of 1 was examined in human eosinophils, which are known to endogenously express the CRTH2 receptor. Compound 2 was not pursued further in native cells because of its low antagonistic potency on CRTH2. PGD2 is known to elicit eosinophil shape change (Hirai et al., 2001; Stubbs et al., 2002; Bohm et al., 2004), a response that can be abrogated in the presence of the CRTH2 antagonist ramatroban (Bohm et al., 2004). The inhibitory effect of ramatroban was confirmed in the eosinophil shape-change assay by a 30-fold rightward shift of the PGD2 dose-response curve (Fig. 5). In contrast, 1 seemed to selectively suppress the efficacy but not the potency of PGD2-dependent eosinophil shape change. It should be emphasized that the inhibitive effects of both ramatroban and 1 were specifically mediated by CRTH2 because neither compound displayed inhibition of eosinophil shape change elicited by eotaxin/CCL11, which acts through the chemokine CCR3 receptor.

Effects of 1 and 2 on PGD2-mediated stimulation of the CRTH2 receptor in different functional assays. Effect of 1 (A) and 2 (B) on PGD2-mediated stimulation of [35S]GTPγS binding in membranes from CHO-K1 cells stably transfected with CRTH2. PGD2 concentration-response curves in the presence of various concentrations of 1 (C) or ramatroban (D) in HEK293 cells transiently transfected with the CRTH2 receptor in inositol phosphate accumulation assays. E, inhibition of PGD2-mediated recruitment of βarrestin to the CRTH2 receptor in the presence of ramatroban, 1, and 2 in HEK293 cells stably transfected with βarrestin-GFP2 and CRTH2-Rluc. Antagonistic potency of the compounds was assessed in the presence of 100 nM PGD2. mBRET ratios were calculated as described under Materials and Methods. F, inhibition of U-46619-mediated recruitment of βarrestin to the TXA2 receptor in the presence of ramatroban, 1, and 2 in HEK293 cells stably transfected with βarrestin-GFP2 and TXA2-Rluc. Antagonistic potency of the compounds was assessed in the presence of 300 nM U-46619, the TXA2 agonist. mBRET ratios were calculated as described under Materials and Methods. G and H, inhibition of PGD2-mediated recruitment of βarrestin to the CRTH2 receptor by 1 and 2 in HEK293 cells stably transfected with βarrestin-GFP2 and CRTH2-Rluc pretreated (G) or not (H) with pertussis toxin (PTX). Data shown (A-H) are mean values ± S.E. of representative experiments each conducted in duplicate. At least three additional experiments gave similar results.
Discussion
Many ligands that target 7TM receptors bind to the same site as the endogeneous ligand, defined as the orthosteric binding site. However, receptors can also be influenced by ligands that bind to a topographically distinct allosteric site on the same receptor (Christopoulos and Kenakin, 2002; Soudijn et al., 2002; Jensen and Spalding, 2004). Orthosteric ligands do not necessarily have exactly overlapping binding domains but overlap sufficiently in their binding sites to be incapable of occupying a receptor at the same time. Therefore, orthosteric ligands are competitive in equilibrium binding assays. Allosteric ligands have little or no overlap with the orthosteric site. Hence, both allosteric and orthosteric ligands can concomitantly occupy the receptor. In this study, we report the identification of two indole derivatives structurally closely related to the orthosteric ligands indomethacin and ramatroban (Fig. 1), but unlike indomethacin or ramatroban, they are capable of binding CRTH2 simultaneously with its endogeneous ligand PGD2. In particular, compounds 1 and 2 enhanced [3H]PGD2 equilibrium binding in whole cells and retarded [3H]PGD2 dissociation reminiscent of the mode of action of allosteric enhancers (Gao et al., 2002; Lazareno et al., 2002; Figler et al., 2003; Muth et al., 2003; Avlani et al., 2004; Mohr et al., 2004). However, careful examination of their nature of interaction with CRTH2 using a combination of binding and functional assays revealed a quite unusual mechanism of action: 1) unlike many classic allosteric enhancers, both compounds failed to affect [3H]PGD2 binding in CRTH2 membrane preparations; 2) compound 1 not only increased the affinity of PGD2 for CRTH2 but seemed to create additional PGD2 binding sites; and 3) despite their ability to enhance the PGD2 binding capacity of CRTH2, they did not enhance the effects of PGD2 in functional assays.

Ramatroban and compound 1 inhibit flow cytometric shape-change responses of eosinophils to PGD2 but not to eotaxin/CCL-11. A, samples of polymorphonuclear leukocytes were pretreated with the antagonists or their vehicle and then stimulated with PGD2. Eosinophils were identified according to their autofluorescence, and shape-change responses were quantified as a percentage of the maximal response to PGD2 in the absence of an antagonist. Whereas ramatroban shifted the concentration-response curve to PGD2 rightward by a factor of 30, compound 1 reduced only the efficacy of PGD2. B, compound 1 does not affect eotaxin/CCL-11-induced eosinophil shape change. Data are shown as mean ± S.E., n = 4 to 6.
It seems that the intracellular composition of living cells and/or the fluidity and lipid composition of cell membranes play an active role in the PGD2-CRTH2-modulator interaction, because 1 and 2 did not show any effect in membrane preparations. The ability of 7TM receptors to interact with a large number of membrane-associated and intracellular proteins other than G proteins, kinases, or arrestins has been well-documented (Milligan and White, 2001), and the investigation on 7TM receptor dimers or oligomers (Terrillon and Bouvier, 2004) clearly establishes that such interactions may affect ligand binding and function. We have not yet sought to address an apparent cause for the discrepancies in whole-cell versus membrane binding assays in this study, and we are not aware of specific accessory proteins for CRTH2 nor of the propensity for the receptor to act as dimers or oligomers—all factors that may well influence the nature and extent of binding to (or signaling via) the CRTH2 receptor. Future studies, however, are warranted to address these issues.
To shed more light on the mode of interaction of 1 and 2 with CRTH2, we investigated the consequences of simultaneous CRTH2 occupation by PGD2 and the respective indole derivatives in functional signaling assays. In assays measuring PGD2-mediated stimulation of [35S]GTPγS binding or inositol phosphate production, cellular responses related to the activation of heterotrimeric G proteins, both compounds were unable to modulate the PGD2 cellular response (Fig. 4, A-C). The lack of modulatory efficacy in GTPγS assays, which require cell-membrane preparations, is congruent with the inability of both compounds to enhance [3H]PGD2 binding in membrane-based binding assays. However, their inability to affect PGD2-induced inositol phosphate production in whole cells is clearly indicative of a lack of modulatory efficacy in a classic G protein-dependent signaling assay. On the other hand, both compounds showed antagonistic efficacy in assays measuring the physical interaction between the ligand-activated CRTH2 receptor and βarrestin in both the absence and presence of a functional CRTH2-Gαi/o signaling pathway (Fig. 4, E, G, and H). The differential effects of the compounds on PGD2 binding capacity on the one hand and signaling on the other hand suggests that the CRTH2 receptor may be physically altered by the compounds such that enhanced PGD2 binding is offset by reduced receptor functionality, potentially caused by a “biased” conformational constraint on CRTH2 signaling. This conformational constraint could be evoked by the binding of a compound to any domain on the receptor that is not identical with but conformationally linked to the orthosteric binding site. It will be interesting to explore whether this second binding site is located on a single CRTH2 monomeric receptor or in a protomer of a potential CRTH2 homodimer, a question that is currently being addressed in our laboratory. To the best of our knowledge, this is the first report showing that antagonists may be endowed with the ability to discriminate between different receptor-mediated signal transduction pathways.
In turn, such biased antagonism also impacts the determination of antagonist affinities to a given receptor as the basis for receptor classification. Affinity of antagonists for a particular receptor has customarily been calculated from its ability to block agonist responses (Arunlakshana and Schild, 1959). Provided there is no change in the chemical nature of the antagonist or receptor, this affinity should remain constant for a given antagonist-receptor interaction, regardless of which agonists are present or what downstream signaling events are monitored. Therefore, differences in antagonist affinity have long been the basis for characterization of receptors and their subtypes (Arunlakshana and Schild, 1959; Black et al., 1972). Our data clearly add a layer of complexity to receptor classification on the basis of antagonist affinities and highlight potential pitfalls in interpreting Schild-type analyses derived from a single functional readout such as inhibition of arrestin translocation.
Arrestin proteins are well-known for their role in agonist-mediated desensitization and internalization of 7TM receptors (Ferguson, 2001; Luttrell and Lefkowitz, 2002; Gainetdinov et al., 2004; Gurevich and Gurevich, 2004). They have mainly been viewed as a regulatory component in a sequential pathway involving an activated receptor → G protein coupling → receptor phosphorylation → G protein uncoupling → arrestin recruitment. The finding that the indole derivatives 1 and 2 are capable of inhibiting arrestin translocation, whereas lacking any detectable effect on CRTH2-dependent G protein signaling provides additional support for the notion that arrestin proteins can function as independent signaling modules in an alternative G protein-independent pathway (Azzi et al., 2003; Baker et al., 2003; Wei et al., 2003). The observation that antagonists can cause markedly dissimilar degrees of inhibition of different effector pathways is vastly different from classic receptor theory that proposes a unique active conformation responsible for G protein signaling and subsequent arrestin recruitment. Our findings rather support the concept of ligand-specific receptor conformations (Kenakin, 2001, 2003, 2004) (that ligands are capable of selecting between different receptor-mediated pathways and thus activate or even inhibit certain pathways preferentially over others).
The results of the present study also have important implications for the detection and validation of biased antagonists. Whereas functional assays that are not biased toward detecting orthosteric ligands would be capable of identifying agents that modify receptor function in a pathway-specific manner, binding assays with binding phenomena such as those observed in our study would imply allosteric enhancer type of agents. It is apparent, therefore, that only a combination of two different functional assays monitoring G protein-dependent and -independent signaling pathways can validate such an unusual mechanism of action. It is important to note, however, that the ability of compounds to increase agonist binding may be indicative of either allosteric enhancement or biased antagonism.
Although the potency of our indoles is not sufficient to be explored in ex vivo settings, compound 1 was tested for its ability to interfere with PGD2-mediated CRTH2-dependent shape change of eosinophils that endogeneously express CRTH2. When applied at a concentration of 10 μM (maximally effective in inhibiting arrestin translocation in the BRET assay) (Fig. 4E), 1 significantly inhibited efficacy but not potency of the PGD2-mediated shape-change response. Inhibition of the maximum PGD2 efficacy was not caused by nonspecific interference with eosinophil shape change because the indole derivative lacked any significant effect on eosinophil shape change elicited by the CCR3 agonist eotaxin/CCL11. These data suggest that a decrease of PGD2 efficacy in eosinophil shape-change assays is the functional correlate of inhibition of arrestin recruitment in HEK293 cells and define a role for arrestin as a signaling molecule contributing to the complex phenomenon of eosinophil shape change. It will be interesting to determine whether decreased efficacy of PGD2-induced shape change in eosinophils affects their normal chemotactic movement and thus their recruitment to sites of inflammation, and we are currently exploring this question in our laboratory.
In summary, we reported herein the identification of two indole derivatives that selectively inhibit a specific signaling event of CRTH2 and thus provide the first example of antagonists capable of discriminating between different effector pathways. Such biased antagonists may serve as valuable tools for dissecting the contribution of individual signaling pathways of receptors in physiological and pathophysiological processes in vitro and ex vivo. We anticipate that the study of such unusual ligands will become of progressively greater importance to the drug discovery process because of the availability of a wide range of different 7TM receptor screening technologies derived from G protein-dependent and -independent signaling pathways.
Acknowledgments
We thank Prof. Jesus Gomeza for critical proofreading of the manuscript and helpful discussions, Helle Zancho Andresen for expert technical assistance, and Kate Hansen for excellent cloning support.
Footnotes
-
This work was supported by the European Community's Sixth Framework Programme (grant LSHB-CT-2003-503337).
-
ABBREVIATIONS: 7TM, seven transmembrane; CRTH2, chemoattractant receptor-homologous molecule expressed on Th2 cells; PGD2, prostaglandin D2; ELISA, enzyme-linked immunosorbent assay; BRET, bioluminescence resonance energy transfer; βarr2-GFP2, green fluorescent protein-β-arrestin2 fusion protein; CRTH2-Rluc, R. reniformis luciferase-CRTH2 fusion protein; HBSS, Hanks' balanced salt solution; 1, 1-(4-ethoxyphenyl)-5-methoxy-2-methylindole-3-carboxylic acid; 2, Nα-tosyltryptophan; DMSO, dimethyl sulfoxide; MAP, mitogen-activated protein; HEK, human embryonic kidney; CHO, Chinese hamster ovary; SPA, scintillation proximity assay; h, human; BSA, bovine serum albumin; PBS, phosphate-buffered saline; PTX, pertussis toxin; PCR, polymerase chain reaction; TXA2, thromboxane A2; ICI118551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol; SR121463B, benzamide, N-(1,1-dimethylethyl)-4-((cis-5′-ethoxy-4-(2-(4-morpholinyl)ethoxy)-2′-oxospiro(cyclohexane-1,3′-(3H)indol)-1′ (2′H)-yl)sulfonyl)-3-methoxy; U73122, 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; U-46619, 9,11-dideoxy-9α, 11α-methanoepoxy-prostaglandin F2α; [35S]GTPγS, guanosine 5′-O-(3-[35S]thio)triphosphate.
-
↵1 Current address: Ernest Gallo Clinic and Research Center, University of California, San Francisco, Emeryville, California.
- Received December 20, 2004.
- Accepted May 3, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}