


Abstract
Seven transmembrane receptors (7TMRs), commonly referred to as G protein-coupled receptors, form a large part of the “druggable” genome. 7TMRs can signal through parallel pathways simultaneously, such as through heterotrimeric G proteins from different families, or, as more recently appreciated, through the multifunctional adapters, β-arrestins. Biased agonists, which signal with different efficacies to a receptor's multiple downstream pathways, are useful tools for deconvoluting this signaling complexity. These compounds may also be of therapeutic use because they have distinct functional and therapeutic profiles from “balanced agonists.” Although some methods have been proposed to identify biased ligands, no comparison of these methods applied to the same set of data has been performed. Therefore, at this time, there are no generally accepted methods to quantify the relative bias of different ligands, making studies of biased signaling difficult. Here, we use complementary computational approaches for the quantification of ligand bias and demonstrate their application to two well known drug targets, the β2 adrenergic and angiotensin II type 1A receptors. The strategy outlined here allows a quantification of ligand bias and the identification of weakly biased compounds. This general method should aid in deciphering complex signaling pathways and may be useful for the development of novel biased therapeutic ligands as drugs.
Introduction
For more than two decades, it has been appreciated that a 7TMR can signal through parallel pathways simultaneously, such as through heterotrimeric G proteins from different families (Abramson et al., 1988; Fargin et al., 1989). It was soon discovered that ligands can have different efficacies for these different signaling pathways (Kenakin, 1995), a characteristic referred to as biased agonism or functional selectivity (Roth, 2009). Compared with “balanced agonists” that signal with equal efficacy to available downstream pathways, biased agonists have different efficacies for signaling to different G proteins (Kenakin, 1995) or to G proteins and the multifunctional adapter proteins β-arrestins (Wei et al., 2003; Gesty-Palmer et al., 2006). Unlike heterotrimeric G proteins, which traditionally act through the activation of second messengers such as cAMP, diacylglycerol, or calcium, β-arrestins act as scaffolds for a number of signaling proteins, such as mitogen-activated protein kinases and E3 ubiquitin ligases (DeWire et al., 2007). Biased agonists are currently being developed as tools to dissect the signaling complexity downstream of 7TMRs and as novel therapeutics, because they seem to have different functional and physiological consequences from conventional balanced agonists (Rajagopal et al., 2010). For example, a β-arrestin-biased ligand of the parathyroid hormone receptor results in increased bone density without activating treatment-limiting catabolic pathways (Gesty-Palmer et al., 2009), and the novel AT1R agonist Sar-Arg-Val-Tyr-Ile-His-Pro-d-Ala-OH (TRV120027) selectively signals via β-arrestins, leading to increased cardiac performance with a reduction in blood pressure (Violin et al., 2010).
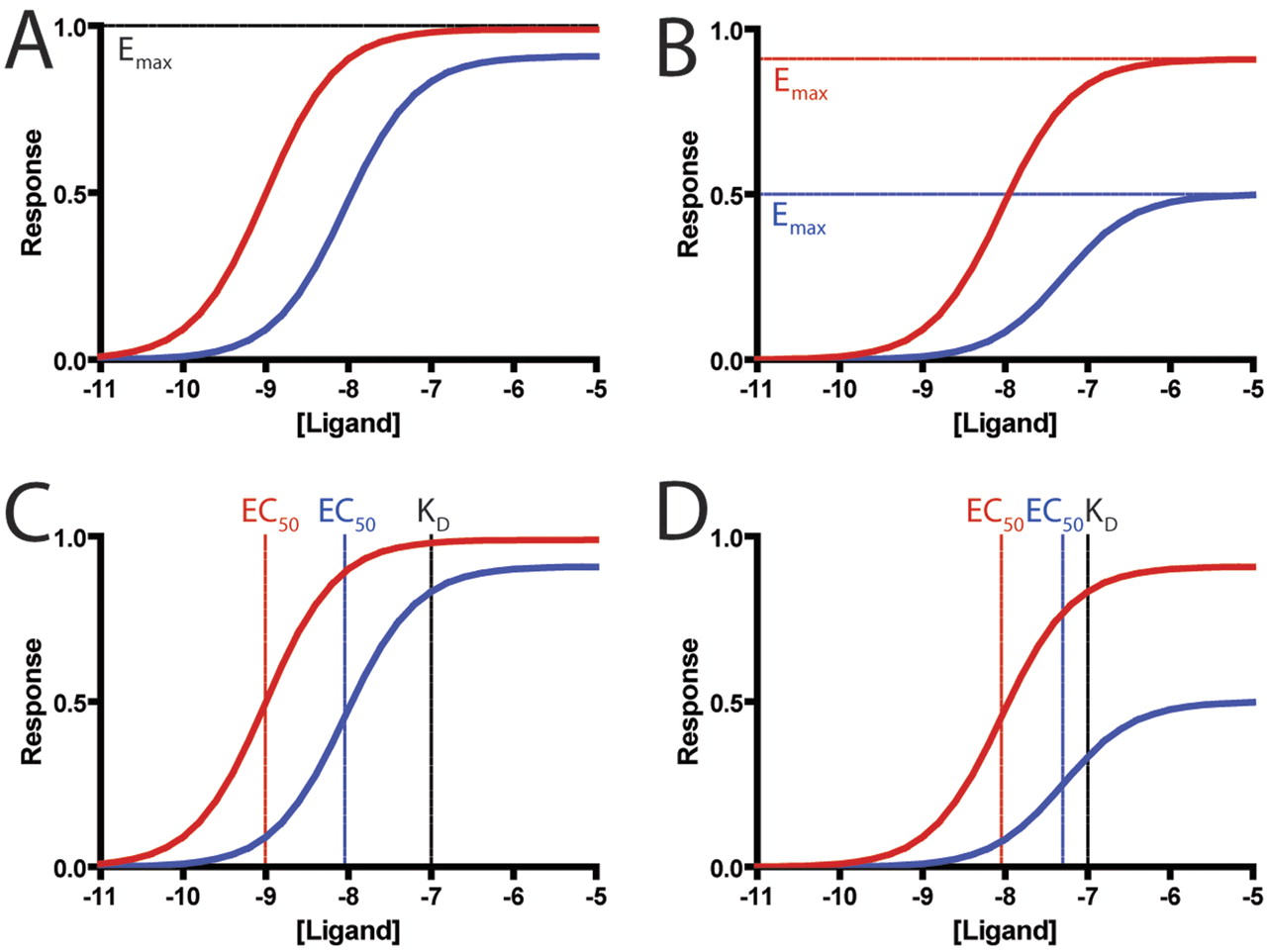
At this time, there are no widely accepted methods for quantifying ligand bias, and most groups have relied on comparing the maximal effects (Emax) and potencies (EC50) of ligands for different signaling pathways (Galandrin and Bouvier, 2006). However, these parameters cannot account for differences in receptor reserve and amplification of different assays (Rajagopal et al., 2010). In assays with significant amplification, such as second-messenger assays (e.g., cAMP formation), both full and partial agonists can reach the same maximal response (Fig. 1A), whereas in assays with little amplification, such as assays that monitor recruitment of β-arrestin to a receptor by enzyme complementation (Eglen et al., 2007), partial agonists have significantly lower maximal responses than full agonists (Rajagopal et al., 2010) (Fig. 1B). Therefore, a partial agonist that reaches maximal effect in one assay and half-maximal effect in another assay would be incorrectly identified as being biased compared with a full agonist, which reaches maximal response in both assays. A comparison of potencies is likewise limited by differences in receptor reserve between assays; as shown, the difference in potencies between the full agonist and partial agonist may be smaller in assays with less receptor reserve (Fig. 1, C and D) (Rajagopal et al., 2010). Recent studies that have attempted to identify biased agonists using such comparisons (Galandrin and Bouvier, 2006; Molinari et al., 2010) may be confounded by these problems, although a reversal in rank order of efficacies or potencies would be evidence for ligand bias (Berg et al., 1998; Kenakin, 2007). More recently, a few approaches have been proposed for overcoming these problems (Figueroa et al., 2009; Gregory et al., 2010; Kenakin and Miller, 2010; Koole et al., 2010), but they have not been tested rigorously against one another and may have limitations (see below). With the rising interest in the development of biased agonists, a robust method for identifying weakly biased ligands and for quantifying ligand bias in 7TMR drug development is sorely needed.

Limitations of classic pharmacological parameters in quantifying bias. A, in an assay with significant receptor reserve, such as that for second messengers, both full (red, τ = 100) and partial (blue, τ = 10) agonists reach close to a maximal response. B, in assays with little to no receptor reserve, such as those based on translocation or recruitment, full agonists (red, τ = 10) reach higher maximal responses than partial agonists (blue, τ = 1). Potencies are similarly affected by receptor reserve, because in assays with high levels of receptor reserve (C), a full agonist (τ = 100) would have a greater leftward shift compared with a partial agonist (τ = 10) from its dissociation constant. However, in assays with lower levels of receptor reserve (D), these shifts are do not correlate in a linear fashion (full agonist, τ = 10; partial agonist, τ = 1). Simulated data were generated using the operational model (see text) with the dissociation constant set to 100 nM.
Here, we modify these approaches to develop a general method for identifying biased ligands and validate it at two well characterized 7TMR drug targets, the β2AR and AT1AR. This method uses complementary approaches that are based on comparisons of the following: 1) responses at the same ligand concentrations (equimolar) (Gregory et al., 2010); 2) ligand concentrations that result in equiactive responses (Figueroa et al., 2009); and 3) estimates of coupling efficiency derived from the operational model (Black and Leff, 1983; Evans et al., 2011; Kenakin and Miller, 2010) using experimentally determined dissociation constants. The first two approaches can allow the identification of weakly biased ligands with concentration-response data alone but are not as robust as the operational model, which, although requiring an experimentally determined dissociation constant, allows an estimate of ligand efficacy and a calculation of ligand bias. Thus, these complementary approaches can serve in a general strategy for the development of biased ligands.
Materials and Methods
Materials.
The β2AR ligands isoproterenol, epinephrine, dobutamine, dichloroisoproterenol, fenoterol, salbutamol, norepinephrine, formoterol, clenbuterol, salmeterol, and pindolol were all obtained from Sigma-Aldrich (St. Louis, MO). The AT1AR ligands angiotensin II, Sar1Gly4Gly8 (SGG), S1C4, and A1 were custom synthesized by Genscript (Piscataway, NJ). The ligands Sar-Arg-Val-Tyr-Tyr-His-Pro-NH2 (TRV120026), Sar-Arg-Val-Tyr-Val-His-NH2 (TRV120055), Asp-Arg-Val-Tyr-Ile-His-Pro-Gly (TRV120056), NMAla-Arg-Val-Tyr-Ile-His-Pro-d-Ala (TRV120044), Sar-Arg-Val-Tyr-Arg-His-Pro-NH2 (TRV120045), and NMAla-Arg-Val-Tyr-Ile-His-Pro-Ala (TRV120034) were custom-synthesized (Sar denotes sarcosine, NMAla denotes N-methyl-l-alanine, and NH2 denotes an amino group at the C terminus) by Trevena, Inc. (King of Prussia, PA). Bright-Glo and Glosensor reagents were obtained from Promega (Madison, WI). Reagents for the IP-One HTRF assay were obtained from Cisbio Bioassays (Bedford, MA). Reagents for the DiscoveRx PathHunter β-arrestin assay were obtained from DiscoveRx (Fremont, CA). The Tango construct for the β2AR was provided by Gilad Barnea and Richard Axel.
β-Arrestin Recruitment Assays.
For the β2AR, β-arrestin recruitment to receptor was assessed by the Tango assay, as described previously by Barnea et al. (2008). In this assay, the C terminus of the human β2AR is replaced with the C-terminal tail of the V2 vasopressin receptor tail (to increase signal-to-noise ratio) followed by a Tobacco Etch Virus (TEV) protease cleavage site and a tTA transcription factor. This construct was stably transfected in HEK293 cells along with a construct encoding β-arrestin 2 fused to TEV protease. Upon ligand stimulation, the recruitment of β-arrestin to the receptor results in the cleavage tTA from the receptor. The tTA translocates to the nucleus, in which it transcribes a stably expressing luciferase reporter gene. HEK293 cells stably transfected with these constructs were seeded at 2.5 × 104 cells per well in a 96-well plate. The next day, compounds diluted in phosphate-buffered saline were added to the wells to their final concentration followed by incubation at 37°C for 14 to 20 h. The next day, the plate was cooled to room temperature, and an equal amount of Bright-Glo luciferase assay reagent (Promega) was added to each well. After 5 min, luminescence was read in a NOVOstar microplate reader (BMG Labtech, Durham, NC). To ensure that the results obtained using this technology were not an artifact of the overnight incubation with ligand or the V2R tail, we also used the PathHunter β-arrestin assay from DiscoveRx (see below), which uses the human β2AR (with a Prolink peptide added to the C terminus) with a shorter incubation time with ligand (∼30 min), the representative data of which are shown in Supplemental Fig. S5.
For the AT1AR, we used the PathHunter β-arrestin assay from DiscoveRx and read for chemiluminescent signaling on a PheraStar reader (BMG Labtech) as described previously (Violin et al., 2010). In brief, complementary halves of β-galactosidase were genetically fused to the carboxyl termini of the human AT1R and β-arrestin2. When cotransfected, the two fusion proteins serve as a proximity sensor; when β-arrestin 2 translocates to active receptor, the β-galactosidase fragments interact to form a functional enzyme, which is detected by a chemoluminescent substrate.
cAMP Assay.
The GloSensor cAMP biosensor (Promega) uses a modified form of firefly luciferase containing a cAMP-binding motif (Fan et al., 2008). Upon cAMP binding, a conformational change leads to enzyme complementation and incubation with a luciferase substrate results in a luminescence readout. Analysis of cAMP accumulation was performed in HEK293 cells stably transfected with the Glosensor construct and the human β2AR. Cells were seeded in 96-well white, clear-bottomed plates at 8 × 104 cells/well, in minimal essential medium supplemented with 10% fetal bovine serum [10% (v/v)]. The next day, the GloSensor reagent [Promega; 4% (v/v)] was incubated at room temperature for 2 h. Cells were then stimulated with a range of β2 AR agonists for 5 min, and increases in luminescence were read on a NOVOstar microplate reader (BMG Labtech). These assays were repeated in the Tango cell lines used for the β-arrestin recruitment assays with transient transfection of the Glosensor construct, which demonstrated the same behavior, albeit with poorer signal-to-noise ratio (Supplemental Fig. S6).
Inositol 1-Phosphate Assay.
Inositol 1-phosphate (IP1), a downstream metabolite of inositol trisphosphate, which itself is downstream of signaling by Gq, was detected by the IP-One Tb HTRF kit (Cisbio Bioassays) as described previously (Violin et al., 2010). Plates were read on a PheraStar reader using a time-resolved fluorescence ratio (665/620 nm).
Angiotensin II Type IA Receptor Competition Membrane Radioligand Binding Assays.
HEK293 cells with stable expression of the rat (r) AT1 receptor were harvested by centrifugation at 400g for 30 min at 4°C, washed once with a balanced salt solution, repelleted, and the pellet was flash-frozen in liquid nitrogen. The cell pellets were stored at −80°C until processed for membranes. Pellets were resuspended in buffer (50 mM HEPES, 2 mM EDTA, pH 7.4) containing fresh protease inhibitors, Complete Brand protease tablets from Roche Diagnostics (Indianapolis, IN), and subjected to nitrogen cavitation with a Parr Cell Disruption Bomb (Parr Instrument Co., Moline, IL) at 1000 psi for 20 min on ice. Ruptured cells were sedimented at 500g for 10 min at 4°C, and the supernatant containing cellular membranes was washed twice at 48,000g for 15 min. cell pellets were resuspended at 4°C in 10 volumes of ice-cold buffer A and cavitation, placed on ice. To remove large particles, a low-speed centrifugation (500g for 30 min at 4°C) was performed, followed by high-speed centrifugation (48,000g for 45 min at 4°C), resuspension in buffer plus protease inhibitor cocktail, and a final high-speed centrifugation at (48,000g for 45 min at 4°C). A Dounce homogenizer was used to resuspend the final pellet using ice-cold buffer. The membrane suspension was passed through a 23-gauge needle, and aliquots were made and stored at −80°C. Total protein concentration of the membrane preparation was determined with a Coomassie Plus Reagent Kit from Pierce Biotechnology (Rockford, IL) using bovine serum albumin as the standard.
Membranes were diluted in assay buffer [50 mM HEPES, 150 mM NaCl, 5 mM MgCl2, Gpp(NH)p 10 μM, pH 7.2, at 23°C] to a concentration of 1 to 3 μg of protein/well. Assays were initiated by the addition of 94 μl of membrane suspension to 200 μl of [125I]Sar1Ile8-angiotensin II (specific activity, 2200 Ci/mmol; PerkinElmer Life and Analytical Sciences, Waltham, MA), at 0.4 to 1 times Kd and various concentrations of inhibitors in buffer plus a cocktail of protease inhibitors and 0.02% bovine serum albumin to reduce nonspecific radioligand binding. Compounds were diluted in dimethyl sulfoxide and tested at a final concentration of 1% dimethyl sulfoxide (determined to be nondetrimental to the assay). Competition binding with compounds (11-point concentrations) was performed in polypropylene 96-well plates (Corning Life Sciences, Lowell, MA). Nonspecific binding was defined in the presence of 10 μM losartan. Competition assays were performed at 23°C for 4 h to allow adequate time for compounds and radioligand to reach equilibrium for binding. The separation of bound from free radioligand was accomplished by rapid vacuum filtration of the incubation mixture over GF/B unifilter (polyethylenimine-treated) plates (PerkinElmer Life and Analytical Sciences) using a Brandel cell harvester (Brandel Inc., Gaithersburg, MD). Filters were washed two times with 0.3 ml of ice-cold phosphate-buffered saline, pH 7.0, containing 0.01% Triton X-100. Radioactivity on the filters was quantified using a MicroBeta TriLux Liquid Scintillation Counter (PerkinElmer Life and Analytical Sciences).
Data Analysis.
For radioligand binding, calculation of apparent binding affinities, Ki = IC50/(1+ [radioligand]/Kd) was performed using the nonlinear iterative curve-fitting computer program Prism (GraphPad Software Inc., San Diego, CA). All fitting using the equiactive approach and operational model was performed using Prism. For the β2AR, reported dissociation constants for ligands from Del Carmine et al. (2002) were used.
Equiactive Comparison.
The equiactive comparison is analogous to the method used by Furchgott (1966) to determine the dissociation constant of agonists. In this approach, the concentrations of ligand required for an equiactive response for pathway 1 ([A1]) and 2 ([A2]) are extrapolated from fits of each concentration-response curve (Fig. 2B). A linear relationship between the inverse of these concentrations is then given by the equation (see Supplemental Methods)
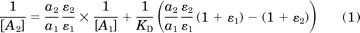 A bias factor, which quantifies the relative stabilization of one signaling state over another compared with the reference agonist, can then be calculated as
A bias factor, which quantifies the relative stabilization of one signaling state over another compared with the reference agonist, can then be calculated as
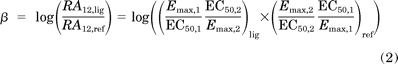
Operational Model.
We chose to use the operational model of Black and Leff (1983) to quantify the effective signaling by receptors. In the operational model, the response of the system to ligand stimulation is based on receptor occupancy alone, because the ligand/receptor complex is coupled to downstream signaling pathways without any allosteric component. The response of the system is then related to ligand concentration (when the Hill coefficient is 1):
 where Em is the maximal response of the system to a full agonist, KD is the agonist dissociation constant, and τ is “coupling efficiency” between the agonist/receptor complex and its downstream signaling partners. This coupling efficiency τ can be considered to be composed of two components (τ = τ*ε), where the τ* term accounts for the amplification inherent to the downstream signaling pathway that is the same for all ligands in the same assay, and the other component (ε) accounts for a ligand's efficacy at generating a signaling-competent agonist/receptor conformation. The ability of an agonist to signal to downstream pathways can then be compared with a reference agonist by the effective signaling (σlig).
where Em is the maximal response of the system to a full agonist, KD is the agonist dissociation constant, and τ is “coupling efficiency” between the agonist/receptor complex and its downstream signaling partners. This coupling efficiency τ can be considered to be composed of two components (τ = τ*ε), where the τ* term accounts for the amplification inherent to the downstream signaling pathway that is the same for all ligands in the same assay, and the other component (ε) accounts for a ligand's efficacy at generating a signaling-competent agonist/receptor conformation. The ability of an agonist to signal to downstream pathways can then be compared with a reference agonist by the effective signaling (σlig).
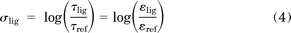 A bias factor, β, equal to the distance from the point (σpath1, σpath2) for a ligand to the line of unity for balanced ligands, can then be calculated as the difference between the effective signaling factors (Fig. 2C) in relation to balanced agonists:
A bias factor, β, equal to the distance from the point (σpath1, σpath2) for a ligand to the line of unity for balanced ligands, can then be calculated as the difference between the effective signaling factors (Fig. 2C) in relation to balanced agonists:
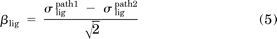
Results
Development of Approaches for the Quantification of Ligand Bias.
We used three general approaches to identify biased ligands (Fig. 2). The first of these is a qualitative approach to identify biased ligands originally proposed by Christopoulos and coworkers (Gregory et al., 2010), which we refer to as an “equimolar comparison.” As shown in Fig. 2A, data for a single ligand are collected in two different assays, such as those for G protein and β-arrestin signaling. The responses of these two different assays at the same concentration of ligand are then plotted against each other; therefore, the shape of this curve is a direct comparison of the signaling through the two different pathways. The shape of this curve may vary depending on the assays compared; for example, two assays based on biosensors for the same second messenger may have different sensitivities to the messenger, one with nanomolar and the other with picomolar sensitivity. If their concentration-response data were plotted against each other, a hyperbolic curve would be obtained, suggesting bias toward the assay with picomolar sensitivity, although no true underlying bias would actually be present. Therefore, to identify biased ligands using this method, the shape of the equimolar curve for a test ligand must be qualitatively compared with that of a reference balanced agonist (Fig. 2A, right). In the example shown, the test agonist is biased toward response 1 compared with the reference agonist (dashed line), which by definition is balanced.

Approaches to quantifying ligand bias. A, in the equimolar comparison, data for a single ligand are collected in two different assays (left). The responses of these two different assays at the same concentration of ligand (middle) are then plotted against each other (right). B, in the equiactive comparison, concentration-response data are fit to a logistic equation, yielding EC50 and Emax (middle). This allows the calculation of a bias factor (right). C, in the operational model, the data are fit to the equation proposed by Black and Leff (1983) (left). From the coupling coefficient, τ, the effective signaling of each ligand in each assay can be calculated (middle), which then allows a calculation of a bias factor (right).
The second approach is a quantitative “equiactive comparison” between two different assays for a ligand (Fig. 2B). This is analogous to pharmacological methods for the estimation of agonist affinity (Furchgott, 1966), but by comparing the various assays downstream of the receptor, a quantification of bias can be obtained. In most cases, this comparison can be performed using a simplified formula with intrinsic relative activities proposed by Ehlert (2008), which can be calculated from maximal effects and potencies (Figueroa et al., 2009) (Fig. 1B, middle) (see Supplemental Materials). A “bias factor” (denoted β) is calculated as the logarithm of the ratio of intrinsic relative activities for a ligand at two different assays compared with a reference agonist (Fig. 2B, right; see Materials and Methods). This bias factor is an estimate for the molecular efficacy of pathway 1 versus pathway 2 on a logarithmic scale (e.g., a bias factor of 1 between two pathways means that a ligand is 10 times better at generating the active receptor conformation for one pathway over the other pathway compared with the reference balanced agonist):
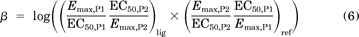 where P1 and P2 denote signaling through pathways 1 and 2, respectively.
where P1 and P2 denote signaling through pathways 1 and 2, respectively.
The third approach is based on classic pharmacological models that were originally developed to account for receptor reserve and shifts in agonist concentration-response curves, the first of which was proposed by Stephenson (1956). We chose to use the operational model of Black and Leff (1983), which allows the calculation of a coupling efficiency to each downstream signaling pathway, and has been proposed as a method to quantify bias (Evans et al., 2011; Kenakin and Miller, 2010). To calculate this coupling efficiency, referred to as τ, concentration-response data are fitted by eq. 7 (see Materials and Methods) using the dissociation constant of the ligand for the receptor from a separate binding experiment (McPherson et al., 2010) (Fig. 2C, left). By comparing these coupling efficiencies to that of a reference compound, the effective signaling (σ) by a ligand in each assay can be calculated (eq. 8). A comparison of effective signaling between different pathways can be performed by the calculation of bias factors (β) (eq. 9), equal to the distance from the point (σpath1, σpath2) to the line of unity for each ligand (resulting in division by the square root of 2), thereby allowing the identification of biased ligands (Fig. 2C, right).


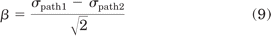 If there are errors in the dissociation constants used in the operational model, such as those associated with different conditions used for ligand binding and functional assays, they would be expected to largely cancel out in a calculation of bias factors as the higher or lower effective signaling associated with those errors should affect estimations of both pathways similarly. If the dissociation constants are left-shifted relative to the EC50 values, an observation that cannot be accounted by any pharmacological model, it should be obvious from the poor fits to the data (which were not observed in this study).
If there are errors in the dissociation constants used in the operational model, such as those associated with different conditions used for ligand binding and functional assays, they would be expected to largely cancel out in a calculation of bias factors as the higher or lower effective signaling associated with those errors should affect estimations of both pathways similarly. If the dissociation constants are left-shifted relative to the EC50 values, an observation that cannot be accounted by any pharmacological model, it should be obvious from the poor fits to the data (which were not observed in this study).
Identification of Biased Ligands at the β2AR.
The β2AR is a prototype for 7TMRs and is a drug target in the treatment of heart failure and asthma. At this receptor, the identification of a partially β-arrestin-biased agonist may allow for the development of more strongly biased agonists with possible therapeutic utility. Upon stimulation by its endogenous agonists epinephrine and norepinephrine, the β2AR signals to G proteins, which increase cAMP formation by adenylate cyclase, and β-arrestins, which signal to a wide range of intracellular targets (DeWire et al., 2007). No strongly biased ligands have been identified at this receptor, although the “β blocker” carvedilol does lead to weak β-arrestin recruitment and signaling in the absence of G protein activation (Wisler et al., 2007). Other studies at this receptor have also identified potentially β-arrestin-biased agonists using direct comparisons of pharmacological or biophysical parameters (Galandrin and Bouvier, 2006; Drake et al., 2008; Reiner et al., 2010).
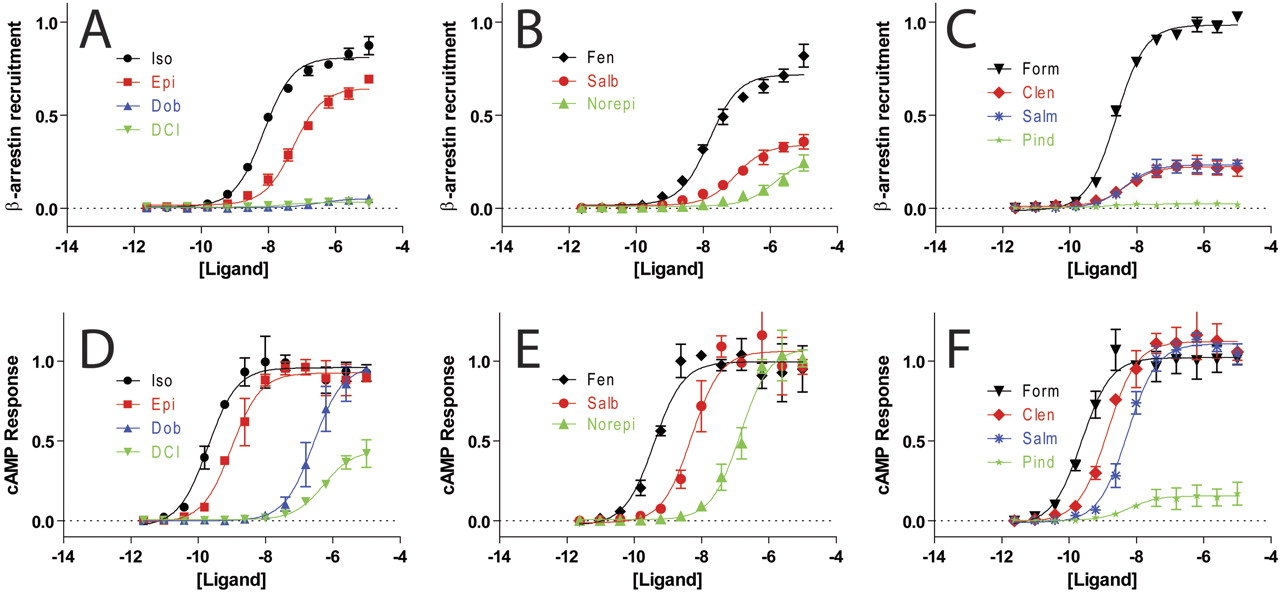
We collected concentration-response data for β-arrestin recruitment, using an assay based on release of a transcription factor upon β-arrestin recruitment to a modified receptor (Barnea et al., 2008), and cAMP generation, using a luminescence-based cAMP biosensor (Fan et al., 2008), for a panel of clinically used β2AR ligands (Fig. 3, A–F). There is a suggestion of bias in these data because formoterol is more potent than isoproterenol in the β-arrestin recruitment assay, whereas the two drugs are equipotent in the cAMP assay (Supplemental Fig. S1). However, an equimolar comparison does not demonstrate any significantly biased compounds (Figs. 4A; Supplemental Fig. S2) because of the large difference in amplification between the cAMP and β-arrestin recruitment assays that results in strongly hyperbolic equimolar comparison curves for all compounds. The concentration-response data were fit well by logistic equations (Supplemental Table S1), and bias factors were calculated using an equiactive comparison (Fig. 4B; Table 1). Although the equimolar comparison was unable to identify any biased ligands, the equiactive analysis identifies a number of potentially β-arrestin biased compounds: pindolol (Pin), dichloroisoproterenol (DCI), salmeterol (Slm), and formoterol (For) (p < 0.05 by unpaired t test). However, for the weak partial agonists pindolol and DCI, the change in bias factor is driven by differences in the EC50 between the cAMP and β-arrestin assays derived from poor fits (Fig. S3). The fits for formoterol and salmeterol do not suffer from this problem, and their calculated bias factors probably represent a true difference in efficacies in the G protein- and β-arrestin-mediated pathways.

Concentration-response for β-arrestin recruitment and cAMP generation at the β2AR. Normalized β-arrestin recruitment (A–C) and cAMP generation (D–F) for isoproterenol (Iso), epinephrine (Epi), dobutamine (Dob), and DCI (A and D); B and E, fenoterol (Fen), salbutamol (Salb), and norepinephrine (Norepi); C and F, formoterol (Form), clenbuterol (Clen), salmeterol (Salm), and pindolol (Pind). (β-arrestin and cAMP signals normalized to formoterol, n = 3, error bars denote S.E.M.).

Identification of weakly β-arrestin-biased ligands at the β2AR. A, an equimolar comparison between the G protein and β-arrestin-mediated assays does not demonstrate any significant bias. Fenoterol (black), salbutamol (red), norepinephrine (blue), and formoterol (green). B, bias factors from an equiactive comparison demonstrate bias for DCI, pindolol (Pin), Slm, and For (p < 0.05 by t test). C, comparison of effective signaling (σ) in β-arrestin recruitment and cAMP generation for a panel of ligands compared with the reference agonist epinephrine. The red line is the theoretical line of balanced signaling. D, bias factors calculated from the operational model. Only For and Slm are significantly biased (p < 0.05 by t test).
Bias factors for ligands at the β2AR
The column “Bias” denotes whether a statistically significant difference in bias compared to the reference balanced agonist epinephrine is present (either toward G protein or β-arrestin), whereas “Non” denotes an insignificant difference from the balanced agonist.
The operational model was then used to fit these data and calculate relative signaling efficacies compared with epinephrine, which was chosen as the reference compound because it is an endogenous agonist that activates the receptor physiologically. A comparison of the effective signaling in each pathway (σpathway) of the panel of ligands to epinephrine is shown in Fig. 4C and Table 1. Balanced compounds, with similar bias to epinephrine, would be expected to lie on a line of unity in this analysis (red line, Fig. 4C). Epinephrine, considered a full agonist in most studies, is actually less effective in stabilization of the G protein- and β-arrestin signaling states than the synthetic agents fenoterol and isoproterenol, neither of which seem biased. Bias factors analogous to those calculated from the equiactive comparison were then calculated (Fig. 4D; Table 1). Here, formoterol and salmeterol are again identified as having bias toward β-arrestin recruitment (p < 0.05 by unpaired t test). Pindolol and DCI, identified as biased compounds in the equiactive comparison, are not significantly biased in this analysis.
It is noteworthy that the three different approaches for quantifying bias yielded different results. A major limitation in the equimolar comparison is its inability to identify weakly biased agonists when assays have significantly different levels of amplification. The equiactive comparison performed poorly with data from suboptimal fits of weak partial agonists (Pin and DCI), which display little signaling activity. This problem is less of an issue with the operational model, in which the additional information provided by the dissociation constant improves the quality of these fits and yields a better estimation of the bias factors. Therefore, we conclude that formoterol and salmeterol, two long-acting β agonists in our panel of ligands, are β-arrestin-biased agonists of the β2AR. These compounds were not identified as biased in a previous analysis of β2AR ligands (Drake et al., 2008), a finding that is probably due to differences in the assays used for assessment of G protein and β-arrestin signaling and the method for quantifying bias. In that earlier study, both signaling parameters had significant kinetic components, with β-arrestin signaling quantified by the rate of β-arrestin recruitment to the receptor as measured by fluorescence resonance energy transfer and G protein signaling quantified by the integrated signal of a cAMP-binding fluorescent biosensor over time. In this study, the assays used have significantly higher levels of amplification and are measured at a single late time point. Also of note, norepinephrine, which was identified as a biased agonist in a recent publication based on biophysical and signaling experiments performed at saturating doses of ligands (Reiner et al., 2010), does not display any significant signaling bias compared with epinephrine. Carvedilol, a weakly β-arrestin-biased agonist, was not tested in these assays because it is an inverse agonist of G protein signaling (Wisler et al., 2007) and, therefore, by definition is biased.
Some rather counterintuitive findings arise from this type of analysis compared with one based on the classic pharmacologic parameters of maximal responses and potencies. In a comparison of maximal responses, it would seem that dobutamine would be a strongly cAMP-biased agonist, reaching a maximal response in the cAMP assay (Emax ∼96%) but only very limited activity (Emax < 5%) in the β-arrestin recruitment assay. This finding, however, is wholly due to the weak partial agonism of dobutamine, which can still lead to a maximal response in the assay with significant receptor reserve and amplification (cAMP formation) but results in a very weak response in an assay with little receptor reserve (β-arrestin recruitment). Thus, within the errors of this experiment, the response of dobutamine is no different from a low dose (∼10 nM) of the reference agonist epinephrine. However, no concentration of epinephrine could result in the pattern of cAMP formation and β-arrestin recruitment of a truly biased agonist, such as formoterol.
Identification of Biased Ligands at the AT1AR.
The AT1AR is notable among 7TMRs in that a number of well characterized β-arrestin-biased agonists have been described at this receptor. These include SGG and Sar1Ile4Ile8 angiotensin II (SII) (Holloway et al., 2002). SII recruits β-arrestin and leads to β-arrestin-mediated extracellular signal-regulated kinase phosphorylation in the absence of significant G protein activation. SII is also capable of enhancing the contraction of isolated cardiac myocytes (Rajagopal et al., 2006), as does a more potent β-arrestin-biased agonist, TRV120027, which has been found to reduce blood pressure and increase cardiac performance in rats (Violin et al., 2010). We chose 10 derivatives of angiotensin II (AngII) to test whether those compounds had more bias than the initially described compound SGG. We used an assay for β-arrestin recruitment based on enzyme complementation (Fig. 5, A–C) and an assay for Gq signaling based on IP1 formation (Fig. 5, D–F). It is noteworthy that from a comparison of representative concentration-response curves, a number of compounds seem to be biased, with partial activity with respect to β-arrestin recruitment and little IP1 formation.
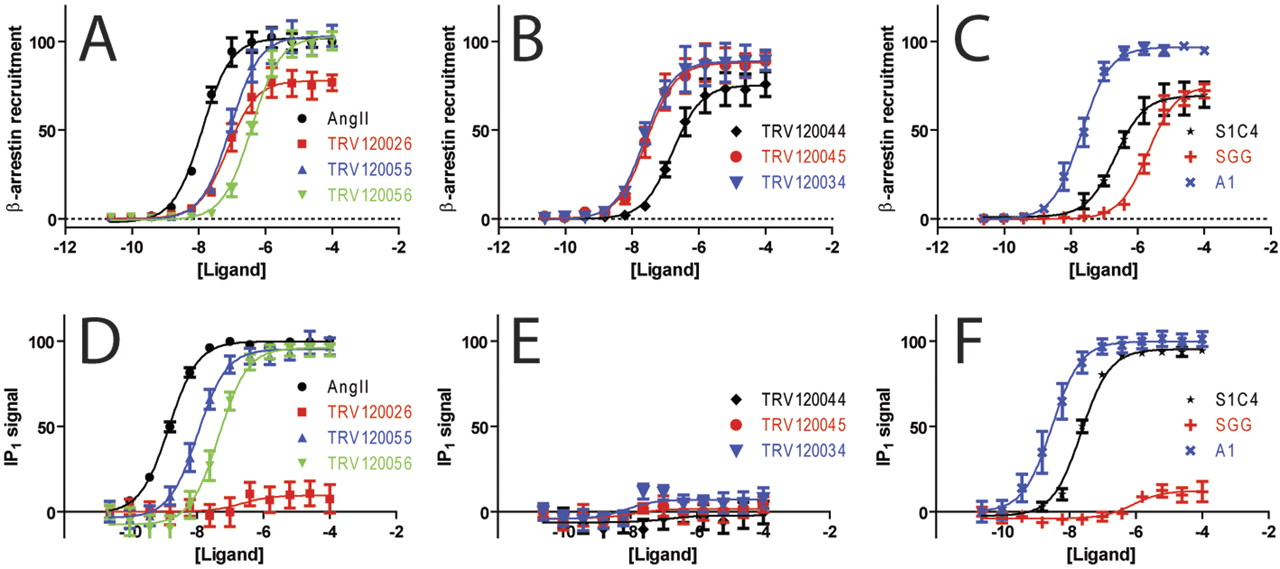
Concentration-response for β-arrestin recruitment and IP1 formation at the AT1AR. Normalized β-arrestin recruitment (A–C) and IP1 generation (D–F) for Angiotensin II (AngII), TRV120026 (red), TRV120055 (blue) and TRV120056 (green) (A and D); TRV120044 (black), TRV120045 (red), and TRV120034 (blue) (B and E); S1C4 (black), S1G4G8 (red), and A1 (blue) (C and F). (β-arrestin and IP1 signals normalized to AngII, n = 3, error bars denote S.E.M.).
The equimolar comparison clearly identifies such compounds (TRV120026, TRV120034, TRV120045, TRV120044, and SGG) as β-arrestin-biased ligands, whereas the other compounds seem to be balanced (TRV120055, TRV120056, A1, and S1C4) (Fig. 6A; Supplemental Fig. S4). For example, the SGG and TRV120044 compounds are shifted to the left portion of the plot, whereas the balanced agonists AngII and TRV120055 both have similar hyperbolic shapes consistent with increased amplification in the IP1 assay compared with the β-arrestin recruitment assay (Fig. 6A). The plots for these two β-arrestin-biased compounds suggest that TRV120044 (red) has more β-arrestin bias than SGG (green), although it is difficult to ascertain in such a qualitative analysis. Bias factors for all of the compounds using the equiactive approach were then calculated (Fig. 6B). Consistent with the equimolar comparison, the TRV120026, TRV120034, TRV120044, TRV120045, and SGG compounds all had bias factors consistent with β-arrestin bias, although the large errors for a number of these compounds led to the differences being statistically insignificant. This was due to the poor fits of the IP1 concentration-response data, in which many of the compounds displayed little to no signaling activity.
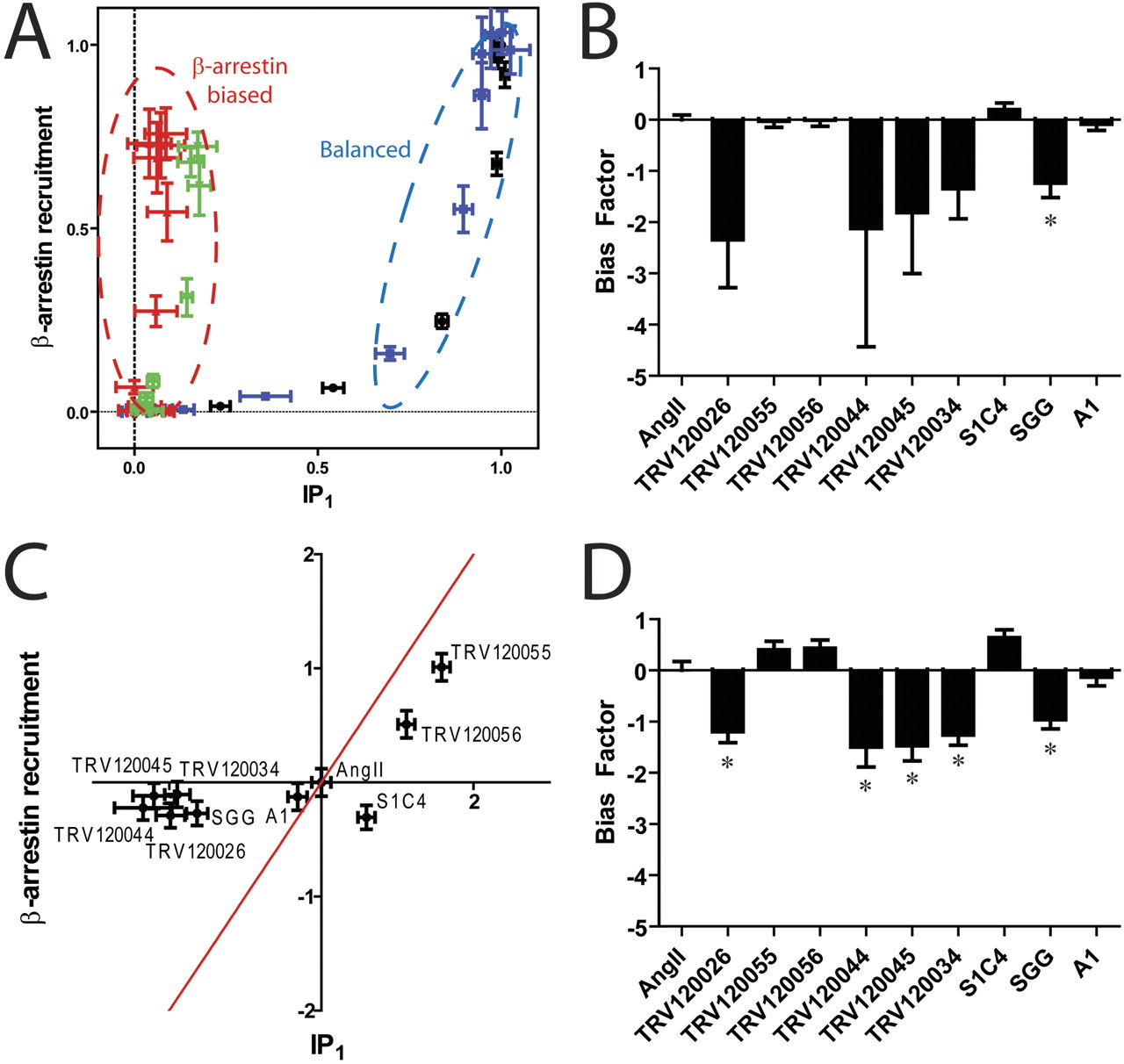
A group of angiotensin II analogs at the AT1AR are significantly β-arrestin-biased. A, the equimolar comparison identifies the ligands SGG (green) and TRV120044 (red) as β-arrestin-biased compared with the reference agonist angiotensin II (black) or TRV120055 (blue). A complete equimolar analysis for all compounds is shown in Supplemental Fig. S4. B, bias factors calculated using the equiactive model for the set of AT1AR ligands. Because of large errors, only SGG is identified as a biased ligand. C, effective signaling via G proteins and β-arrestins compared with the endogenous agonist AngII. TRV120026, TRV120034, TRV120044, TRV120045, and SGG are significantly β-arrestin-biased, whereas all the other compounds appear balanced (red line). D, bias factors calculated using the operational model. TRV120044 and TRV120045 have nearly an order of magnitude more bias compared with one of the initially described ligands, SGG (*, p < 0.05 by t test).
We then compared effective signaling for the G protein and β-arrestin-mediated pathways as calculated by the operational model (Fig. 6C) using experimentally determined dissociation constants from radioligand competition binding (Table S3). Again, the compounds separate into two groups, the β-arrestin-biased compounds displaying preserved β-arrestin signaling in the absence of G protein signaling, and a number of balanced compounds that signal through both pathways. This was confirmed by a calculation of bias factors derived from the operational model (Fig. 6D), which have an excellent correlation with the bias factors calculated using the equiactive approach (Table 2). Although some of the synthetic compounds do not seem to display any significant bias, such as TRV120055 and TRV120056, other compounds have nearly an order of magnitude more bias than the initially described β-arrestin-biased agonist SGG (Holloway et al., 2002). In this case, all three approaches yielded similar results.
Bias factors for ligands at the AT1AR
The column “Bias” denotes whether a statistically significant difference in bias compared to the reference balanced agonist angiotensin II is present (either toward G protein or β-arrestin), whereas “Non” denotes an insignificant difference from the balanced agonist.
Discussion
In this work, we develop a general method for the quantification of ligand bias by using three different approaches, each with its own strengths and weaknesses. Both the equimolar and equiactive approaches are free of the assumptions inherent in pharmacological models (e.g., a 1:1 receptor/agonist complex mediates that signaling and that the effects are due to a receptor/ligand complex at equilibrium). Therefore they can be used more generally (e.g., for analyzing bias in systems with receptor dimers or allosteric modulators). Although the equimolar comparison is intuitively appealing and graphically displays different levels of bias, it is unable to identify weakly biased ligands when assays with markedly different levels of amplification are compared, and, more generally, it is unable to quantify bias. The equiactive comparison allows for a quantification of bias; however, the resulting bias factors are prone to error with partial agonists or strongly biased compounds because of the poor fits of the concentration-response data with weak signal-to-noise levels. In contrast, these large errors are not as problematic in the operational model, in which the additional information from a separate ligand binding experiment constrains the fits and yields a better estimate of bias. This model not only allows for quantification of bias but also yields an estimate of efficacy, the effective signaling (σ). Therefore, we conclude that the operational model gives the best approach to quantifying bias, although a good estimate of bias can be obtained using the equiactive comparison if the dissociation constant for a ligand is not known.
Several approaches have been proposed previously to quantify ligand bias in an effort to overcome the limitations associated with an analysis of classic pharmacologic parameters. Some methods are qualitative, such as “bias plots” (Gregory et al., 2010) or a comparison of rank order of potencies (Kenakin, 1995), whereas others are quantitative, such as comparisons of transduction ratios (Evans et al., 2011; Gregory et al., 2010; Kenakin and Miller, 2010; Koole et al., 2010) or intrinsic relative activities (Ehlert, 2008; Figueroa et al., 2009). The qualitative approaches to identify biased ligands are inherently limited in their scope, whereas the current quantitative approaches have theoretical or practical limitations. “Transduction ratios” (Figueroa et al., 2009; Evans et al., 2011; Gregory et al., 2010; Kenakin and Miller, 2010; Koole et al., 2010), defined as τ/KA derived from the operational model (where KA denotes the dissociation constant), have been used to estimate ligand bias. We chose not to use this approach for a number of reasons. First, the parameters of interest in assessing bias are the ligand's different efficacies through different signaling pathways, which is quantified by the ligand's coupling efficiency (τ) for the different pathways (Black and Leff, 1983; McPherson et al., 2010) and not by its dissociation constant, KA (see Supplemental Materials). Second, in these studies, the effective dissociation constant is derived directly from the concentration-response data themselves (Evans et al., 2011; Gregory et al., 2010; Koole et al., 2010), which may differ for the same ligand in different signaling assays because of the formation of different receptor ternary complexes with G proteins or β-arrestins in each assay (De Lean et al., 1980; Colquhoun, 1985). In addition, the detailed method for fitting data using the transduction ratio approach has yet to be published (Evans et al., 2011). In fitting our data, we chose to use a dissociation constant determined from competition radioligand binding experiments under conditions that would limit the formation of a receptor ternary complex, which should allow for a separation of affinity and efficacy (Kenakin, 1999) in our analysis. Even with these differences, the bias factors calculated from transduction ratios (Evans et al., 2011) should be similar to those from our operational analysis, because the dissociation constant terms would largely cancel out.
Over the past few years, there has been an explosion in publications describing the identification of biased agonists at a wide variety of 7TMRs (Whalen et al., 2011). In such studies, it is important to optimize experimental conditions to avoid the false identification of biased ligands because of the differences in compound stability or variations in cell types and other conditions used for different assays. Many biased ligands have been identified in screening, whereas for other well known drug targets such as the β2AR, strongly biased agonists have yet to be identified. However, most presumably biased compounds have been identified based on comparisons of classic pharmacological parameters such as EC50 and Emax, which are prone to errors in interpretation in the setting of receptor reserve. Therefore, it is still unclear the extent to which these ligands are biased. On the other hand, it is likely that there are a number of weakly biased ligands that have yet to be identified because of the inability to properly quantify ligand bias. Here, we have demonstrated that weakly biased ligands, which could serve as tool compounds to dissect receptor biology or as lead compounds in the drug development process, can be identified using these approaches. It is noteworthy that the weakly biased ligands identified at the β2AR, formoterol and salmeterol, are used clinically, suggesting that a number of drugs that are used in the clinic today may also be similarly biased. The ability to quantify such signaling bias may facilitate the mechanistic understanding of both desirable and undesirable properties of such therapeutics.
Authorship Contributions
Participated in research design: Rajagopal, Ahn, Rominger, Gowen-McDonald, DeWire, Violin, and Lefkowitz.
Conducted experiments: Rajagopal, Ahn, Rominger, Gowen-McDonald, Lam, and DeWire.
Performed data analysis: Rajagopal, Ahn, Rominger, and Gowen-McDonald.
Wrote or contributed to the writing of the manuscript: Rajagopal, Ahn, DeWire, Violin, and Lefkowitz.
Acknowledgments
We thank Tommaso Costa, Terry Kenakin, Arthur Christopoulos, and Ryan Strachan for valuable discussions. We thank Gilad Barnea and Richard Axel for providing the β2AR Tango construct. We thank Donna Addison and Quivetta Lennon for secretarial assistance.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported in part by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL16037, HL70631, HL07101-34].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.072801.
-
ABBREVIATIONS:
- 7TMR
- seven transmembrane receptor
- β2AR
- β2 adrenergic receptor
- AT1AR
- angiotensin II type 1A receptor
- HEK
- human embryonic kidney
- IP1
- inositol 1-phosphate
- Pin
- pindolol
- DCI
- dichloroisoproterenol
- Slm
- salmeterol
- For
- formoterol
- SGG
- Sar1Gly4Gly8 angiotensin II
- SII
- Sar1Ile4Ile8 angiotensin II
- TRV120027
- Sar-Arg-Val-Tyr-Ile-His-Pro-d-Ala-OH
- TRV120026
- Sar-Arg-Val-Tyr-Tyr-His-Pro-NH2
- TRV120055
- Sar-Arg-Val-Tyr-Val-His-NH2
- TRV120056
- Asp-Arg-Val-Tyr-Ile-His-Pro-Gly
- TRV120044
- N-methyl-l-alanine-Arg-Val-Tyr-Ile-His-Pro-d-Ala
- TRV120045
- Sar-Arg-Val-Tyr-Arg-His-Pro-NH2
- TRV120034
- N-methyl-l-alanine-Arg-Val-Tyr-Ile-His-Pro-Ala
- TEV
- Tobacco Etch Virus.

- Received April 6, 2011.
- Accepted May 24, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}