


Abstract
Dual orthosteric agonists of metabotropic glutamate 2 (mGlu2) and mGlu3 receptors are being developed as novel antipsychotic agents devoid of the adverse effects of conventional antipsychotics. Therefore, these drugs could be helpful for the treatment of psychotic symptoms associated with Alzheimer's disease (AD). In experimental animals, the antipsychotic activity of mGlu2/3 receptor agonists is largely mediated by the activation of mGlu2 receptors and is mimicked by selective positive allosteric modulators (PAMs) of mGlu2 receptors. We investigated the distinct influence of mGlu2 and mGlu3 receptors in mixed and pure neuronal cultures exposed to synthetic β-amyloid protein (Aβ) to model neurodegeneration occurring in AD. The mGlu2 receptor PAM, N-4′-cyano-biphenyl-3-yl)-N-(3-pyridinylmethyl)-ethanesulfonamide hydrochloride (LY566332), devoid of toxicity per se, amplified Aβ-induced neurodegeneration, and this effect was prevented by the mGlu2/3 receptor antagonist (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)glycine (LY341495). LY566332 potentiated Aβ toxicity regardless of the presence of glial mGlu3 receptors, but it was inactive when neurons lacked mGlu2 receptors. The dual mGlu2/3 receptor agonist, (−)-2-oxa-4-aminobicyclo[3.1.0]exhane-4,6-dicarboxylic acid (LY379268), was neuroprotective in mixed cultures via a paracrine mechanism mediated by transforming growth factor-β1. LY379268 lost its protective activity in neurons grown with astrocytes lacking mGlu3 receptors, indicating that protection against Aβ neurotoxicity was mediated entirely by glial mGlu3 receptors. The selective noncompetitive mGlu3 receptor antagonist, (3S)-1-(5-bromopyrimidin-2-yl)-N-(2,4-dichlorobenzyl)pyrrolidin-3-amine methanesulfonate hydrate (LY2389575), amplified Aβ toxicity on its own, and, interestingly, unmasked a neurotoxic activity of LY379268, which probably was mediated by the activation of mGlu2 receptors. These data indicate that selective potentiation of mGlu2 receptors enhances neuronal vulnerability to Aβ, whereas dual activation of mGlu2 and mGlu3 receptors is protective against Aβ-induced toxicity.
Introduction
Current antipsychotic drugs have profoundly changed the treatment of psychotic disorders over the past 50 years. Nevertheless, these drugs and, in particular, second-generation antipsychotics have not fulfilled initial expectations, and several challenges persist in long-term management of psychotic disorders, such as the treatment of negative symptoms and cognitive deficits. Clinical trials have also raised questions about the perceived advantages of second-generation antipsychotics over first-generation antipsychotics, considering both the efficacy and safety of these drugs (Leucht et al., 2009). Furthermore first- and second-generation antipsychotics are known to increase the risk of cerebrovascular events in patients with dementia (Gill et al., 2005; Mazzucco et al., 2008).
Therefore, new antipsychotic compounds with improved efficacy and safety are needed as an alternative to current antipsychotic drugs. Neuroprotective strategies have been proposed as a new approach for preventing clinical deterioration in the course of schizophrenia (Lieberman, 2007). New antipsychotic drugs with a neuroprotective activity might also be useful for the treatment of psychosis in neurodegenerative disorders, such as Alzheimer's disease (AD) with severe psychosis (PAD).
Metabotropic glutamate (mGlu) receptors have been studied as potential new drug targets for the treatment of schizophrenia (Conn et al., 2009). mGlu receptors play a central role in neuronal and glial functions as well as in the modulation of neuronal excitability and synaptic transmission (reviewed by Niswender and Conn, 2010). A randomized, three-armed, double-blind, placebo-controlled study has demonstrated that (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039), a dual orthosteric agonist of mGlu2 and mGlu3 receptors, has a good efficacy in treating positive and negative symptoms in schizophrenic patients (Patil et al., 2007). It is noteworthy that patients treated with LY404039 do not differ from placebo-treated patients with respect to extrapyramidal motor symptoms or weight gain, suggesting that drugs targeting group II mGlu receptors have a better profile of tolerability compared with conventional antipsychotic drugs (Patil et al., 2007). This makes mGlu2/3 receptor agonists potential candidates for the treatment of PAD, providing that these drugs have a favorable impact on neurodegeneration associated with AD. Animal studies suggest that the antipsychotic activity of dual mGlu2/3 receptor agonists is mediated largely by the activation of mGlu2 receptors and is mimicked by selective positive allosteric modulators (PAMs)/enhancers of mGlu2 receptors (Fell et al., 2008; Fraley, 2009). Phase II clinical trials with mGlu2 receptor PAMs were expected to start in 2010 for the treatment of schizophrenia and anxiety. For an optimal use of group II mGlu receptor ligands in the treatment of PAD, it is critical to establish whether dual activation of mGlu2/3 receptors and selective activation of mGlu2 receptors differentially affect the pathophysiology of AD. Soluble aggregates of β-amyloid (Aβ) are widely considered as the primary cause of all pathological hallmarks of AD, including synaptic dysfunction, formation of neurofibrillary tangles, and neuronal death (Hardy, 2009; reviewed by Querfurth and LaFerla, 2010). Lee et al. (2004) have found that mGlu2 receptors show an increased expression and are colocalized with neurofibrillary tangles in the hippocampus of AD patients. Based on more recent data obtained in cell cultures (Lee et al., 2009), the same group speculates that activation of mGlu2 receptors supports neuronal survival and may therefore be protective in the AD brain. However, this contrasts with our own data showing that the protective activity of mGlu2/3 receptor agonists against excitotoxic neuronal death is entirely mediated by mGlu3 receptors, and activation of mGlu2 receptors might be harmful to neurons (Corti et al., 2007).
Here, we examine the distinct influence of mGlu2 and mGlu3 receptors on Aβ neurotoxicity, concluding that dual mGlu2/3 receptor agonists are better candidates than selective mGlu2 receptor PAMs for clinical use in patients with AD.
Materials and Methods
Drugs.
(−)-2-Oxa-4-aminobicyclo[3.1.0]exhane-4,6-dicarboxylic acid (LY379268), (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)glycine (LY341495), N-4′-cyano-biphenyl-3-yl)-N-(3-pyridinylmethyl)-ethanesulfonamide hydrochloride (LY566332), and (3S)-1-(5-bromopyrimidin-2-yl)-N-(2,4-dichlorobenzyl)pyrrolidin-3-amine methanesulfonate hydrate (LY2389575) were kindly provided by Eli Lilly and Company (Indianapolis, IN). (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) and 6,7-dinitroquinoxaline-2,3-dione (DNQX) were purchased from Tocris Bioscience (Bristol, UK). All other chemicals were purchased as specified.
Characterization of Compound LY2389575 as a Negative Allosteric Modulator of mGlu3 Receptors.
To study the pharmacological profile of LY2389575 we carried out fluorometric imaging plate reader (FLIPR) assays for mGlu3 and other human mGlu receptors. Individual AV-12 cell lines containing the rat excitatory amino acid transporter 1 and stably expressing the human mGlu1, mGlu2, mGlu3, mGlu4, mGlu5, mGlu7, mGlu8, or GABAB receptors were used for these studies. For mGlu2, mGlu3, mGlu4, mGlu7, mGlu8, and GABAB, clones also expressed the Gα15 subunit to force coupling to phospholipase C, resulting in the ability to measure receptor activation by fluorometric calcium response assay with a FLIPR instrument. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose and pyridoxine hydrochloride supplemented with 5% heat inactivated, dialyzed fetal bovine serum, 1 mM sodium pyruvate, 10 mM HEPES, 1 mM l-glutamine, and varying amounts of selection agents (geneticin, hygromycin B, zeocin, and blasticidin), depending on the cell line (all media from Invitrogen, Milan, Italy). Confluent cultures were passaged biweekly using an enzyme-free dissociation solution (Millipore Bioscience Research Reagents, Temecula, CA). All cell lines were cultured at 37°C; most were grown in 6.5% CO2 (mGlu1 and mGlu5 were grown in 5% CO2). Cells were harvested 18 to 24 h before assay and dispensed into 96-well, black-walled, poly-d-lysine-coated plates. Cell densities and plating medium varied for each receptor. For mGlu3, cells were seeded at 115,000 cells per well in medium containing only 125 μM l-glutamine (freshly added). The cell culture medium was removed, and the cells were incubated with 8 μM Fluo-3AM (50 μl/well) for 90 to 120 min (depending on the cell line) at 25°C. The dye solution was removed and replaced with assay buffer (50 μl/well) comprised of Hanks' balanced salt solution supplemented with 20 mM HEPES. A single-addition FLIPR assay generating a 10-point concentration response curve for the agonist glutamate was conducted before each experiment. The results were analyzed by Prism v4.03 (GraphPad Software, Inc., San Diego, CA) to calculate the concentrations of glutamate needed to induce the EC90 (antagonist assay) and EC10 (potentiator assay) responses. The compound was diluted in dimethyl sulfoxide using a 3-fold dilution series and tested at each mGlu receptor in a two-addition FLIPR assay using a 10-point concentration response profile starting at a final concentration of 25 μM for the agonist assay and 12.5 μM for the potentiator and antagonist assays (final concentration of dimethyl sulfoxide, 0.625%).
LY2389575 was added to the cells in the FLIPR, and data were collected every second for the first 30 s and then every 3 s for a total of 90 s to detect agonist activity, immediately followed by an EC90 or EC10 value of glutamate in assay buffer. After the second addition, data were collected every second for 29 images and then every 3 s for 15 images. The maximal response was defined as that induced by ECmax (100 μM glutamate). The compound effect was measured as maximal − minimal peak heights in relative fluorescent units corrected for basal fluorescence measured in the absence of glutamate. Agonist effects were quantified as the percentage of stimulation induced by compound alone relative to the maximal glutamate response. Antagonist effects were quantified by calculating the percentage inhibition of the EC90 glutamate response caused by the compound. Potentiation effects were quantified as the percentage of increase in the presence of an EC10 response in glutamate relative to the ECmax response. All data were calculated as relative IC50 or EC50 values using a four-parameter logistic curve-fitting program (ActivityBase v5.3.1.22; IBS, Alamenda, CA).
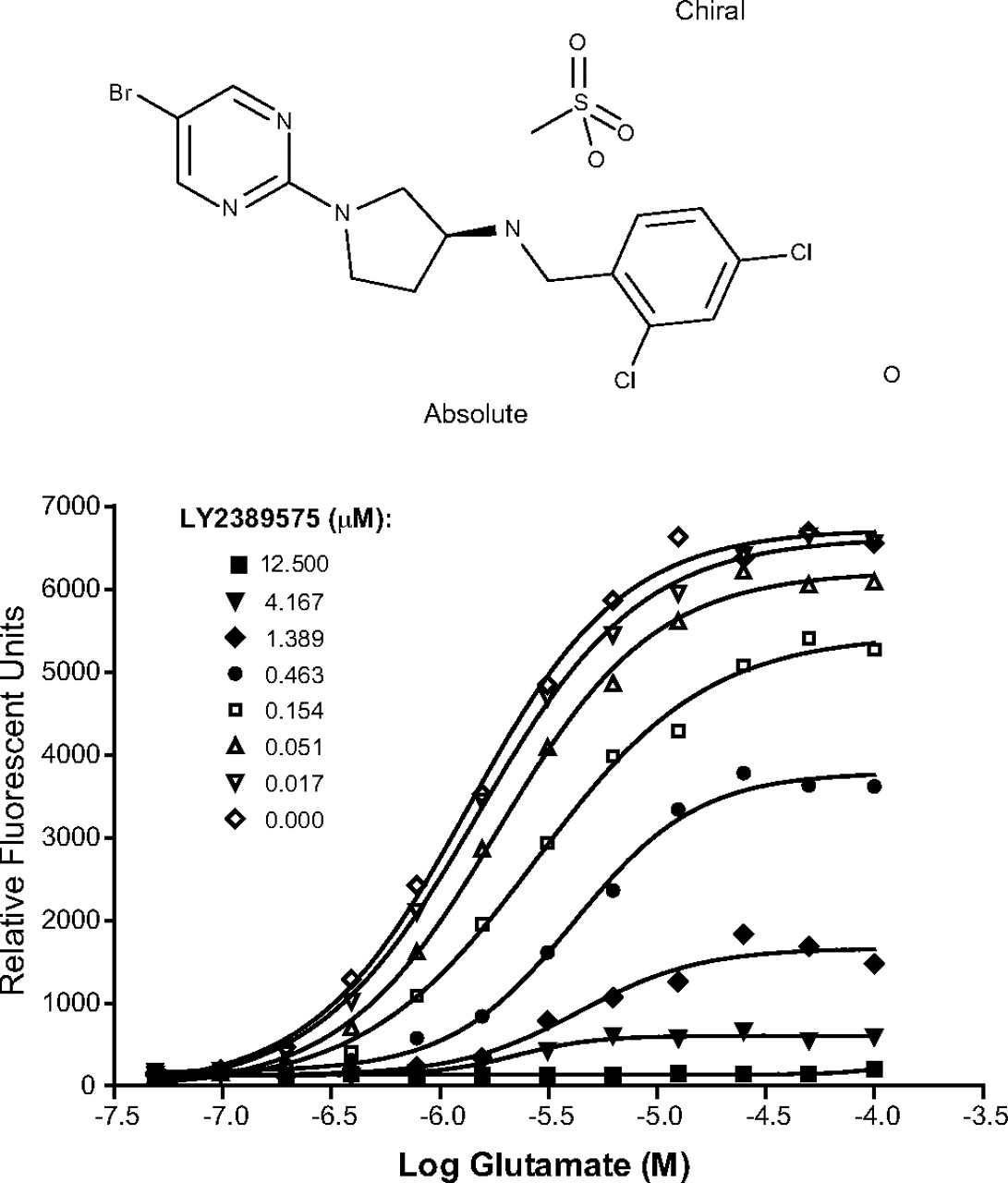
The FLIPR assay system was also used to determine whether the compound-receptor interaction was competitive or allosteric in nature. In these experiments, a 12-point glutamate dose-response curve was generated using the antagonist mode in the absence and presence of increasing concentrations of LY2389575. The response curves were visualized using Prism. Figure 1 shows that LY2389575 behaved as a noncompetitive antagonist of mGlu3 receptors with an IC50 value of 190 ± 26 nM and an efficacy of 100 ± 0.68% (means + S.E.M.). LY2389575 did not affect responses mediated by all other mGlu receptor subtypes or GABAB receptors (Table 1).

Chemical structure of LY2389575 (top) and glutamate curve shift in the presence of LY2389575 in the FLIRP assay using mGlu3 receptor clones expressing the Gα15 subunit (bottom). See Materials and Methods for a detailed description of the assay.
Summary of potency values (EC50 or IC50; nanomolar) for LY2389575 as an agonist, antagonist, or potentiator across a panel of human cloned mGlu and GABAB receptors using the FLIPR assay format
Mean EC50 or IC50 ± S.E.M. Entries of > indicate that the response to LY2389575 was <50% at the highest concentration used in the assay. Values typically represent three to five replicates.
Animals.
Animal experimental procedures were carried out in accordance with the directives of the Italian and European Union regulations for the care and use of experimental animals (DL116/92) and approved by the Italian Ministry of Health. Animals (Morini, Reggio Emilia, Italy) were kept under environmentally controlled conditions (ambient temperature = 22°C, humidity = 40%) on a 12-h light/dark cycle with food and water ad libitum. mGlu2 receptor knockout mice, mGlu3 receptor knockout mice, and double mGlu2/mGlu3 receptor knockout mice with a CD1 genetic background were kindly provided by Eli Lilly and Company. All mice undergoing experimental procedures were individually genotyped for the mGlu2 and mGlu3 receptor gene by polymerase chain reaction (PCR).
Pure Cultures of Cortical Astrocytes.
Cortical glial cells were prepared from 1- to 3-day-old Sprague-Dawley rats. After removal of meninges and isolation of cortices, cells were dispersed by mechanical and enzymatic dissociation using a 0.25% solution of trypsin (Invitrogen). Cells were plated onto 75-mm2 flasks and maintained in DMEM, supplemented with 10% fetal calf serum, penicillin/streptomycin (100 U/ml-100 μg/ml), and glutamine (2 mM). All medium constituents were from Invitrogen, and all plastic materials were from Corning Life Sciences (Lowell, MA). Confluent cultures at 8 to 10 days in vitro were shaken overnight at 37°C to remove microglia and oligodendrocytes. Astrocytes were collected by trypsin digestion, seeded onto 35- or 100-mm dishes, and used for experiments 6 to 8 days after replating.
Astrocyte cell cultures were also prepared from wild-type and mGlu3(−/−) mice 1 to 3 days after birth. Dissociated cortical cells were grown in 15-mm multiwell vessels (Falcon Primaria, Lincoln Park, NJ) using a plating medium of MEM-Eagle's salts supplemented with fetal bovine serum (10%), horse serum (10%), glutamine (2 mM), and glucose (final concentration 21 mM). Cultures were kept at 37°C in a humidified CO2 atmosphere until they reached confluence (10–14 days in vitro). Confluent cultures were then used as a support for mixed cultures.
Pure Cultures of Rat Cortical Neurons.
Cultures of pure cortical neurons were obtained from rats at embryonic day 15. In brief, cortices were dissected in Ca2+/Mg2+-free buffer and mechanically dissociated. Cortical cells were plated at a density of 2 × 106/dish on 35-mm dishes (Nalge Nunc International, Rochester, NY) precoated with 0.1 mg/ml poly-d-lysine (Sigma-Aldrich, Milan, Italy) in DMEM/Ham's F12 (1:1) supplemented with the following components: bovine serum albumin (10 mg/ml), insulin (10 μg/ml), transferrin (100 μg/ml), putrescine (100 μM), progesterone (20 nM), selenium (30 nM), glutamine (2 mM), glucose (6 mg/ml), and penicillin/streptomycin (100 U/ml-100 μg/ml). Cytosine-d-arabinofuranoside (10 μM) was added to the cultures 18 h after plating to avoid the proliferation of non-neuronal elements and was kept for 3 days before medium replacement. This method yields >99% pure neuronal cultures, as judged by immunocytochemistry for glial fibrillary acidic protein and flow cytometry for neuron-specific microtubule-associated protein 2 (Copani et al., 1999).
Mixed Cultures of Rat Cortical Cells.
Cells dissected from the cortices of rat embryos and dissociated as described above were grown into DMEM/F12 (1:1) supplemented with horse serum (10%), fetal calf serum (10%), glutamine (2 mM), and glucose (6 mg/ml). After 7 to 10 days in vitro, glial cell division was halted by exposure to cytosine-d-arabinoside (10 μM) for 3 days, and cells were shifted into a maintenance serum-free medium. Mature cultures contained approximately 35 to 40% neurons and 60 to 65% of glial fibrillary acidic protein plus astrocytes.
Mixed Cultures of Mouse Cortical Cells.
Mixed cultures of cortical cells, containing both neurons and astrocytes, were also prepared from wild-type, mGlu2(−/−), mGlu3(−/−), and double mGlu2(−/−)/mGlu3(−/−) fetal mice (14–16 days of gestation). In brief, mice were killed by cervical dislocation under chloroform anesthesia, and dissociated cortical cells were plated in 15-mm multiwell vessels (Falcon Primaria) on a layer of confluent astrocytes (10–14 days in vitro), prepared from wild-type and mGlu3(−/−) mice, using a plating medium of MEM-Eagle's salts (supplied glutamine free) supplemented with heat-inactivated horse serum (10%), fetal bovine serum (10%), glutamine (2 mM), and glucose (final concentration 21 mM). Cultures were kept at 37°C in a humidified 5% CO2 atmosphere. After 3 to 5 days in vitro, non-neuronal cell division was halted by 1- to 3-day exposure to cytosine-β-arabinoside (10 μM), and cultures were shifted to a maintenance medium identical to plating medium but lacking fetal serum. Subsequent partial medium replacement was carried out twice a week. Only mature cultures (13–14 days in vitro) were used for the experiments.
Handling of Aβ.
Different lots of Aβ(25-35) (Bachem, Bubendorf, Switzerland) were tested, and the same batch was used throughout the entire study to rely on a consistent profile of toxicity. Peptides were solubilized in sterile, double distilled water at an initial concentration of 2.5 mM and stored frozen at −20°C. Aβ(25-35) was used at a final concentration of 25 μM in the presence of the glutamate receptor antagonists MK-801 (1 μM) and DNQX (30 μM) to avoid the potentiation of endogenous glutamate toxicity.
Aβ(1-42) (Bachem) and oligomers were obtained using a well established protocol (Lambert et al., 1998) with some modifications. The full-length peptide was dissolved in trifluoroacetic acid at a concentration of 1 mg/ml and sonicated for 10 min. Trifluoroacetic acid was removed by gentle streaming of argon. Aβ(1-42) was then dissolved in 1,1,1,3,3,3-hexa-fluoro-2-propanol and incubated at 37°C for 1 h. After argon streaming, peptides were dissolved again in 1,1,1,3,3,3-hexa-fluoro-2-propanol, lyophilized, and then resuspended in 5 mM anhydrous dimethyl sulfoxide before dilution to 100 μM in ice-cold DMEM-F12. According to Lambert protocol, the peptide suspension was allowed to oligomerize overnight at 4°C. High-mass oligomers known to be toxic (>50 kDa) were isolated from the peptide suspension by the use of 50-kDa cutoff filters (Giuffrida et al., 2009). Aβ(25-35) or Aβ(1--42) were added to cultures for 24 h in the absence or presence of mGlu receptor ligands.
Assessment of Neuronal Injury.
In mixed cortical cultures, neuronal injury was estimated by examination of the cultures by phase-contrast microscopy 24 h after the incubation with Aβ. Neuronal damage was quantitatively assessed in all experiments by estimation of dead neurons by trypan blue staining. Stained neurons were counted from three random microscopic fields per well. In pure neuronal cultures, neuronal injury was assessed by the 3-[4,5-dimethylthioazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay. Cultures were incubated with MTT (0.9 mg/ml final concentration; Sigma-Aldrich) for 2 h at 37°C. A solubilization solution containing 20% SDS was then added for an additional 1 h, and formazan production was evaluated in a plate reader (absorbance = 560 nm).
Determination of Transforming Growth Factor-β1 Levels in the Astrocyte Medium.
Astrocyte-conditioned medium was collected and subjected to acid treatment procedure. Samples were acidified to a pH of approximately 2.6 with 1 N HCl for 15 min at room temperature, then neutralized to approximately pH 7.6 with 1 N NaOH. Levels of TGF-β1 released into the medium were measured by enzyme-linked immunosorbent assay using the TGF-β1 Emax ImmunoAssay System (Promega, Madison, WI), based on an antibody sandwich format, strictly following the manufacturer's instructions. In brief, 96-well plates were coated overnight at 4°C with primary monoclonal anti-TGF-β1 antibody. A blocking solution was added for 35 min at 37°C before incubation with samples and standards for 90 min at room temperature, to allow binding of soluble TGF-β1. A primary polyclonal anti-TGF-β1 antibody was then added for 2 h to bind captured TGF-β1. Finally, specifically bound polyclonal antibody was detected by incubation for 2 h with a horseradish peroxidase-conjugated secondary antibody. Wells were extensively washed between each step. After a final 10-min incubation with a chromogenic substrate solution, the resulting redox reaction was stopped by acidification with 1 N HCl, and absorbance was immediately measured at 450 nm. The assay is sensitive in the range of 32 to 1000 pg/ml.
Gene Expression Analysis by Real-Time RT-PCR.
Total RNA was isolated from cultured astrocytes treated with LY379268 (1 μM) ± LY341495 (1 μM), using TRIzol reagent (Invitrogen) according to the manufacturer's protocol and retrotranscribed into cDNA by using SuperScript III Reverse Transcriptase (Invitrogen). Real-time RT-PCR was performed on the ABI Prism7500 Sequence Detection System (Applied Biosystems, Foster City, CA). PCR was performed by using Power SYBR Green PCR Master Mix Kit (Applied Biosystems) according to the manufacturer's instructions. Thermal cycler conditions were as follows: 5 min at 50°C, 2 min at 95°C, 40 cycles of denaturation (45 s at 95°C), and combined annealing/extension (1 min at 59°C). The sequences of β-actin and TGF-β1 primers used were: β-actin forward 5′-ctggctcctagcaccatga-3′ and reverse 5′-tagagccaccaatccacaca-3′; TGF-β1 forward 5′-atacgcctgagtggctgtct-3′ and reverse 5′-tgggactgatcccattgatt-3′. Concentrations of mRNA were calculated from serially diluted standard curves simultaneously amplified with the samples and normalized with respect to β-actin mRNA levels.
Results
Rat cortical cultures, both pure neuronal and mixed neuron-glial types, were challenged either with soluble aggregates of the active Aβ fragment, Aβ(25-35), (Giuffrida et al., 2007) or oligomers of the full-length Aβ(1-42) (Giuffrida et al., 2009). Aβ(25-35) and Aβ(1-42) were used at concentrations of 25 μM and 100 nM, respectively, which induced a similar extent of neuronal death (35–45%) over a 24-h period. Because Aβ is able to potentiate glutamate toxicity (Koh et al., 1990; Copani et al., 1991), experiments were carried out in the presence of a cocktail of ionotropic glutamate receptor antagonists (1 μM MK-801 + 30 μM DNQX) to exclude the contribution of endogenous excitotoxicity to the overall process of neuronal death.
We first tested the effects of the selective mGlu2 receptor enhancer LY566332 (Johnson et al., 2005; Rorick-Kehn et al., 2005) in combination with either Aβ(25-35) or Aβ(1-42) (Fig. 2). LY566332 was not toxic per se up to a concentration of 10 μM (see legend of Fig. 2); however, it potentiated the toxicity of Aβ peptides in both mixed (Fig. 2, A and C) and pure (Fig. 2, B and D) neuronal cultures. LY566332-induced potentiation of Aβ toxicity was prevented by the coapplication of the preferential mGlu2/3 receptor antagonist LY341495 (Schoepp et al., 1999), indicating that the enhancer amplified the action of endogenous glutamate at mGlu2 receptors in both mixed and pure neuronal cultures (Fig. 2, A, C, and D).

The selective mGlu2 receptor PAM, LY566332, amplifies Aβ neurotoxicity in culture. A, data obtained in mixed rat cortical cultures challenged with Aβ(25-35) are shown. LY566332 and LY341495 were coincubated with Aβ(25-35) for 24 h. Toxicity was assessed by cell counting after trypan blue staining. Cell counts was performed in three random microscopic fields/well. In control cultures, the number of dead neurons was 41 ± 2.6 (n = 12). This number increased to 92 ± 10 after incubation with Aβ(25-35) (n = 12). The number of dead neurons was 50 ± 2.3 in cultures treated with 10 μM LY566332 (n = 9). Values are expressed as percentage of Aβ(25-35) toxicity and are means ± S.E.M. of 9 to 12 determinations. *, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus Aβ(25-35) alone. B, Aβ(25-35) toxicity in pure cultures of rat cortical neurons is shown, where values of the MTT assay are expressed as optical density (O.D.) per culture dish and are means ± S.E.M or six to nine determinations. p < 0.05 (one-way ANOVA + Fisher's PLSD) versus the respective control values (*) or versus values obtained with Aβ(25-35) in the absence of LY566332 (#). C and D, data in mixed (C) and pure (D) neuronal cultures challenged with full-length Aβ(1-42) are shown. In C, addition of Aβ(1-42) increased the number of dead neurons from 42 ± 3.8 to 110 ± 15. Values are means ± S.E.M. of nine determinations. *, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus Aβ(1-42) alone. In D, values are means ± S.E.M. of six to nine determinations. p < 0.05 (one-way ANOVA + Fisher's PLSD) versus controls (Ctrl) (*) or versus Aβ(1–42) alone (#). LY341495 had no effect on neuronal viability in the absence of Aβ (not shown).
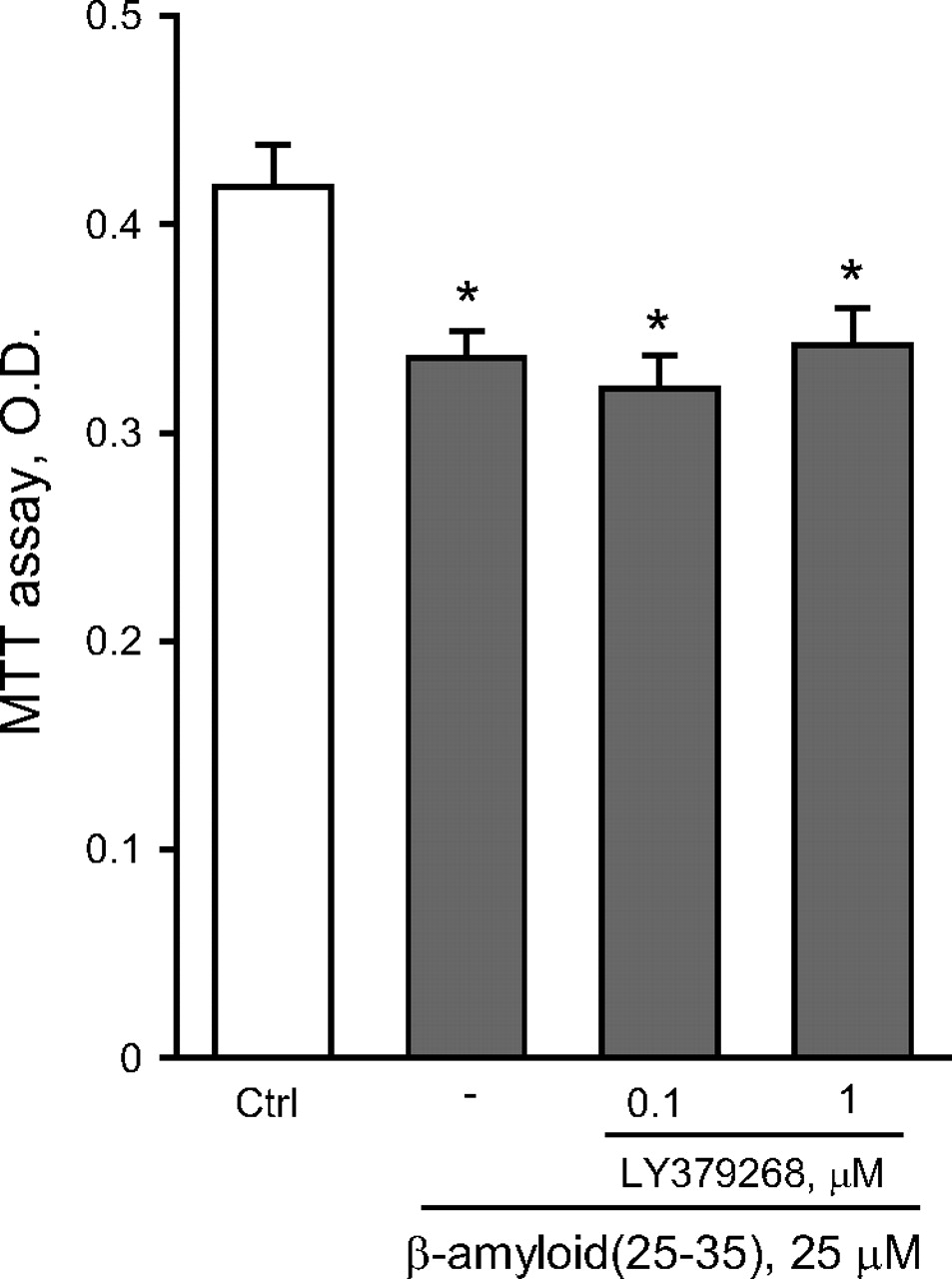
To compare the effects of an isolated activation of mGlu2 receptors versus a combined activation of mGlu2 and mGlu3 receptors, we used the orthosteric mGlu2/3 receptor agonist LY379268. As opposed to LY566332, in mixed cultures LY379268 reduced Aβ(25-35)-induced neurodegeneration in a concentration-dependent manner, and protection was prevented by LY341495 (Fig. 3A). These data indicated that, at least in mixed cultures, activation of mGlu3 receptors by LY379268 could result into neuroprotection against Aβ. In mixed cultures, the mGlu3 receptor NAM, LY2389575 (see Fig. 1 and Table 1), abolished the protective activity of 1 μM LY379268 in the concentration range of 0.1 to 1 μM and amplified Aβ toxicity on its own at the highest concentration (Fig. 3B). At these concentrations, LY2389575 was not toxic in the absence of Aβ (see legend of Fig. 3). It is noteworthy that LY379268 lost its neuroprotective activity in pure neuronal cultures (Fig. 4), indicating that activation of glial mGlu3 receptors could be required for neuroprotection.

Activation of mGlu3 receptors protects neurons against Aβ toxicity in mixed rat cortical cultures. Mixed cortical cultures were challenged with Aβ(25-35) in the absence or presence of the dual orthosteric mGlu2/3 receptor agonist, LY379268, applied alone or combined with the mGlu2/3 receptor antagonist, LY341495 (A) or with the mGlu3 receptor NAM, LY2389575 (B). Values are expressed as percentage of Aβ(25-35) toxicity and are means ± S.E.M. of 12 to 15 (A) or 9 (B) determinations. In A, *, p < 0.05 versus Aβ(25–35) alone; in B, p < 0.05 versus the respective values obtained in the absence of LY379268 (*) or values obtained with Aβ(25–35) alone (#). LY2389575 did not affect neuronal viability in the absence of Aβ (number of dead neurons in three microscopic fields: 41 ± 3, and 42 ± 1 in control cultures and in cultures treated with 1 μM LY2389575, respectively; n = 3 per condition). LY379268 and LY341495 were also devoid of activity on their own.
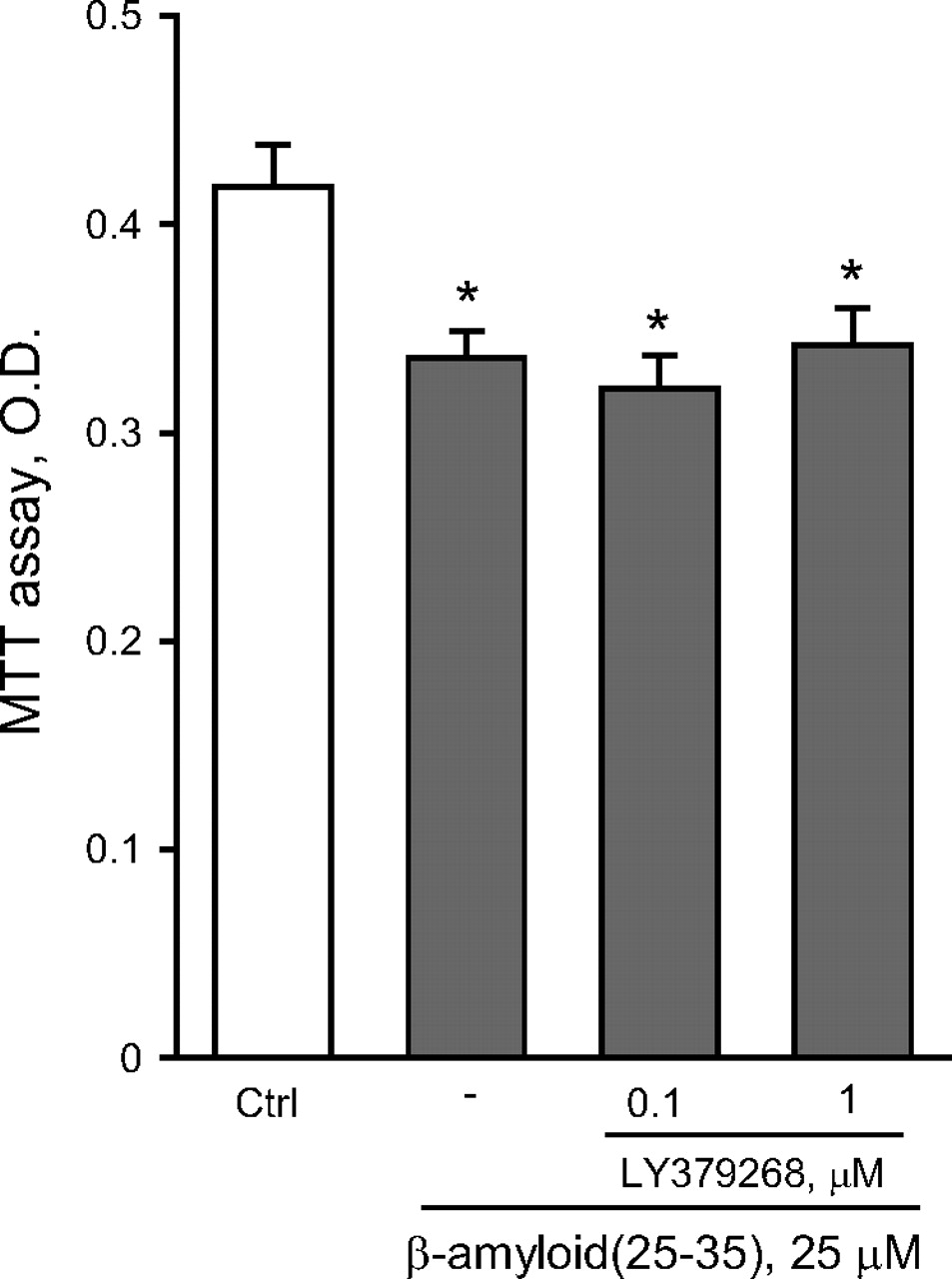
The dual orthosteric mGlu2/3 receptor agonist, LY379268, is not neuroprotective in pure cultures of rat cortical neurons challenged with Aβ(25-35). Values are means ± S.E.M. of six to nine determinations. *, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus control (Ctrl) cultures. LY379268 had no effect on neuronal viability on its own (not shown).
To ascertain the contribution of glial mGlu3 receptor activation to the effect of LY379268, pure neuronal cultures were exposed to a glial conditioned medium (GCM) (i.e., medium collected from cultures of cortical astrocytes 24 h after a transient exposure to 1 μM LY379268 or vehicle) before the treatment with Aβ(25-35). GCM collected from untreated astrocytes reduced spontaneous death occurring in pure neuronal cultures, but it was not sufficient to protect neurons against Aβ toxicity (Fig. 5A), which was prevented instead by the GCM collected from astrocytes challenged with LY379268 (Fig. 5A). Application of LY341495 to cultured astrocytes prevented not only the neuroprotective effect of the LY379268 challenge, but also the basal prosurvival effect of GCM (Fig. 5A). Similar data were obtained treating cultured astrocytes with the mGlu3 receptor NAM, LY2389575 (Fig. 5B). All together, these data indicate that the basal prosurvival effect of GCM and the protective effect of the GCM collected by astrocytes treated with LY379268 are mediated by the activation of glial mGlu3 receptors. The GCM collected from untreated astrocytes contained a fair amount of TGF- β1 (∼50 pg/ml) (Fig. 5E), an established neuroprotective factor against Aβ toxicity (reviewed by Caraci et al., 2009), which is known to be released by astrocytes in response to mGlu2/3 receptor agonists (Bruno et al., 1998). Astrocytes challenged with LY379268 released a greater amount of TGF-β1 (Fig. 5D), probably resulting from an increased transcription of the growth factor (Fig. 5D). A neutralizing antibody specific for TGF-β1 (2 μg/ml) added to the glial medium abolished the basal prosurvival effect of GCM, as well as the protective activity of the GCM derived from LY379268-treated astrocytes against Aβ (Fig. 5C). Similar data were obtained by supplementing the GCM with the specific inhibitor of type-1 TGF-β receptor, 4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]benzamide (SB431542) (Fig. 5C). Finally, TGF-β1 antibodies prevented the neuroprotective activity of LY379268 applied to mixed cultures challenged with either Aβ(25-35) (Fig. 6A) or Aβ(1-42) (Fig. 6B). Consistent with the indication that protection against Aβ neurotoxicity is selectively mediated by glial mGlu3 receptors, LY379268 lost its protective activity in murine mixed cortical cultures in which astrocytes were obtained from mGlu3 receptor knockout mice (Fig. 7A). As expected, the mGlu3 receptor NAM, LY2389575, was able to amplify Aβ toxicity and abolish the neuroprotective activity of LY379268 only in cultures containing wild-type astrocytes (Fig. 7A). Finally, the selective mGlu2 receptor PAM, LY566332, potentiated Aβ toxicity regardless of the presence of glial mGlu3 receptors (Fig. 7, B and C). Instead, potentiation of Aβ toxicity by LY566332 was lost in mixed cultures in which neurons were obtained from double mGlu2 and mGlu3 receptor knockout mice (Fig. 7B) or mGlu2 receptor knockout mice (Fig. 7C), indicating once more that the isolated activation of neuronal mGlu2 receptors was toxic to neurons.
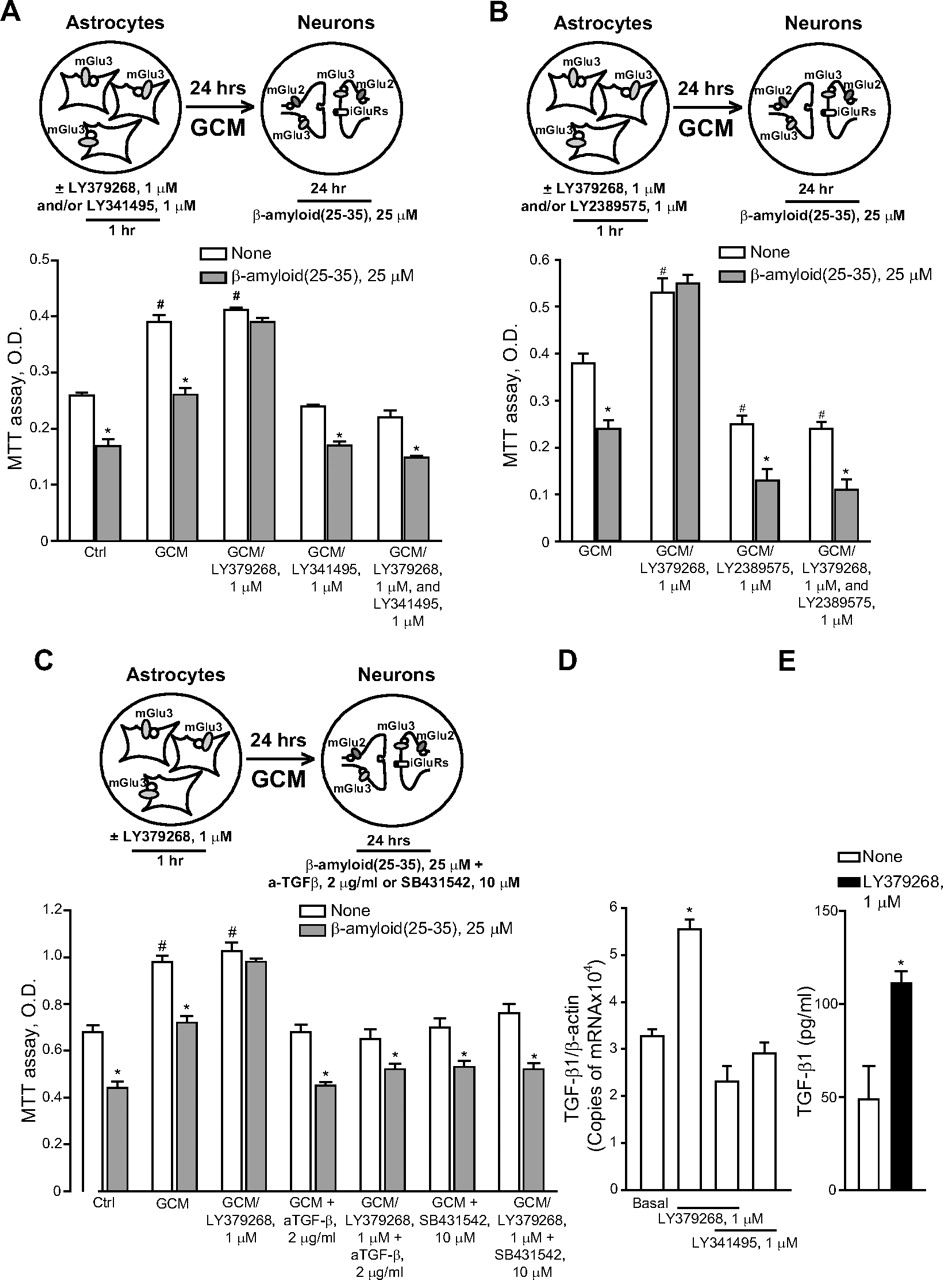
Activation of glial mGlu3 receptors protects neurons against Aβ toxicity via a paracrine mechanism mediated by TGF-β. A to C, pure cultures of rat cortical neurons were exposed to GCM and then challenged with Aβ(25-35). GCM alone = medium collected from untreated rat cultured cortical astrocytes; GCM/LY379268 and/or LY341495 or LY2389575 = medium collected from cultured astrocytes transiently exposed to LY379268 and/or LY341495 or LY2389575; GCM and GCM/LYs + anti-TGF-β1 neutralizing antibodies (aTGF-β) or SB431542 = astrocyte medium supplemented with anti-TGF-β1 antibodies or SB431542 just before being transferred into pure neuronal cultures. Values are means ± S.E.M. of 9 to 12 (A and C) or 5 to 6 (B) determinations. In A and C, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus the respective values obtained in the absence of Aβ (none) (*) or versus control (Ctrl)/none values (#). In B, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus the respective values obtained in the absence of Aβ (none) (*) or versus Ctrl values obtained with GCM alone (#). D, TGF-β1 mRNA levels in cultured astrocytes transiently exposed to LY379268 and/or LY341495 24 h before are shown. Values were normalized by β-actin mRNA levels and are means + S.E.M. of six determinations. *, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus values obtained in control astrocytes (Ctrl). E, levels of TGF-β1 in the medium of cultured astrocytes transiently exposed to LY379268 24 h before are shown. Values are means + S.E.M. of six determinations. *, p < 0.05 (Student's t test) versus values measured in the medium of untreated control astrocytes.

aTGF-β abolish the neuroprotective action of LY379268 in mixed cultures of rat cortical cells challenged with either Aβ(25-35) (A) or Aβ(1-42) (B). Values are means ± S.E.M. of nine (A) or six (B) determinations. *, p < 0.05 (one-way ANOVA + Fisher's PLSD) versus the respective values obtained in the absence of LY379268. aTGF-β had no effect on neuronal viability in the absence of Aβ (not shown).

Influence of mGlu2/mGlu3 receptor ligands on Aβ(25-35) toxicity in mixed cultures of cortical cells prepared from wild-type, mGlu3(−/−), mGlu2(−/−), or double mGlu2(−/−)/mGlu3(−/−) mice. A, LY379268 and LY2389575 were tested in wild-type cultures and cultures in which the astroglial component was prepared from mGlu3(−/−) mice and the neuronal component was from wild-type mice. B, LY566332 and LY341495 were tested in wild-type cultures, cultures with mGlu3(−/−) astrocytes and wild-type neurons, and cultures with mGlu3(−/−) astrocytes and double mGlu2(−/−)/mGlu3(−/−) neurons. C, LY566332 was tested in wild-type cultures, cultures with mGlu3(−/−) astrocytes and wild-type neurons, and cultures with mGlu3(−/−) astrocytes and mGlu2([minus]/[minus]) neurons. Values are means ± of six to nine (A), five to six (B), and six (C) determinations. p < 0.05 (one-way ANOVA + Fisher's PLSD) versus the respective values obtained with Aβ(25-35) alone (*), the respective values obtained with Aβ(25-35) + LY2389575 (§), or the respective values obtained in control cultures with wild-type astrocytes and wild-type neurons (#).
Discussion
Oligomers of Aβ(1-42), the most toxic forms of amyloid deposits, cause neuronal dysfunction and death (reviewed by Ashe and Zahs, 2010). Neuronal cultures challenged with synthetic analogs of human Aβ provide a widely accepted and reliable model of neurodegeneration occurring in AD. We used either the shortest neurotoxic fragment of Aβ, Aβ(25-35), or isolated oligomers (>50 kDa) of full-length Aβ(1-42). As a dual orthosteric mGlu2/3 receptor agonist, we used compound LY379268, which shares the same pharmacological properties of the clinically active compound, LY404039, and relieves psychotic-like symptoms in animal models of schizophrenia (Cartmell et al., 2000; Swanson and Schoepp, 2003; Carter et al., 2004). To dissect the individual role of mGlu2 and mGlu3 receptors we have used compound LY566332 as a selective mGlu2 receptor PAM, and compound LY2389575, which was proven to behave as a selective mGlu3 receptor NAM. By definition, a PAM amplifies receptor activation in the presence of an orthosteric agonist, whereas a NAM inhibits receptor activation regardless of the agonist concentration. Both LY566332 and LY2389575 influenced Aβ toxicity on their own, suggesting that the process of neuronal death is under the influence of endogenous glutamate-activating group II mGlu receptors. Data indicate that LY566332 enhanced Aβ toxicity by amplifying the endogenous activation of mGlu2 receptors in neurons. Accordingly, potentiation of neurotoxicity was abrogated by the mGlu2/3 receptor antagonist, LY341495, and was not seen when cultured neurons lacked mGlu2 receptors. This is the first direct clear-cut evidence that pharmacological activation of mGlu2 receptors acts synergistically with a neurotoxin in causing neuronal death and discourages the development of mGlu2 receptor PAM for the treatment of psychiatric symptoms in AD or other neurodegenerative disorders. In contrast, Lee et al. (2009) suggest that activation of mGlu2 receptors may have a neuroprotective role in AD. However, this suggestion was based on the ability of LY379268 to activate the extracellular signal-related kinase pathway and tau phosphorylation in cultured neurons and to protect mGlu2 receptor-expressing cells against oxidative stress. Although the data of Lee et al. (2009) are interesting, they cannot be easily compared with our data obtained with subtype-selective ligands in neurons challenged with β-amyloid.
It should be highlighted that in our hands LY566332 was not toxic on its own and might therefore be safe in the absence of an associated neuronal insult. Whether or not selective activation of mGlu2 receptors affects the intrinsic neurodegeneration associated with schizophrenia (reviewed by Lieberman, 1999; Pérez-Neri et al., 2006; Rund, 2009) is an important question that warrants further investigation.
Activation of glial mGlu3 receptors was protective against Aβ neurotoxicity to an extent sufficient to overcome the toxic activity of mGlu2 receptors when cultures were treated with the dual agonist, LY379268. LY379268, which was otherwise highly protective, became neurotoxic in the presence of the mGlu3 receptor NAM, LY2389575. Orthosteric mGlu2/3 receptor agonists might therefore be optimal candidates for the treatment of PAD, relieving psychotic symptoms (via the mGlu2 receptor) and protecting neurons against Aβ toxicity (via the mGlu3 receptor) at the same time. Results of experiments in which the GCM was transferred to pure neuronal cultures indicate that pharmacological activation of glial mGlu3 receptors was protective against Aβ neurotoxicity via a paracrine mechanism mediated by TGF-β1. Endogenous activation of mGlu3 receptors in astrocytes enhanced neuronal survival, but was insufficient to afford protection against Aβ neurotoxicity. Only when glial mGlu3 receptors were pharmacologically activated by LY379268 the amount of TGF-β1 secreted in the GCM became sufficient to protect neurons against Aβ toxicity. This implies that glial mGlu3 receptors are not saturated by the endogenous glutamate and are able to respond to a pharmacological agonist by stimulating the synthesis and secretion of TGF-β1. A paracrine mechanism of neuroprotection mediated by the mGlu3/TGF-β1 axis has already been described in cellular models of excitotoxic neuronal death (Bruno et al., 1998; D'Onofrio et al., 2001; Corti et al., 2007). However, this mechanism gains more strength if related to Aβ neurotoxicity. TGF-β1 inhibits the activation of the abnormal cell cycle induced by Aβ in neurons (Herrup et al., 2004) by inducing cyclin-dependent kinase inhibitors, such as p21 and p27, and, in addition, protects neurons against Aβ toxicity by activating an antiapoptotic cascade mediated by the phosphatidylinositol-3-kinase pathway (reviewed by Caraci et al., 2009). A reduced expression of type-2 TGF-β1 receptor is specifically found in the AD brain, and reduction correlates with the cognitive decline in AD patients (Tesseur et al., 2006). A polymorphism in the coding region of the TGF-β1 gene has been associated with conversion of mild cognitive impairment to AD (Arosio et al., 2007). Thus, activation of glial mGlu3 receptors affects central mechanisms of Aβ-induced neurodegeneration and represents a novel interesting target for the treatment of AD/PAD.
We strongly recommend the use of dual mGlu2/3 receptor agonists and not mGlu2 receptor PAMs for the treatment of psychiatric symptoms occurring in neurodegenerative disorders. The lack of weight gain with group II mGlu receptor ligands (Patil et al., 2007) is particularly advantageous in AD patients, which have an increased risk of metabolic syndrome and cerebrovascular events in response to conventional antipsychotic medication (Bullock, 2005; Herrmann and Lanctôt, 2005; Recupero and Rainey, 2007; Mazzucco et al., 2008; Trifirò et al., 2009). With mGlu2 receptor PAMs, however, this advantage will be frustrated if these drugs amplify neurodegeneration associated with AD. The mechanism by which activation of mGlu2 receptors enhances Aβ neurotoxicity remains to be established. It has been suggested that mGlu2 receptors inhibit GABA release, thereby facilitating excitotoxic neuronal death in cultured cortical cells (Corti et al., 2007). This mechanism cannot be applied here because we have eliminated the endogenous excitotoxic component from Aβ neurotoxicity with a cocktail of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/N-methyl-d-aspartate receptor antagonists (see above). In cultured neural stem cells, mGlu3 receptors negatively regulate the activity of bone morphogenetic protein receptors, which share the same transduction mechanisms of TGF-β receptors (Ciceroni et al., 2010). It will be interesting to examine whether neuronal mGlu2 receptors functionally interact with receptors that support neuronal survival, such as TGF-β receptors.
Finally, the potential use of mGlu2/3 receptor ligands in AD should be examined in a broader context. Kim et al. (2010) have found that pharmacological activation of group II mGlu receptors with the old-generation agonist, (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV), enhances the production of Aβ(1-42) in isolated intact nerve terminals prepared from AD mutant mice, thereby suggesting the use of mGlu2/3 receptor antagonists for the treatment of AD. These drugs, however, might enhance Aβ-induced neurodegeneration by limiting the protective activity of glial mGlu3 receptors.
Authorship Contributions
Participated in research design: Caraci, Bruno, Drago, Sortino, Copani, and Nicoletti.
Conducted experiments: Caraci, Molinaro, Giuffrida, Riozzi, Traficante, Cannella, and Merlo.
Contributed new reagents or analytic tools: Wang, Heinz, Nisenbaum, and Britton.
Performed data analysis: Battaglia and Copani.
Wrote or contributed to the writing of the manuscript: Nicoletti.
Footnotes
This work was supported by the Italian Ministero dell'Istruzione, dell'Università e della Ricerca [Grant 200728AA57] (to F.N).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.067488.
-
ABBREVIATIONS:
- AD
- Alzheimer's disease
- PAD
- AD with severe psychosis
- mGlu
- metabotropic glutamate
- PAM
- positive allosteric modulator
- NAM
- negative allosteric modulator
- LY379268
- (−)-2-oxa-4-aminobicyclo[3.1.0]exhane-4,6-dicarboxylic acid
- LY341495
- (2S,1′S,2′S)-2-(9-xanthylmethyl)-2-(2′-carboxycyclopropyl)glycine
- LY566332
- N-4′-cyano-biphenyl-3-yl)-N-(3-pyridinylmethyl)-ethanesulfonamide hydrochloride
- LY2389575
- (3S)-1-(5-bromopyrimidin-2-yl)-N-(2,4-dichlorobenzyl)pyrrolidin-3-amine methanesulfonate hydrate
- LY404039
- (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid
- MTT
- 3-[4,5-dimethylthioazol-2-yl]-2,5-diphenyl tetrazolium bromide
- MK-801
- (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (dizocilpine maleate)
- DNQX
- 6,7-dinitroquinoxaline-2,3-dione
- ANOVA
- analysis of variance
- PLSD
- protected least significant difference
- DMEM
- Dulbecco's modified Eagle medium
- TGF-β
- transforming growth factor-β
- aTGF-β
- anti-TGF-β1 neutralizing antibodies
- Aβ
- β-amyloid
- GCM
- glial conditioned medium
- FLIPR
- fluorometric imaging plate reader
- PCR
- polymerase chain reaction
- SB431542
- 4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]benzamide
- DCG-IV
- (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine.
- Received July 13, 2010.
- Accepted November 29, 2010.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}