


Abstract
Although G protein-coupled receptors are often categorized in terms of their primary coupling to a given type of Gα protein subunit, it is now well established that many show promiscuous coupling and activate multiple signaling pathways. Furthermore, some agonists selectively activate signaling pathways by promoting interaction between distinct receptor conformational states and particular Gα subunits or alternative signaling proteins. We have tested the capacity of agonists to stimulate Ca2+ release, cAMP accumulation, and changes in extracellular acidification rate (ECAR) at the human α1A-adrenoceptor. Signaling bias factors were determined by novel application of an operational model of agonism and compared with the reference endogenous agonist norepinephrine; values significantly different from 1.0 indicated an agonist that promoted receptor conformations distinct from that favored by norepinephrine. Oxymetazoline was a full agonist for ECAR and a partial agonist for Ca2+ release (bias factor 8.2) but failed to stimulate cAMP production. Phenylephrine showed substantial bias toward ECAR versus Ca2+ release or cAMP accumulation (bias factors 21 and 33, respectively) but did not display bias between Ca2+ and cAMP pathways. Cirazoline and N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulfonamide (A61603) displayed bias toward cAMP relative to Ca2+ release (bias factors of 7.4 and 8.6). It is noteworthy that epinephrine, a second endogenous adrenoceptor agonist, did not display bias relative to norepinephrine. Our finding that phenylephrine displayed significant signaling bias, despite being highly similar in structure to epinephrine, indicates that subtle differences in agonist-receptor interaction can affect conformational changes in cytoplasmic domains and thereby modulate the repertoire of effector proteins that are activated.
Introduction
Although G protein-coupled receptors (GPCRs) are traditionally categorized as Gs-, Gq-, or Gi/o-coupled, studies of multiple signaling outputs often indicate that a G protein-coupled receptor has the capacity to interact with more than one G protein subtype as well as alternative signaling or effector proteins such as arrestins. This raises the possibility that ligands may display functional selectivity, promoting differential coupling of receptors to Gα subunits or G protein-independent pathways (for review, see Galandrin et al., 2007; Kenakin, 2007; Audet and Bouvier 2008; Evans et al., 2010; Kenakin and Miller, 2010). For example, antipsychotic drugs acting at the dopamine D2 receptor display functional selectivity with respect to Gi/o-mediated decreases in cAMP, compared with receptor recruitment of arrestin-3 (Masri et al., 2008). All clinically effective antipsychotics block arrestin-3 recruitment, despite ranging from partial agonists to inverse agonists when tested for inhibition of cAMP.
In some cases, agonists with only subtle differences in structure still display functional selectivity. Among a series of phenethylamines active at the β2-AR, cyclopentylbutanephrine, α-ethylnoradrenaline, and isoetharine are partial agonists for cAMP production relative to isoproterenol but act as full agonists for arrestin-3 recruitment (Drake et al., 2008). These three compounds share an ethyl group on the αC atom, near the NH3+ group that interacts with Asp113 (3.32). There may be a steric effect of the α-ethyl group that compromises receptor conformational changes linked to G protein activation, without affecting receptor phosphorylation or arrestin binding. In cardiac myocytes, the S,R-isomer of fenoterol stimulates substantially higher activation of Gαi2 than the R,R-isomer, whereas (R,R)-fenoterol produces higher activation of Gαs than (S,R)-fenoterol (Woo et al., 2009). This differential Gs/Gi coupling is apparent in functional assays including myocyte contractility and Erk1/2 phosphorylation, and the authors suggest that agonists selectively able to stimulate β2-AR Gs coupling, without stimulating β2-AR Gi coupling or β1-AR activation, may have considerable therapeutic benefit.
In matched cells expressing a given receptor, functional selectivity can be unambiguously demonstrated when two ligands promote a reversal of efficacy or potency between two pathways; that is, drug A has higher efficacy/potency than drug B for pathway 1 but a lower efficacy/potency than drug B for pathway 2 (Kenakin, 2007; Urban et al., 2007; Kenakin and Miller, 2010). However, such observations do not provide capacity for quantification and statistical analysis, and there are reported instances in which drugs do not display reversal of potency or efficacy but clearly promote distinct receptor conformations (for example, Galandrin et al., 2008). The operational model of agonism, originally derived by Black and Leff (1983), can provide a means of quantifying signaling bias by comparing τ/KA ratios among agonists (Figueroa et al., 2009; Gregory et al., 2010; Kenakin and Miller, 2010; Koole et al., 2010). In this model, the parameter τ encompasses receptor density and the efficacy of the agonist, and KA is the equilibrium dissociation constant of the agonist-receptor complex. The composite parameter, τ/KA, is thus a convenient “transduction ratio” (Kenakin and Miller, 2010) that can account for functional selectivity in the activity of drugs with high affinity/low efficacy or low affinity/high efficacy.
In the current study, we have used this approach to calculate bias factors for agonists acting at the human α1A-adrenoceptor (AR). The three α1-ARs preferentially stimulate coupling to Gq, activating phospholipase C, increasing intracellular levels of inositol (1,4,5)-triphosphate and intracellular Ca2+, and releasing diacylglycerol that in turn activates protein kinase C isoforms. Studies in tissues that endogenously express α1-ARs, or in recombinant cells expressing each of the α1-AR subtypes, have also demonstrated activation of phospholipase A2, phospholipase D, adenylate cyclase, and mitogen-activated protein kinases (Perez et al., 1993; Graham et al., 1996; Zhong and Minneman, 1999; Piascik and Perez, 2001). We have examined the ability of α1A-ARs stably expressed in CHO-K1 cells to stimulate Ca2+ release, cAMP accumulation, and changes in extracellular acidification rate (ECAR) upon agonist exposure. We compared the endogenous agonist norepinephrine, another two phenethylamines (methoxamine and phenylephrine), and three imidazolines, cirazoline, oxymetazoline, and A61603 (Fig. 1). All six agonists stimulated release of intracellular Ca2+ and ECAR measured in a cytosensor microphysiometer. Oxymetazoline was a partial agonist for Ca2+ release and a full agonist in the cytosensor yet failed to stimulate cAMP production even at high concentrations. By calculating bias factors for the agonists relative to norepinephrine, we have found several clear and quantitative indications of functional selectivity. In contrast to this group of compounds, the two endogenous agonists norepinephrine and epinephrine showed no bias relative to each other.

Chemical structures of α1A-AR agonists used in the study. A, phenethylamines. B, imidazolines.
Materials and Methods
Materials.
Sources of drugs were as follows: [125I]HEAT (2-[β- (4-hydroxy-3-[125I]iodophenyl) ethylamino-methyl]tetralone) (ProSearch, Melbourne, Australia); [3H]prazosin (PerkinElmer Life and Analytical Sciences, Waltham, MA); (−)-norepinephrine bitartrate, phenylephrine, and phentolamine HCl (Sigma, St. Louis, MO); methoxamine, oxymetazoline, cirazoline, and N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulfonamide (A61603; Tocris Bioscience, Ellisville, MO); Dulbecco's modified Eagle's medium/Ham's F12 50:50 mix, fetal bovine serum, glutamine, penicillin/streptomycin (ThermoTrace Pty., Ltd., Noble Park, VIC, Australia); Platinum Pfx DNA polymerase, Lipofectamine, phenol red-free OptiMEM, and Fluoro-4 (Invitrogen, Carlsbad, CA); and G418 and IBMX (Sigma). Cytosensor consumables were from Selby Biolab (Clayton Nth, VIC, Australia), except for Costar transwell cups (Corning Life Sciences, Lowell, MA).
Cloning of the Human α1A-AR.
Plasmid containing the human α1A-4-AR cDNA was a gift from Dr. Thomas Chang (Roche Bioscience, Palo Alto, CA). The coding region of this receptor was subcloned into the multiple cloning site of pcDNA3.1. The α1A-4-AR cDNA was converted to the α1A-1-AR as follows. A silent mutation was introduced into the α1A-4-AR construct using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) to generate a BsrGI restriction site (TGT ACC→TGT ACA) corresponding to Cys419 Thr420, immediately upstream of the point at which the four α1A-AR splice variants differ (Chang et al., 1998). The 3′ end of the α1A-1-AR coding region was generated by polymerase chain reaction on human genomic DNA, using Platinum Pfx high-fidelity DNA polymerase. The forward primer incorporated the BsrG1 site (underlined; 5′ AATCCTCCTGTACAACAGCCCGGGTGAG 3′) and the reverse primer included the α1A-1-AR termination codon and a downstream XbaI site (underlined; 5′ CCTCTGCATCTAGACTGTCCTAGACTTCCTC 3′). The α1A-1-AR polymerase chain reaction fragment was digested with BsrG1 and XbaI, then ligated into pcDNA3.1-α1A-4-AR plasmid from which the corresponding cassette had been removed. The complete α1A-1-AR insert and junctions with pcDNA3.1 were checked by DNA sequencing on both strands (Micromon; Monash University, Parkville, VIC, Australia).
Cell Culture and Production of Clones Stably Expressing α1A-ARs.
Chinese hamster ovary (CHO-K1) cells were grown in a 50:50 Dulbecco's modified Eagle's medium/Ham's F12 medium supplemented with 10% (v/v) fetal bovine serum (FBS), glutamine (2 mM), penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37°C with 5% CO2. Transfections were performed using Lipofectamine, and the transfected cells were selected in media containing 800 μg/ml G418 and maintained in media containing 400 μg/ml G418. Clonal cell lines were isolated by screening G418-resistant colonies and preliminary screening of clones was made using a single point [3H]prazosin (2000 pM) whole-cell binding assay. Suitable clones were amplified and receptor levels determined in saturation binding assays using 125I-HEAT.
Radioligand Binding Studies.
Cells were harvested as described previously by Hutchinson et al., (2002). In brief, cells from a 75-cm2 flask were washed twice with HEPES-buffered saline and scraped from flasks with lysis buffer (25 mM Tris, pH 7.5 at room temperature, 1 mM EDTA, 200 μg/ml bacitracin, 10 μg/ml leupeptin, 2.5 μg/ml pepstatin A, and 2.5 μg/ml aprotinin). Cells were homogenized with a Dounce homogenizer and centrifuged at low speed (1000g, 10 min) to remove cell debris. Supernatants were pooled and centrifuged (39,000g, 15 min, 4°C). The pellet was homogenized in binding buffer (50 mM Tris, pH 7.4, 5 mM MgCl2, 1 mM EDTA, 200 μg/ml bacitracin, 10 μg/ml leupeptin, 2.5 μg/ml pepstatin A, and 2.5 μg/ml aprotinin) and frozen at −70°C until required. Membranes (10–20 μg of protein) were incubated with 125I-HEAT (20–1200 pM) in a total volume of 100 μl for 90 min at room temperature (21°C). Phentolamine (100 μM) was used to define nonspecific binding. Competition binding experiments were conducted using a range of concentrations of unlabeled ligand and 125I-HEAT (500 pM), also at room temperature for 90 min (Sharpe et al., 2003). Reactions were terminated by rapid filtration through GF/C filters presoaked for 30 min in 0.5% polyethylenimine using a Packard Filtermate cell harvester. Filters were washed three times with wash buffer (50 mM Tris, pH 7.4, 4°C), dried, and 25 μl of Microscint O (PerkinElmer Life and Analytical Sciences) added and radioactivity counted on a Packard TopCount. Protein concentrations were quantitated using the Lowry et al. (1951) assay. All experiments were performed in duplicate using five different membrane preparations.
Measurement of Intracellular Free Ca2+ Concentration.
CHO-K1 cells expressing the α1A-AR were seeded at 5 × 104 cells per well in 96-well plates overnight. On the day of the experiment, the media were removed and cells were washed three times in a modified Hanks' balanced saline solution (HBSS; 150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2·2H2O, 10 mM d-glucose, 10 mM HEPES, 2.2 mM CaCl2·2H2O, and 2 mM probenecid, pH 7.4) containing 0.5% (w/v) bovine serum albumin. In light-diminished conditions, cells were treated with fluoro-4 [0.1% (v/v) in modified HBSS, 1 h, 37°C]. Excess fluoro-4 not taken up by the cells was removed by washing twice in modified HBSS and then incubated for a further 30 min before the assay plate was transferred to a FlexStation (Molecular Devices, Sunnyvale, CA). Real-time fluorescence measurements were recorded every 1.7 s over 200 s, with drug additions occurring after 17 s, at an excitation wavelength of 485 nm and reading emission wavelength of 520 nm. All experiments were performed in duplicate. Agonist responses represent the difference between basal fluorescence and peak [Ca2+]i measurements expressed as a percentage of the response to calcimycin (A23187) (1 μM) in each experiment.
cAMP Accumulation Studies.
Cells were seeded into 96-well plates at 104 cells/well in media with 0.5% (v/v) FBS the night before the experiment. On the day of the experiment, the media were replaced with stimulation buffer (1 mg/ml BSA, 0.5 mM IBMX, and 5 mM HEPES, pH 7.4, in Hanks' balanced salt solution, 50 μl/well). Cells were exposed to agonists (diluted to 2× final concentration in stimulation buffer, 50 μl/well) for 30 min at 37°C, then reactions were terminated by addition of 100 μl of lysis buffer [1 mg/ml BSA, 0.3% (v/v) Tween 20, 5 mM HEPES, and 1 mM IBMX, pH 7.4]. cAMP was assayed using αScreen in an EnVision plate reader (Elmer Life and Analytical Sciences, VIC, Australia). Responses to forskolin (10−4 M) were determined in parallel with agonist-stimulated cAMP accumulation for each batch of cells, and results are expressed as a percentage of the response to forskolin. All experiments were performed in duplicate, and n is the number of independent experiments.
Cytosensor Microphysiometer Studies.
The cytosensor microphysiometer (Molecular Devices) is a light-addressable silicon sensor-based device that measures increases in the metabolic activity of isolated cells as increases in ECAR (McConnell et al., 1992). Cells were seeded into 12-mm Costar transwell cups (3-μm pore size; Corning Life Sciences) at 5 × 105 cells per cup in media with 0.5% FBS and allowed to grow overnight. The next day, the assembled cups were placed in the sensor chambers in the cytosensor and perfused with modified RPMI 1640 medium (Molecular Devices) at 100 μl/min at 37°C. After initial set-up, cells were superfused with media for 2 h to stabilize baseline extracellular acidification rates, and then cumulative concentration-response curves were constructed for each agonist. Each concentration was perfused for 14 min, consisting of seven 2-min pump cycles during which the flow was stopped for the last 40 s and the acidification rate measured for 30 s. The maximum reading from the seven-cycle recording corresponding to each drug concentration was expressed as a percentage of the basal acidification rate measured over a period of 10 min before the addition of agonist. All drugs were diluted in medium and concentration-response experiments were performed in the presence of (−) propranolol (10−6 M) to block any contributions from endogenous β2-AR. Results were expressed as percentage of basal ECAR, and n values represent cells grown in different flasks before plating into the transwell cups.
Data Analysis.
All values are expressed as mean ± S.E.M. of n. Data were analyzed using nonlinear curve fitting (Prism ver. 5.02; GraphPad Software, San Diego, CA) to obtain pEC50 values for the cytosensor microphysiometer, cAMP accumulation, and [Ca2+]i assays, or pKi, pKD, and Bmax values from radioligand binding assays. To quantify signaling bias, agonist concentration response curves were analyzed by nonlinear regression using an operational model of agonism (Black and Leff, 1983; Gregory et al., 2010) in a method similar to that described by Figueroa et al. (2009), to define τ/KA ratios for each agonist, for each pathway:
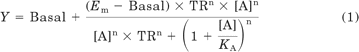 where Em is the maximal possible response of the system (not the agonist), Basal is the basal level of response in the absence of agonist, KA denotes the functional equilibrium dissociation constant of the agonist for the ground (G protein-uncoupled) state of the receptor, n is the slope of the transducer function that links occupancy to response, and TR (transduction ratio) is the ratio of τ/KA, where τ is an index of the coupling efficiency (or efficacy) of the agonist—this parameter incorporates the affinity of the agonist for the active state of the receptor that triggers signaling, as well as the efficiency of coupling of the receptor to its cognate G protein(s) and subsequent cellular stimulus-response transduction mechanisms (Leff and Harper, 1989). Note that KA values are derived directly from the nonlinear regression analysis of agonist concentration-response curves. The estimated TR values were used in the comparison of functional selectivity mediated by each agonist across the various pathways, as described under Results. A detailed exploration of this method for determining agonist signaling bias will be presented separately (T. Kenakin, R. Novick, C. Watson, V. Muniz-Medina, and A. Christopoulos, manuscript in preparation).
where Em is the maximal possible response of the system (not the agonist), Basal is the basal level of response in the absence of agonist, KA denotes the functional equilibrium dissociation constant of the agonist for the ground (G protein-uncoupled) state of the receptor, n is the slope of the transducer function that links occupancy to response, and TR (transduction ratio) is the ratio of τ/KA, where τ is an index of the coupling efficiency (or efficacy) of the agonist—this parameter incorporates the affinity of the agonist for the active state of the receptor that triggers signaling, as well as the efficiency of coupling of the receptor to its cognate G protein(s) and subsequent cellular stimulus-response transduction mechanisms (Leff and Harper, 1989). Note that KA values are derived directly from the nonlinear regression analysis of agonist concentration-response curves. The estimated TR values were used in the comparison of functional selectivity mediated by each agonist across the various pathways, as described under Results. A detailed exploration of this method for determining agonist signaling bias will be presented separately (T. Kenakin, R. Novick, C. Watson, V. Muniz-Medina, and A. Christopoulos, manuscript in preparation).
Results
Expression of the α1A-AR in CHO-KI Cells.
Based on saturation binding with 125I-HEAT, the α1A-AR clone chosen for these studies displayed a Bmax value of 531 ± 94 fmol/mg protein and a pKD of 9.2 ± 0.09. Further characterization of the α1A-AR was carried out with competition binding experiments using the agonists norepinephrine, methoxamine, phenylephrine, oxymetazoline, cirazoline, and A61603. All of the agonists competed for [125I]HEAT binding, with binding curves fitting a single site isotherm. The rank order of binding affinities for the α1A-AR agonists was oxymetazoline (pKi 7.5 ± 0.2) = A61603 (7.3 ± 0.1) > cirazoline (6.7 ± 0.2) ≫ norepinephrine (5.3 ± 0.1) > phenylephrine (4.9 ± 0.1) = methoxamine (4.8 ± 0.1) (n = 3–7). Note that imidazoline agonists have substantially higher affinity for the α1A-AR than the phenethylamines.
Effects of α1A-AR Agonists on Intracellular Ca2+ Release.
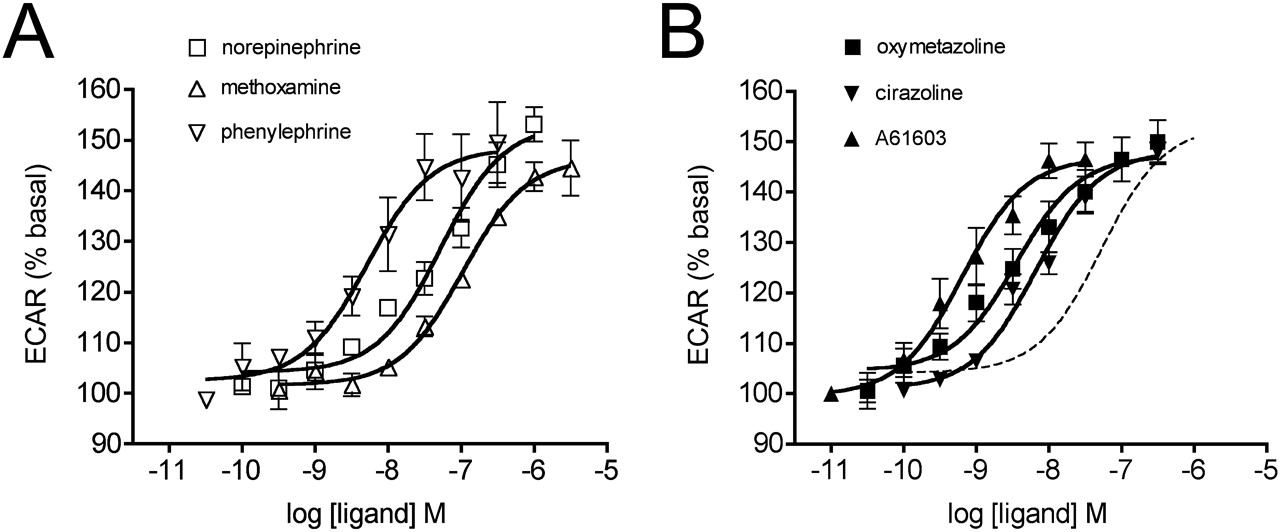
In CHO-K1 cells expressing the α1A-AR, all the agonists stimulated Ca2+ release (Fig. 2, Table 1). Emax values ranged from 97.4% for A61603 (relative to 1 μM A23187) down to 78.4% for oxymetazoline. It is noteworthy that norepinephrine and phenylephrine behaved as high-efficacy, low-affinity agonists, because the ratio of the apparent binding affinity (Ki) to EC50 is approximately 1700. Such a high “amplification” ratio (Strange, 2008) reflects substantially higher agonist affinity for the active state(s) than for the inactive state of the receptor and/or high efficiency of signal transduction for the measured pathway, in this case Ca2+ release. In contrast to norepinephrine and phenylephrine, oxymetazoline and cirazoline display only 36- and 47-fold amplification factors. A61603 and methoxamine differ in pKi for the α1A-AR by more than 2 log units but display similar amplification of the response relative to their binding affinities (250- and 320-fold, respectively).

Concentration-response curves for stimulation of Ca2+ release by the α1A-AR. CHO-KI cells stably expressing the human α1A-AR were treated with 0.1% fluoro-4 at 37°C for 1 h then washed and incubated for a further 30 min. Real-time fluorescence measurements were recorded every 1.7 s, with drug additions occurring after 17 s. Agonist responses represent the difference between basal fluorescence and the peak [Ca2+]i (reached within 20 s of agonist addition), expressed as a percentage of the response to the Ca2+ ionophore A23187 (1 μM). Dose-dependent Ca2+ release was stimulated by phenethylamine (A) and imidazoline (B) agonists. Values are means ± S.E.M. of six independent experiments. In B, the norepinephrine curve is shown as a dashed line for comparison.
Potency (pEC50), maximum response (Emax), and α value (Emax relative to norepinephrine) of agonists at the human α1A-AR
Effects of α1A-AR Agonists on cAMP Accumulation.
All three α1-AR subtypes promote agonist-stimulated Gq activation and Ca2+ release, but only the α1A- and α1B-AR stimulate adenylate cyclase and cAMP production (Shibata et al., 2003). This suggests that the receptor coupling determinants for the Ca2+ and cAMP pathways differ, and thus that the immediate postreceptor step for each of these pathways is distinct. We found that incubation of CHO-α1A-AR cells with norepinephrine, phenylephrine, methoxamine, cirazoline, or A61603 stimulated concentration-dependent increases in cAMP accumulation as shown in Fig. 3. Concentration-response curves were also performed in the presence of inhibiting concentrations of propranolol (10−6 M) to block any endogenous β-ARs. This did not affect cAMP responses, demonstrating that endogenous β-ARs were not responsible for the α1A-AR agonist-stimulated cAMP accumulation (data not shown). In addition, no response was seen in nontransfected CHO-K1 cells stimulated with norepinephrine (data not shown). In contrast to the other agonists, oxymetazoline failed to increase cAMP production above basal levels at concentrations up to 10−4 M. To test for possible physiological antagonism of responses to oxymetazoline mediated by endogenous α2-AR in CHO-K1 cells, experiments were performed in the presence of the α2-AR antagonist rauwolscine (10−7 M). These experiments failed to reveal cAMP accumulation in response to oxymetazoline. To test whether oxymetazoline was directing strong coupling of the α1A-AR to Gi, thereby cancelling out any stimulatory effect on cAMP accumulation, cells were pretreated with pertussis toxin 16 h before agonist stimulation. This likewise failed to reveal any cAMP accumulation in response to oxymetazoline (data not shown). cAMP responses are expressed as a percentage of the control forskolin (10−4 M) response, and Emax and potency values for each agonist are summarized in Table 1. Emax values were similar for the three phenethylamines and A61603, whereas cirazoline was a partial agonist. It is striking that all of the drugs able to activate cAMP accumulation do so with pEC50 values very close to their affinity values.

Concentration-response curves for stimulation of cAMP accumulation by the α1A-AR. CHO-K1 cells stably expressing the α1A-AR were exposed to agonists for 30 min in stimulation buffer containing 0.5 mM IBMX to inhibit phosphodiesterases. Responses to forskolin (10−4 M) were determined in parallel with agonist-stimulated cAMP accumulation for each batch of cells, and results are expressed as a percentage of the response to forskolin. A dose-dependent cAMP response was stimulated by norepinephrine, phenylephrine, and methoxamine (A), cirazoline and A61603 (B), but not oxymetazoline. Values are means ± S.E.M. of four to five independent experiments. In B, the norepinephrine curve is shown as a dashed line for comparison.
Effects of α1-AR Agonists on ECAR.
To examine additional pathways that may be activated by the α1A-AR, the cytosensor microphysiometer was used as a measure of total cellular activity stimulated by agonists. Incubation of cells stably expressing the α1A-AR with agonists stimulated concentration-dependent increases in extracellular acidification rate (ECAR; Fig. 4). Emax values ranged from increases of 146.5 to 152.2% basal (Table 1). In essence, all of the compounds tested were full agonists relative to norepinephrine, representing a substantial change in the behavior of oxymetazoline and cirazoline compared with Ca2+ release or cAMP responses. As seen in the Ca2+ release assay, there were substantial differences in the degree of amplification displayed by each agonist, varying from 2350-fold for phenylephrine down to 9-fold for oxymetazoline. Unlike Ca2+ release, however, norepinephrine promoted only 105-fold amplification, approximately 20-fold lower than that seen with phenylephrine.

Concentration-response curves for changes in extracellular acidification rate measured in the cytosensor microphysiometer. Cells stably expressing the α1A-AR were grown overnight in transwell cups then placed in cytosensor chambers and perfused with modified low-buffered RPMI 1640 medium. After 2 h pre-equilibration, the basal acidification rate was measured over a period of 10 min, and then successive agonist concentrations were added at 14-min intervals. The maximum reading from the seven-cycle recording corresponding to each drug concentration was expressed as a percentage of the basal acidification rate. Changes in ECAR were stimulated by phenethylamine (A) and imidazoline (B) agonists in a dose-dependent manner. Values are means ± S.E.M. of four independent experiments. In B, the norepinephrine curve is shown as a dashed line for comparison.
Analysis of Functional Selectivity at the α1A-AR.
Our data indicated that there are interesting differences in agonist activity between the three measured signaling outputs. We thus carried out a further analysis of this data to test the hypothesis that at least some agonists display functional selectivity; that is, they preferentially induce or stabilize different receptor conformations that in turn couple to two or more signaling pathways (Kenakin, 2007; Urban et al., 2007; Evans et al., 2010; Kenakin and Miller, 2010). One hallmark of functional selectivity is that two agonists show a reversal in efficacy between two signaling assays, although a reversal in potency between two agonists can also be accommodated only by the involvement of distinct receptor conformations (Kenakin, 2007). Among the data presented in Table 1, there were no reversals in Emax between agonists; however, there were several instances of a reversal of potency. As shown in Fig. 2, oxymetazoline stimulated a small cAMP response at 1 mM and thus exhibited very low potency even though no pEC50 value could be derived. Oxymetazoline had higher potency for Ca2+ release than phenylephrine but clearly lower potency for cAMP. Likewise, oxymetazoline had higher potency for both Ca2+ release and ECAR than methoxamine but lower potency for cAMP accumulation. It is noteworthy that phenylephrine had higher potency for ECAR than norepinephrine but lower potency for Ca2+ release.
Although these reversals in potency indicate functional selectivity, there are two major limitations. First, a subjective analysis of the data does not allow us to ascribe statistical significance to bias in drug activity across the three signaling endpoints. Second, although a reversal in efficacy or potency between drugs may be a sufficient condition for demonstrating functional selectivity, there is evidence that such reversals are not a necessary condition (Evans et al., 2010). Thus, we applied an operational model of agonism (eq. 1) to derive quantitative measures of functional selectivity between agonists for the different signaling assays. To exclude possible bias introduced by the cellular host system (as evidenced by the substantial differences in response amplification displayed by norepinephrine across the three assays), the transduction ratios derived from application of the operational model of agonism were normalized to those of a reference agonist, in this case the endogenous agonist, norepinephrine. Under these conditions, if the test agonist and the reference agonist activate the two pathways via a common receptor conformation, the bias factor should be 1.0, irrespective of differences in response amplification between pathways. In contrast, significant deviation of bias factors from 1.0 indicates the involvement of distinct conformations for the different agonists. Table 2 shows the logarithms of the operational model τ/KA ratios for each pathway (logTR), the logTR values normalized to that of norepinephrine (logTRn), and the final bias factor calculations for signaling by the α1A-AR (difference between logTRn values for a given agonist across different pathways; see Supplemental Table 1 for statistical analysis of data).
Calculation of bias factors for phenethylamine and imidazoline agonists at the α1A-AR
LogTR is the log of the transduction ratio (τ/KA), and logTRn values are normalized to the logTR for norepinephrine. Bias factors in bold are significantly different from 1.0. To avoid propagation of error as a result of multiple data manipulation steps, Student's t tests were carried out on the raw logTR data rather than data normalized to norepinephrine (see Supplemental Table 1).
Phenylephrine and oxymetazoline showed significant bias toward ECAR compared with Ca2+ release. In the case of phenylephrine, the bias factor of 21 reflects a high τ/KA ratio for ECAR relative to norepinephrine, despite the lower τ/KA ratio for Ca2+ release. A high bias factor of 8.2 was observed for oxymetazoline because, unlike norepinephrine, this drug displayed the same τ/KA ratio for ECAR and Ca2+ release (log τ/KA values of 8.46 and 8.52, respectively). Phenylephrine also displayed a high bias factor of 33 between ECAR and cAMP accumulation. Again, this reflects the fact that the cAMP τ/KA ratio was substantially lower for phenylephrine than for norepinephrine, whereas the ECAR ratio was higher. The same is true to a lesser extent for methoxamine (bias factor 4.0) but was not seen in the case of the two imidazolines. A different pattern emerged between Ca2+ release and cAMP accumulation. Here, the two phenethylamines (methoxamine and phenylephrine) showed no significant bias relative to norepinephrine, whereas the two imidazolines (cirazoline and A61603) showed clear 7.4- and 8.6-fold bias, respectively, toward cAMP signaling. Given that there was no reversal of potency between norepinephrine and cirazoline or A61603 for cAMP accumulation versus Ca2+ release, this result illustrates the power of being able to quantify functional selectivity using the operational model.
To check that these findings of signaling bias were not artifacts of the particular clonal cell line used or receptor abundance, we repeated the analysis using CHO-K1 cells expressing the α1A-AR with a Bmax of 204 fmol/mg protein. The key finding was still evident in these cells: the bias factors for phenylephrine relative to norepinephrine were 37 for ECAR compared with Ca2+ release and 34 for ECAR versus cAMP, whereas there was no bias between the Ca2+ and cAMP pathways (bias factor 0.9).
Endogenous Agonists for the α1A-AR Do Not Display Functional Selectivity.
Phenylephrine displayed a substantially higher capacity than norepinephrine to activate signaling pathways leading to changes in ECAR, suggesting that it preferentially induces or stabilizes an α1A-AR conformation that couples strongly to one or more of these pathways. In contrast, the lack of bias between cAMP and Ca2+ indicates that the conformation associated with each of phenylephrine and norepinephrine is coupled equally well to pathways mediating Ca2+ release and cAMP accumulation. As shown in Fig. 1, norepinephrine and phenylephrine share the meta-hydroxyl group on the ring structure, and the chiral β-hydroxyl, but there are two key differences in their chemical structure. Norepinephrine has a catechol ring with both the meta- and a para-hydroxyl group, whereas phenylephrine lacks the para-hydroxyl. On the other hand, phenylephrine has a methyl substituent on the active amine group that is not present in norepinephrine. We analyzed signaling bias between norepinephrine and epinephrine because the latter has both the catechol ring and the N-methyl group (Fig. 5A). As shown in Fig. 5, B–D, norepinephrine and epinephrine are full agonists for each of the signaling pathways. Epinephrine always has somewhat higher potency than norepinephrine (Table 3), but there is no signaling bias between these compounds based on τ/KA ratio calculations (Tables 4 and 5).
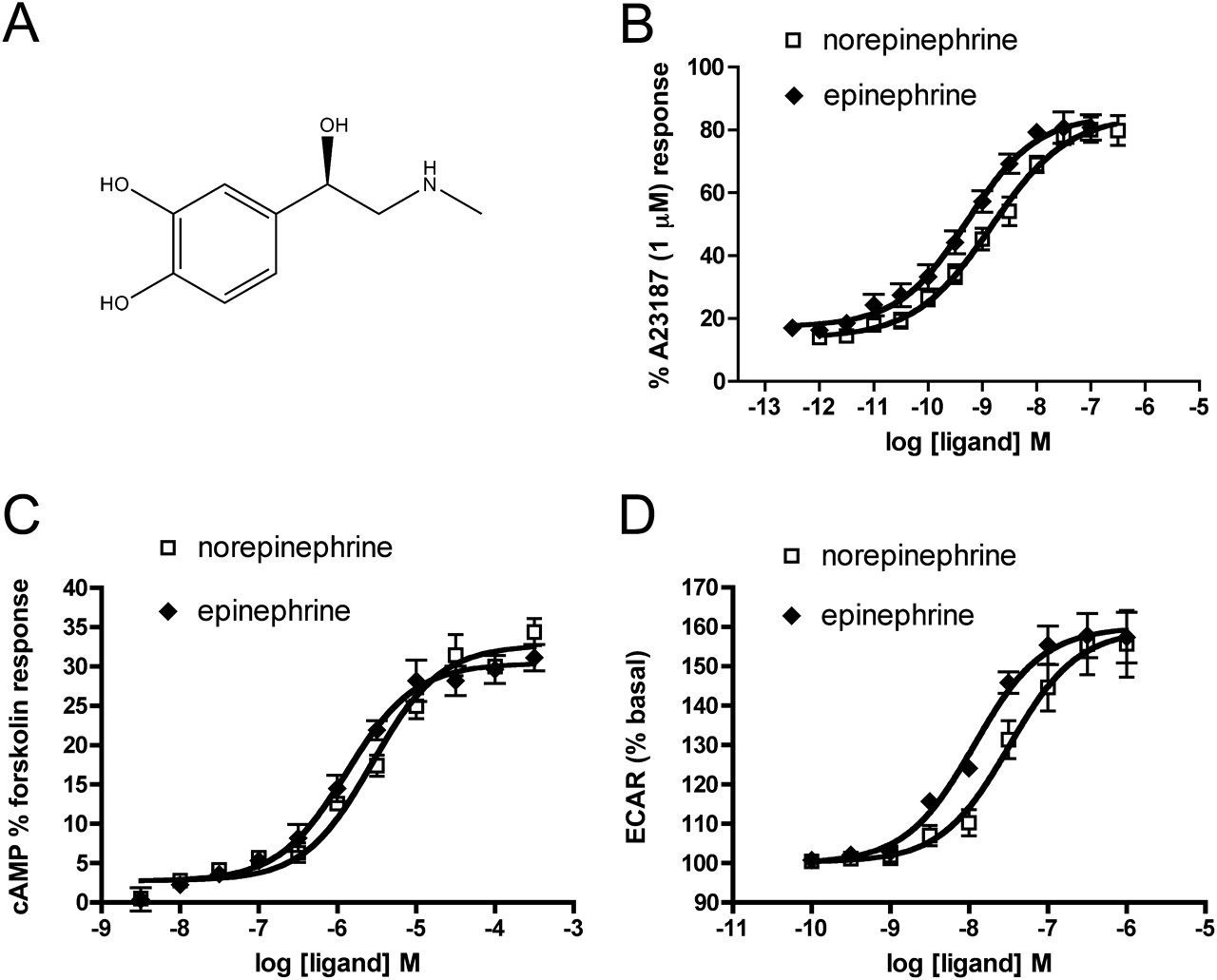
Comparison of the activity of the endogenous agonists norepinephrine and epinephrine at the human α1A-AR. A, structure of epinephrine. B–D, concentration-response curves for Ca2+ release, cAMP, and changes in ECAR were determined as described in the legends to Figs. 2⇑ to 4. Values are means ± S.E.M. of seven to nine independent experiments.
Potency (pEC50), maximum response (Emax), and α value (Emax relative to norepinephrine) of the endogenous agonists norepinephrine and epinephrine at the α1A-AR
Transduction ratios for norepinephrine and epinephrine at the α1A-AR
LogTR is the log of the transduction ratio (τ/KA), and logTRn values are normalized to the logTR for norepinephrine.
Calculation of bias factors for norepinephrine and epinephrine at the α1A-AR
LogTR is the log of the transduction ratio (τ/KA), and logTRn values are normalized to the logTR for norepinephrine. To avoid propagation of error due to multiple data manipulation steps, Student's t tests were carried out on the raw logTR data rather than data normalized to norepinephrine (see Supplemental Table 1). P < 0.05 was considered significant.
Discussion
We show in this study that phenethylamine and imidazoline agonists display functional selectivity at the human α1A-AR. In particular, phenylephrine shows substantial bias toward ECAR versus Ca2+ release or cAMP accumulation compared with norepinephrine but does not display any bias between the Ca2+ and cAMP pathways. The additional endogenous agonist, epinephrine, does not show bias relative to norepinephrine in any of the signaling outputs measured. Our findings indicate no overall class effect between the phenethylamines and imidazolines, because oxymetazoline is unable to stimulate cAMP accumulation, whereas it shows significant bias toward ECAR relative to Ca2+ release. The other two imidazolines, however, both display bias toward cAMP relative to Ca2+ release but no bias toward ECAR. It is noteworthy that we can now draw conclusions about the functional selectivity of α1A-AR agonists because we have an analytical method that provides a numerical measure of bias.
Although many α1A-AR responses, including intracellular Ca2+ release, are produced via the classic Gq pathway, coupling of α1-ARs to additional signaling pathways has been observed in recombinant systems and tissues expressing endogenous receptors (Morgan et al., 1983; Johnson and Minneman, 1986). Although early studies suggested that α1-AR mediated cAMP accumulation was secondary to Gq coupling (Perez et al., 1993), other studies favor α1-AR-Gs coupling. Horie et al. (1995) showed that in CHO-α1B-AR cells, norepinephrine-stimulated cAMP accumulation was inhibited by an antibody against the Gαs C terminus. The phospholipase C inhibitor 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino] hexyl]-1H-pyrrole-2,5-dione (U73122) abolished increases in intracellular Ca2+ but had no effect on cAMP accumulation. In CHO-K1 cells expressing each of the α1-ARs, phenylephrine stimulation of the α1A- and α1B-AR increased inositol phosphate and cAMP accumulation, whereas the α1D-AR activated only inositol phosphate production (Shibata et al., 2003). Thus in CHO-α1D-AR cells, activation of the Gq pathway was not sufficient for stimulation of cAMP. Our data on the α1A-AR also support a dissociation between the Gq and cAMP pathways, in that cirazoline and A61603 promoted significant bias relative to norepinephrine between Ca2+ release and cAMP accumulation. In addition, oxymetazoline was able to stimulate Ca2+ release but failed to increase cAMP accumulation. It is noteworthy that although oxymetazoline, cirazoline, and A61603 share some common structural elements (Fig. 1), they clearly produce distinct α1A-AR conformations that display varying efficiency for cAMP accumulation.
There is increasing understanding of the way in which drugs activate GPCRs derived from recent crystal structures of the β1-AR, β2-AR, and adenosine A2A receptors (Rosenbaum et al., 2007; Jaakola et al., 2008; Warne et al., 2008), biophysical and structure-function studies (Swaminath et al., 2004; Yao et al., 2006; Yao et al., 2009), computer modeling and virtual ligand screening (Katritch et al., 2009; Reynolds et al., 2009). Furthermore, it has been possible to compare the structures of inactive rhodopsin and its active opsin counterpart (Scheerer et al., 2008). Binding of an agonist occurs as a multistage process in which successive conformational changes increase the stability of the drug-receptor interaction (Liapakis et al., 2004; Swaminath et al., 2005). The conformational changes at the extracellular ends of helices III, IV, V, and VII are transmitted via changes in the interactions between key amino acid side chains and water molecules in a hydrogen-bonded network that extends toward the cytoplasmic domain of the receptor (Rosenbaum et al., 2007). The active opsin structure indicates that upon receptor activation, the C terminus of Gα subunits binds within a cavity created by outward tilting of helix VI, altered positioning of helix V, and restructuring of the link between helices VII and VIII (Scheerer et al., 2008).
It is reasonable to suppose that each distinct agonist produces at least subtle differences in the conformation of the ligand binding pocket; however, the key question posed by the demonstration of functional selectivity is whether each agonist also produces a unique conformation at the cytoplasmic face of the receptor. Rosenbaum and coworkers (2007) suggest that the core hydrogen-bonded network allows structural flexibility to the receptor, potentially dampening the capacity of different ligands to induce distinct active conformations. This may be the case, for example, between norepinephrine and epinephrine, which show no bias at the α1A-AR. However our demonstration of functional selectivity between norepinephrine and the other agonists tested is consistent with the idea that the receptor conformational changes induced by distinct compounds can be transmitted to unique active conformations. We suggest that any demonstration of bias for one pair of signaling outputs must indicate the presence of distinct active conformations. For example, there is clear bias between phenylephrine and norepinephrine when we compare ECAR with either Ca2+ release or cAMP accumulation, but not when we compare Ca2+ release and cAMP accumulation with each other. This cannot imply that the active conformations promoted by phenylephrine and norepinephrine are the same but rather that effector proteins for Ca2+ release and cAMP signaling are not sensitive to the conformational difference. In contrast, there is clear bias between two of the imidazolines (cirazoline and A61603) and norepinephrine when we compare Ca2+ release with cAMP accumulation. In this case, the particular receptor conformations induced by cirazoline and A61603 must favor coupling to effectors for the cAMP pathway over Gαq coupling.
Our findings concerning the activity of norepinephrine, epinephrine, and phenylephrine at the α1A-AR are of considerable interest from a structural viewpoint. Phenylephrine but not epinephrine showed substantial bias toward ECAR compared with the other two pathways. Whereas the structure of phenylephrine differs from norepinephrine by two elements, the only difference between epinephrine and phenylephrine is the para-hydroxyl on the catechol ring. Modeling and structure-function studies of the β2-AR have shown that each element within the catecholamine structure interacts with particular amino acid side chains in the ligand-binding pocket (Liapakis et al., 2004; Swaminath et al., 2004). The hydroxyl groups on the catechol ring undergo hydrogen bonding with Ser203 (5.42), Ser204 (5.43), and Ser207 (5.46), the chiral β-hydroxyl interacts with Asn293 (6.55), the aromatic ring undergoes hydrophobic interaction with Phe290 (6.52), and the bioamine −NH3+ group interacts with Asp113 (3.32). In addition, the amine substituent group present in full agonists such as epinephrine and isoproterenol may interact with unidentified residues in TM6 and/or TM7 (Liapakis et al., 2004; Swaminath et al., 2004). The α1A-AR has only two serine residues in TM5, Ser188(5.42) and Ser192(5.46), whereas the α1B-AR and α1D-AR, like the β2-AR, have all three serines. Our data suggest that the absence of the para-hydroxyl in phenylephrine is associated with a key conformational difference in TM5 compared with epinephrine, which has an otherwise identical structure. Although this difference does not affect the capacity of phenylephrine to promote Gαq coupling, it does increase coupling to effectors for ECAR. It would be interesting to test whether a ligand that lacks both the para-hydroxyl and the N-methyl group (norphenylephrine) also shows bias toward ECAR, and to compare the behavior of all these compounds at the α1A-AR and α1B-AR that differ in the number of TM5 serine residues. We also intend to use high-throughput signaling assays to identify the nature of effectors that promote changes in ECAR and are strongly coupled to the α1A-AR in the presence of phenylephrine.
Another major finding from our study is that norepinephrine and epinephrine display no bias in signaling by the α1A-AR for the three pathways studied. This raises the question of whether multiple endogenous agonists have in other cases evolved as a result of their capacity for differential signal transduction at particular receptors. Indeed, we have recently demonstrated signaling bias for multiple endogenous agonists of the GLP1 receptor (Koole et al., 2010), and this may be relevant to other GPCRs that possess multiple endogenous ligands, including the chemokine CXCR2 and CCR7 receptors, cholecystokinin CCK1(A), cholecystokinin CCK2(B), endothelin ETA and ETB, melanocortin MC4, purinergic P2Y2, GLP2, VPAC1, VPAC2 receptors, and calcitonin and related receptors (list derived from http://www.iuphar-db.org/DATABASE/GPCRListForward). The case of adrenoceptors is somewhat different, because both norepinephrine and epinephrine act at nine different receptor subtypes and may display functional selectivity at subtypes other than the α1A-AR. A major functional difference between the two endogenous agonists is that norepinephrine is a neurotransmitter that is released into synapses and neuromuscular junctions, whereas epinephrine is a circulating hormone that acts on the adrenoceptors present in a wide range of peripheral cell types. Selective pressure to maintain the interaction between adrenoceptor subtypes and two distinct agonists may have arisen primarily because of differences in the cell distribution of the target receptors rather than functional selectivity.
In conclusion, we have shown that a series of agonists at the human α1A-AR display functional selectivity across three signaling pathways. The analytical method used has allowed us to derive numerical values for the degree of signaling bias and to test for statistical significance. As shown here, a rigorous description of functional selectivity facilitates correlations between ligand structure and the ability to promote distinct active receptor conformations. More generally, this type of analysis will provide a platform for elucidating the mechanistic basis of functional selectivity at GPCRs by suggesting suitable ligands for receptor crystallization, or classes of ligands for modeling studies.
Authorship Contributions
Participated in research design: Evans, Broxton, Hutchinson, and Summers.
Conducted experiments: Broxton, Merlin, Sato, and Hutchinson.
Contributed new reagents or analytic tools: Christopoulos.
Performed data analysis: Evans and Christopoulos.
Wrote or contributed to the writing of the manuscript: Evans, Broxton, Hutchinson, Christopoulos, and Summers.
Acknowledgments
We thank Maria Papaioannou for excellent technical assistance, Dr. Nathan Hall for helpful discussions, and Dr. Thomas Chang (Roche Bioscience, Palo Alto, CA) for the gift of plasmid containing the human α1A-4-AR cDNA.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was funded by the National Health and Medical Research Council (NHMRC) of Australia [Program Grant 519461]; an NHMRC Senior Research Fellowship (to A.C.); and an NHMRC Career Development Award (to D.S.H.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.067454.
-
ABBREVIATIONS:
- ECAR
- extracellular acidification rate
- IBMX
- 3-isobutyl-1-methylxanthine
- AR
- adrenoceptor
- CHO
- Chinese hamster ovary
- FBS
- fetal bovine serum
- HBSS
- Hanks' balanced saline solution
- A61603
- N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydronaphthalen-1-yl]methanesulfonamide
- A23187
- calcimycin
- U73122
- 1-[6-[[17β-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione
- TM
- transmembrane domain.
- Received July 8, 2010.
- Accepted October 26, 2010.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}