


Abstract
This article describes functional selectivity of agonists and antagonists and distinguishes conventional cell-based functional selectivity, where the strength of signal produces selective signaling in various organs, from true receptor active-state based selectivity, also alternatively referred to in the literature as “stimulus trafficking,” “biased agonism,” and “collateral efficacy.” This latter mechanism of selectivity depends on the ligand-related conformation of the receptor and is not compatible with the parsimonious view that agonists produce a single receptor active state. In addition, protean agonism is described, whereby a ligand produces positive agonism in quiescent systems and inverse agonism in constitutively active systems. This is a special case of active state-based selectivity in which the ligand produces an active state that is of lower efficacy than the natural constitutively active state. It is postulated that receptor active-state based selectivity, unlike cell-based functional selectivity, is controllable through the chemical structure of the ligand and is therefore more likely to be a viable avenue for therapeutic selectivity in the clinic. Reasons are given for differentiating receptor active-state based selectivity from conventional functional organ selectivity.
There is increasing evidence to show that agonists need not simply be mimics of endogenous neurotransmitters and hormones but rather can cause receptors to exercise only portions of their often vast repertoire of behaviors. In other instances, they can emphasize the interaction of the receptor with certain signaling pathways (Fig. 1A). Likewise, data show that antagonists need not function only as eliminators of function but rather can modulate and otherwise edit endogenous signals (Fig. 1B). These effects have been given various names in different contexts from “stimulus-trafficking,” “biased agonism,” “collateral efficacy” to a generally accepted “functional selectivity.” This latter term, although correct, unfortunately encompasses a breadth of effects described in receptor pharmacology, some of which differ from the concept of receptor-based selectivity. Stimulus-trafficking (Kenakin, 1995a) was originally defined to account for receptor behavior that was incompatible with classic receptor theory, which states that a single receptor active state controls all activation behaviors of a receptor. Pharmacological procedures that use agonist potency ratios to classify agonists and receptors are based on this assumption. However, over the past 10 years, observations that some agonists demonstrate different relative potencies for various cellular pathways (actual reversal of relative potencies can be observed) cannot be reconciled with a single receptor active state and require the involvement of multiple agonist-induced receptor active states.
Single Receptor State Receptor Selectivity
Under certain circumstances, a single receptor active state can lead to functional selectivity (a mechanism differentiated in the original definition of stimulus-trafficking as a “strength of signal” mechanism; Kenakin, 1995a). A single activated state can produce selective effects only if the efficiency of coupling is appropriate. Therefore, it is a completely cell-based phenomenon. This well known consequence in single-stimulus systems results in a standard profile whereby a weak partial agonist can produce effect in some (well coupled) tissues and can function as an antagonist (with no direct agonism) in less well coupled tissues. The interplay of weak efficacy with varying levels of affinity can give the overall pattern of organ selectivity, as is seen with the muscarinic agonists carbachol and oxotremorine (see Fig. 2A). Whereas carbachol has low affinity and high efficacy, oxotremorine possesses high affinity and low efficacy. Because potency in well coupled tissues is a complex function of affinity and efficacy and the pEC50 of agonists reflects both, no distinction can be made with respect to the relative contribution of affinity or efficacy to overall potency. In contrast, in less well coupled tissues, the maximal response is solely a function of efficacy, whereas the location parameter of the concentration-response curve (pEC50) is solely the function of affinity. Therefore, a dissociation of potency occurs when an agonist shows partial agonism in a tissue versus when it functions as a full agonist in another tissue. As seen in Fig. 2A, oxotremorine and carbachol are both full agonists for contraction of guinea pig ileum; oxotremorine is the more potent agonist. Diminution of the functional muscarinic receptor density with a controlled treatment with the alkylating agent phenoxybenazamine yields a tissue with fewer receptors that is less responsive to muscarinic agonism. As further seen in Fig. 2A, the tissue now only responds to the higher efficacy agonist (carbachol) and shows no effect to oxotremorine. If the two conditions were observed with two tissues of differing sensitivity to muscarinic agonism, a functional selectivity would be concluded.
This mechanism can lead to more complex signaling events for receptors that pleiotropically couple to multiple mechanisms (i.e., G-proteins) in the cell. Under these conditions, the receptor level can control not only the quantity of observed response but also the quality of response. Figure 2B shows the Gi-protein activating effects of increasing cell surface expression of α2-adrenoceptors on cyclic AMP response; the biphasic response occurs only after sufficient receptor is present to activate both Gi- and Gs-protein (Eason et al., 1992). Similar recruitment of G-protein with increasing receptor density has been shown with calcitonin receptors. In particular, low levels of expression result in solitary coupling to Gs protein [calcitonin activation leads to increased cyclic AMP in human embryonic kidney (HEK) cells]. However, higher levels of receptor expression lead to elevation of cyclic AMP and calcium response mediated by Gq receptors (Kenakin, 1996). Receptor density-linked activation of cellular pathways also has been shown for opioid receptors. In NG108 cells, the opioid agonist [d-Ala2-d-Leu5]-enkephalin produces inhibition of adenylate cyclase and stimulation of high-affinity GTPase. Upon reduction of opioid receptor density through alkylation, the less well coupled GTPase response is eliminated and the sole response becomes adenylate cyclase inhibition (Costa et al., 1988). In each of these cases, the agonists involved demonstrate true organ or assay-dependent selectivity. However, this behavior still is consistent with the production of a single receptor active state by the agonists. The strength of the receptor stimulus and the responsiveness of the cell (as controlled either by receptor density and/or efficiency of receptor coupling) combines to produce the demonstrated selective responses. In no instance does the actual rank order of activity reverse; for this to occur, more than one receptor active state must be involved.

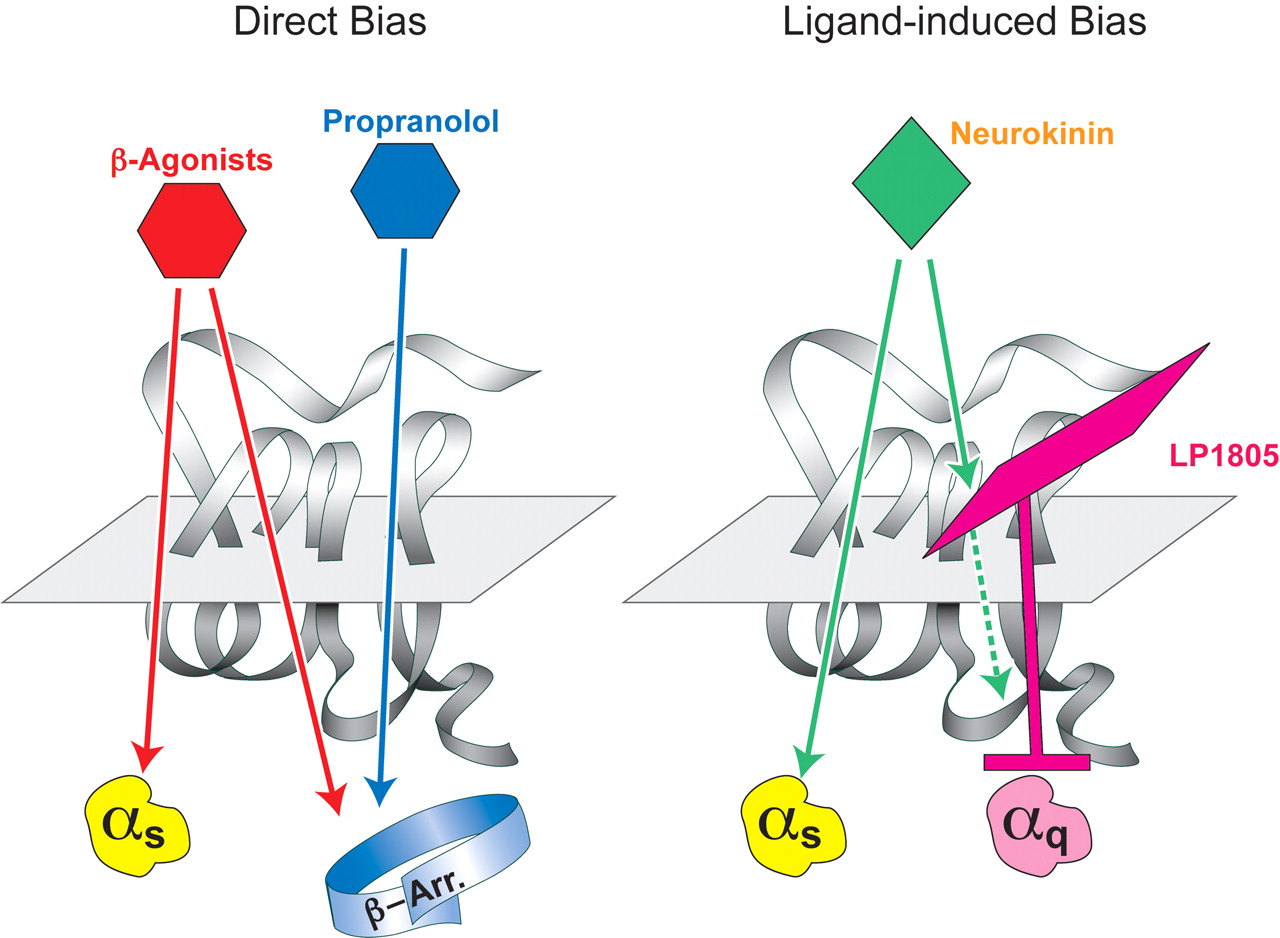
Schematic diagram of directly induced and indirectly induced stimulus bias by a ligand. Direct bias is demonstrated by the effects of β-adrenoceptor ligands. Although standard β-adrenoceptor agonists initiate Gs-protein and β-arrestin signaling, the antagonist propanolol does not activate Gs-proteins (in fact it is an inverse agonist for this signaling pathway) but does activate the G-protein independent β-arrestin signaling pathway (data from Azzi et al., 2003; Baker et al., 2003). Indirect imposition of biased signaling occurs when an allosteric ligand cobinds with the agonist to the receptor to modify the signaling properties of the agonist. This is demonstrated by the modulator LP1805 (N,N-(2-methyl naphthyl-benzyl)-2-aminoacetonitrile) which changes the signaling pattern for the endogenous agonist neurokinin A from activation of Gs and Gq protein to only activation of Gs protein. Data from Maillet et al., 2007.
Receptor-Based Biased Functional Selectivity
Operational theory, as presented by Black and Leff (1983), gives agonist response as: 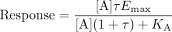 where KA is the equilibrium dissociation constant of the agonist-receptor complex (1/affinity), Emax is the maximal response attainable in the system, and τ is a measure of both the efficacy of the agonist and the sensitivity of the system to agonism. The term τ is the receptor density divided by KE, the equilibrium dissociation constant of the agonist-occupied receptor and the saturable stimulus-response mechanism(s) of the cell. This constant contains both the measure of the overall sensitivity of the cell to agonism and the intrinsic efficacy of the agonist. Thus, a ratio of KE values (actually τ values with a cancellation of the receptor density term) is a system-independent measure of the relative intrinsic efficacies of the two agonists. It is important to note that for a single receptor active state, the KE for a given agonist must be constant for all pathways in a cell. The existence of different τ values for various pathways is not compatible with a single receptor active state for that receptor.
where KA is the equilibrium dissociation constant of the agonist-receptor complex (1/affinity), Emax is the maximal response attainable in the system, and τ is a measure of both the efficacy of the agonist and the sensitivity of the system to agonism. The term τ is the receptor density divided by KE, the equilibrium dissociation constant of the agonist-occupied receptor and the saturable stimulus-response mechanism(s) of the cell. This constant contains both the measure of the overall sensitivity of the cell to agonism and the intrinsic efficacy of the agonist. Thus, a ratio of KE values (actually τ values with a cancellation of the receptor density term) is a system-independent measure of the relative intrinsic efficacies of the two agonists. It is important to note that for a single receptor active state, the KE for a given agonist must be constant for all pathways in a cell. The existence of different τ values for various pathways is not compatible with a single receptor active state for that receptor.
A classic hallmark of trafficking of stimulus is the observation of a reversal of relative potencies of full agonists. Eq. 1 can be used to predict the relative potency of full agonists (as the ratio of molar concentration producing 50% maximal response, EC50). For agonists [A1] and [A2], the ratio of EC50 values is:  From this equation it can be seen that the relative potency depends solely on parameters unique to the agonists and the receptor (namely KA and τ); thus, it is a system-independent parameter. Therefore, if the agonists produce a single receptor active state, the potency ratio for the production of that state must be constant for all pathways mediated by that active state. The corollary to this is that reversal in the potency ratio for different agonist pathways is incompatible with a single receptor active state. This was the experimental basis for proposing stimulus trafficking on theoretical grounds (Kenakin, 1995a); specifically, Spengler et al. (1993) showed that the order of potency for PACAP agonists PACAP(1–27) and PACAP(1–38) reversed for PACAP-mediated elevated cyclic AMP and inositol phosphate production in LLC-PK1 cells. It was shown that PACAP(1–27) was more potent for the cyclic AMP pathway but that the reverse was true for the inositol phosphate pathway.
From this equation it can be seen that the relative potency depends solely on parameters unique to the agonists and the receptor (namely KA and τ); thus, it is a system-independent parameter. Therefore, if the agonists produce a single receptor active state, the potency ratio for the production of that state must be constant for all pathways mediated by that active state. The corollary to this is that reversal in the potency ratio for different agonist pathways is incompatible with a single receptor active state. This was the experimental basis for proposing stimulus trafficking on theoretical grounds (Kenakin, 1995a); specifically, Spengler et al. (1993) showed that the order of potency for PACAP agonists PACAP(1–27) and PACAP(1–38) reversed for PACAP-mediated elevated cyclic AMP and inositol phosphate production in LLC-PK1 cells. It was shown that PACAP(1–27) was more potent for the cyclic AMP pathway but that the reverse was true for the inositol phosphate pathway.
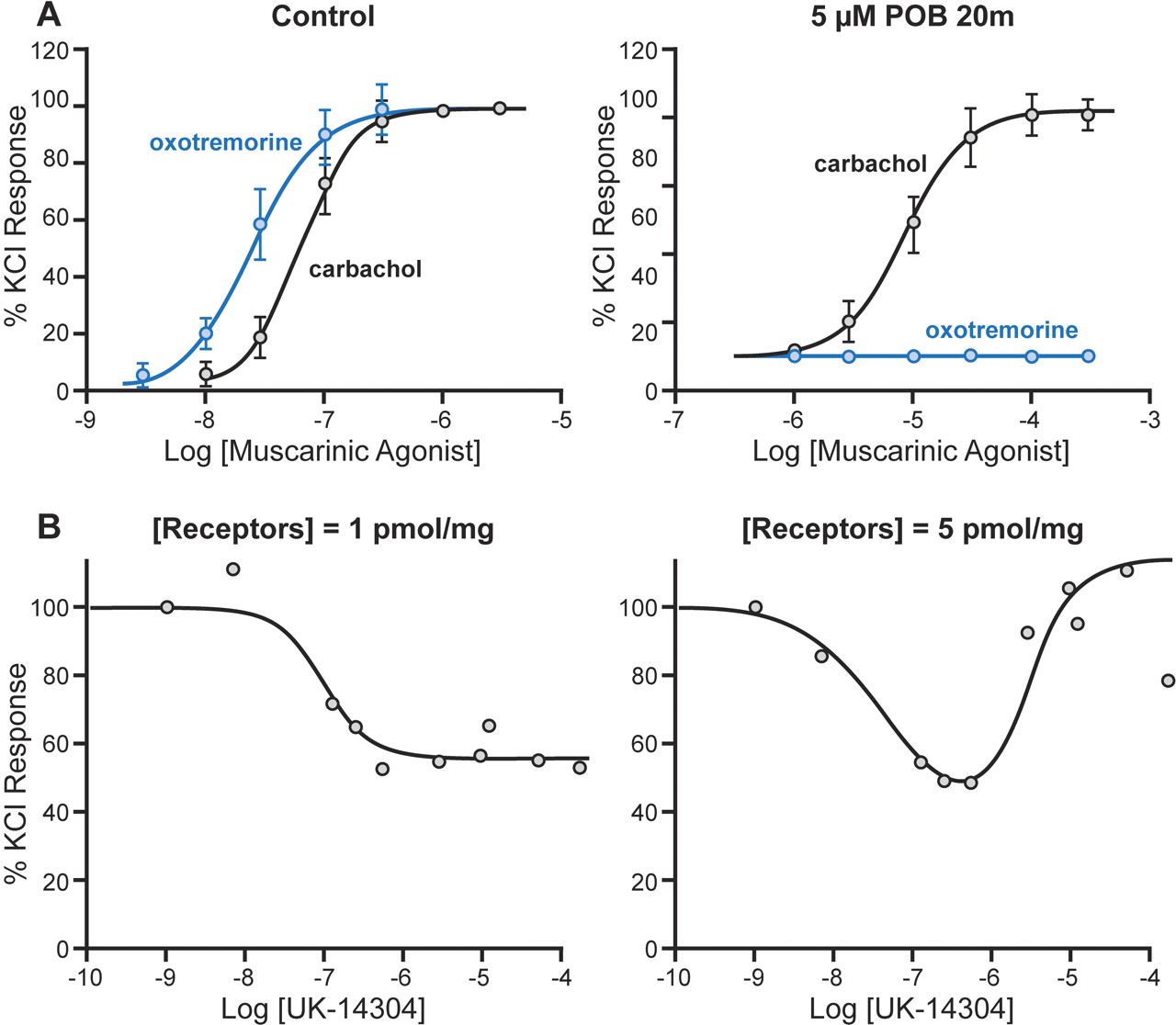
A, relative potency of muscarinic agonists carbachol and oxotremorine producing contraction of guinea pig isolated ileum. Left, relative effects in untreated ileum; right, effects after exposure to the muscarinic receptor alkylating agent phenoxybenzamine (5 μM for 20 min followed by1hof washing). The activation profile changes from oxotremorine > carbachol to carbachol >> oxotremorine after reduction of muscarinic receptor density. Data from Kenakin (1997). B, effects of the α2-adrenoceptor agonist UK-14304 on adenylate cyclase in transfected Chinese hamster ovary cells expressing different levels of α2-C10 receptors. At low expression levels (1 pmol/mg), only Gi-protein-mediated inhibition of adenylate cyclase is observed; at higher receptor levels (5 pmol/mg), a biphasic response is seen with increases due to activation of Gs-protein. New graphs from the data of Eason et al. (1992).
Figure 3A gives an example of data that is incompatible with a single receptor active state. In particular, calcium transient responses to two agonists for the human calcitonin receptor are measured in two types of HEK cells: normal, wild-type HEK cells and those cotransfected with Gαs protein. It can be seen that whereas eel calcitonin is more potent than porcine calcitonin in wild-type cells, these agonists reverse their relative potency in cells enriched in Gαs protein. These data indicate that the agonists produce at least two active states, one of which has a higher affinity for the Gαs subunit (Watson et al., 2000). In general, such reversals of full agonist potency are indicators of heterogeneous receptor active states that result in stimulus trafficking.
Another experimental finding that is incompatible with agonist production of identical receptor active states is a difference in the maximal capability of agonists to stimulate different pathways in cells. From eq. 1, the relative maximal responses for two agonists [A1] and [A2] (i.e., as [A]→∞) is calculated as: 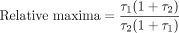
It can be seen from this equation that the relative maximum is strictly a function of the efficacy of the agonist. If, for two given agonists, the ratio of the relative maxima is >1 (MaxA1 > MaxA2), then it can be shown that τ1 > τ2. A change in the relative maximum would necessitate a change in the relative efficacy of the agonists (i.e., a change in the nature of the agonist-activated receptor producing response). Therefore, a reversal of relative maximal responses for two pathways for any two agonists is incompatible with a single receptor active state and strongly indicates that the two agonists produce different primary active states (i.e., true receptor-based functional selectivity). Figure 3B shows an example of such a reversal of the maximal capabilities of serotonin agonists for arachidonic acid and inositol triphosphate production in response to activation of the 5-HT2C receptor (Berg et al., 1998). This effect is incompatible with the idea that theses two agonists produce the same receptor active state for the activation of these cellular pathways.

Reversal of potencies of agonists not compatible with production of a single uniform receptor active state. A, relative potency of eel and porcine calcitonin (calcium response in HEK cells transfected with human calcitonin receptors) in wild-type cells and cells cotransfected with Gαs-protein. It can be seen that enrichment of the Gαs-protein selectively increases the potency of porcine calcitonin to the point where the relative potencies of these full agonists is reversed. New graphs from the data of Watson et al. (2000). B, reversed maximal capability of agonists to produce arachidonate release and inositol phosphate (IP) accumulation through activation of 5-HT2C receptors in Chinese hamster ovary cells. Although 3-trifluoromethylphenylpiperazine (TFMPP) has greater efficacy for IP accumulation than arachidonate release, the reverse is true for (±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI). This type of reversal clearly indicates that these agonists produce different receptor active states. New graphs from the data of Berg et al. (1998).
Protean Agonism
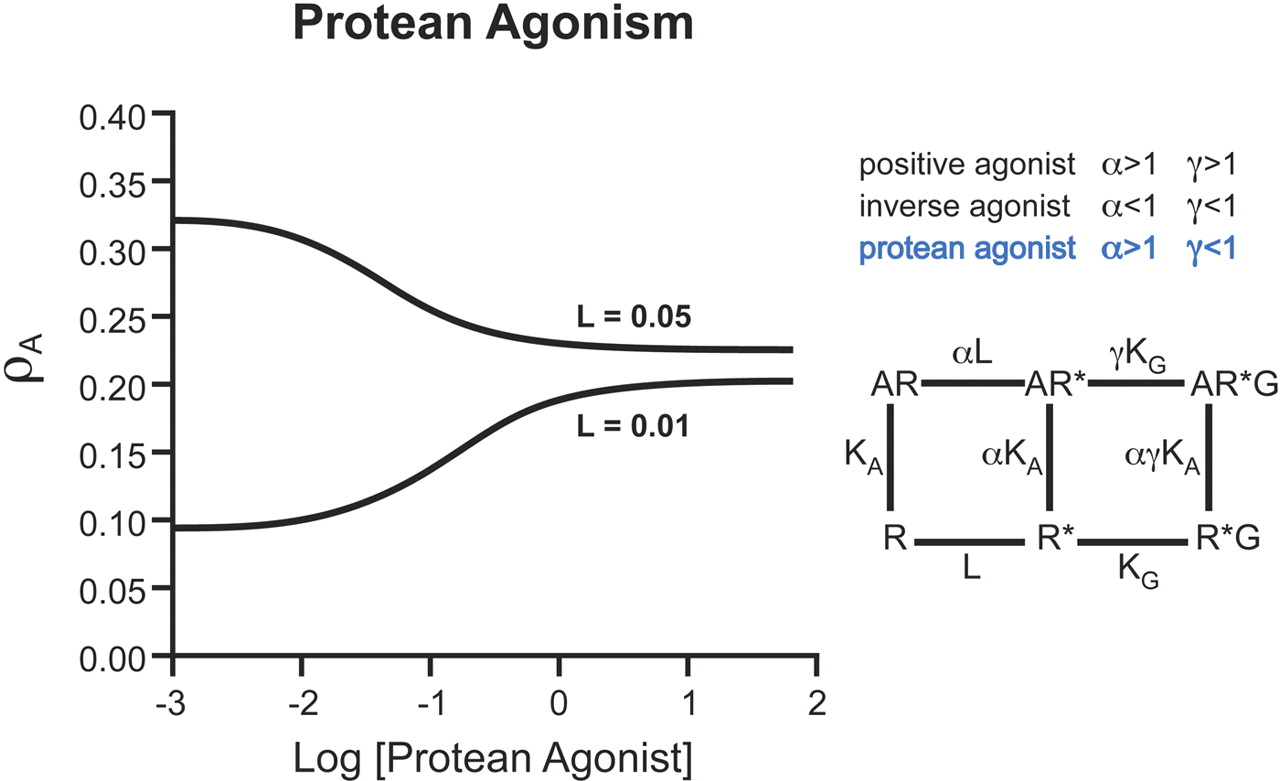
A special case of receptor-based functional selectivity is “protean” agonism. These are ligands that produce a receptor active state that is capable of initiating signal where there is none but from a receptor active state that is less efficacious than the naturally occurring, spontaneously formed constitutive active state (Kenakin,1995b, 2001). In a quiescent system, consisting mainly of receptors in the inactive state, protean agonists produce positive agonism. In contrast, in a constitutively active system consisting of a substantial amount of spontaneously formed receptor active state, protean agonists produce inverse agonism. This is because they convert the efficacious active state to a less efficacious ligand-selective active state (see Fig. 4). Because the ligand effect changes in response to the system, these molecules were named after the Greek sea-god Proteus (son of Poseidon), who could change shape at will depending on his environment and needs (Kenakin, 1995b). Examples of protean agonists, such as dichloroisoproterenol, have been seen experimentally (Chidiac et al., 1996). Protean agonists theoretically should be the best equalizers of endogenous effect because they would reduce effects due to endogenous agonist tone and effects due to constitutive activity; the latter activity would not be observed with normal partial agonists because a partial agonist would be incapable of altering the constitutive receptor activity. It is important to note that the term protean refers to a specialized receptor ligand and not just a generic functionally selective ligand.
Indirect Ligand-Induced Bias
Orthosteric antagonists occlude the agonist binding site, thereby preventing receptor activation. No “texture” in the antagonism is possible with this mechanism because the result is an unresponsive receptor. This is not necessarily true of an allosteric modulator that binds to its own site on the receptor and allows the agonist to bind as well. With this mechanism, the modulator may modify the response to agonist with a range of effects from complete inhibition to potentiation. The effect is caused by a modulator-induced change in the conformation of the receptor; i.e., the modulator stabilizes an allosteric conformation that has modified responsiveness to the agonist. The change in conformation of the receptor also may modify the interaction of the receptor with cellular membrane interactants such as G-proteins, GRKs, β-arrestin(s) and other proteins. Because these molecules bind to different loci on the receptor, the changes in responsiveness need not be uniform (in fact, it might be predicted that the changes should not be). This idea was the basis for using different G-protein enrichment to detect agonist-selective receptor active states shown in Fig. 3A. Because different regions of the receptor were known to interact with various G-proteins, the postulate was that diverse receptor conformations of the receptor would not expose these regions in an identical manner and that the heterogeneity of exposure with dissimilar states would be reflected in variable reliance of response on different G-proteins (this is a case of using the G-protein complement of the cell to detect different conformations). As seen in Fig. 3A, this was confirmed as ligand-directed stimulus trafficking was made obvious by diversity in G-protein content of cells. The corollary to this idea, then, is that a change in conformation will not present identical changes to different signaling partners for the receptor in the cell. Under these circumstances, an allosteric change in receptor conformation could alter the array of responses produced by the agonist (Fig. 1B). For example, neurokinin produces activation of Gs and Gq protein through NK1 receptors. However, the allosteric modulator LP1805 changes this pattern to one of enhanced Gq response and antagonism of Gs activation (Maillet et al., 2007). Likewise, prostaglandin D2 interacts with CRTH2 receptors to activate Gi-protein and β-arrestin. Binding of the modulator sodium tosyltryptophan causes PDG2 to lose its ability to initiate receptor interaction with β-arrestin but not Gi-protein (Mathiesen et al., 2005). In both of these cases, the allosteric modulator imposes collateral efficacy (partial expression of all possible receptor behaviors) onto the natural agonist.

Molecular description of Protean agonism. Ligands that enrich existence of the active state R* (through α and γ>1) produce positive agonism. Likewise, ligands that destabilize R* and shift equilibria toward R (α and γ<1) will produce inverse agonism. Ligands that enrich an active state (α>1) that has a reduced affinity for the G-protein than the spontaneously formed active state (γ<1) will produce agonism in systems not containing spontaneous R* (will be agonists in nonconstitutively active systems). However, this ligand will produce a receptor species less likely than R* to induce response and therefore will be an inverse agonist in constitutively active systems.
Receptor-Based Selectivity
There have been reviews citing many instances of true receptor-based selectivity of trafficking with respect to signaling pathways in cells (Kenakin, 2002a, 2003, 2006; Perez and Karnik, 2005; Urban et al., 2007). The basic difference between this and cell-based selectivity is that the ligand “steers the ship” from the point of view of controlling the effect. In contrast, cell-based selectivity relies on the stoichiometry and sensitivity of the cellular components driving the response. Although any given system can yield therapeutically favorable instances of selectivity, the effect is still under the control of cell physiology and pathology. Because drugs usually are developed in cell systems not controlled by pathological mechanisms and then used in systems that are, it is difficult to correlate cell-based selectivity seen in test systems with corresponding selectivity in the therapeutic system (i.e., there may or may not be correspondence).
A better starting point for the design of therapeutically useful functionally selective drugs is to have the ligand itself direct the stimulus. Under these circumstances, selectivity occurs in all systems with no dependence on the relative emphasis that any given cell places on a signaling pathway. The control of the effect is governed by the differential affinities of the ligand-bound receptor for various cellular pathway effectors. Thus, the activated receptor will ignore some pathways and preferentially activate others. From this standpoint, receptor-based selectivity is unique and should be differentiated from general functional selectivity.
It is useful to mathematically model receptor coupling to extend predictions to multiple effector systems. Thus, a receptor that binds a ligand [A] to form a ligand-bound complex [AR] can go on to bind to any number of “effectors” in the cell (designated E1 to En); these range from different G-proteins to β-arrestin(s) and GRKs. The receptor is conserved in that the limiting constraint on the system is the amount of receptor available to couple to the cellular components; this allows for the modeling of the effects of changing receptor density. Finally, the response can be given as a logistically forced function of the effector complex (for example, for effector Ei, the complex AREi goes on to stimulate a pathway in the cell that leads to response of the form ([AREi]/([AREi] + φi), where φi is a fitting parameter). The addition of this function does not alter the conclusions made from this model but does eliminate the necessity of assuming a one-to-one relationship between response and amount of effector complex. It can be shown that the fractional response for a pathway (designated pathway i) is given by (derived in Appendix): 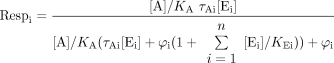 where the size of the effector pool is given by [Ei], and τAi is the efficacy of the agonist for the response pathway. This model allows the prediction of the effects of ligands that produce a single versus multiple active states in systems of varying receptor density and/or receptor coupling efficiencies when there are a number of effectors coupling to the receptor.
where the size of the effector pool is given by [Ei], and τAi is the efficacy of the agonist for the response pathway. This model allows the prediction of the effects of ligands that produce a single versus multiple active states in systems of varying receptor density and/or receptor coupling efficiencies when there are a number of effectors coupling to the receptor.
In comparing two agonists, the ratio of degree of stimulation of a given pathway (for equivalent values of [A]/KA), can be calculated with equations analogous to eqs. 2 and 3 for relative potency and relative maxima (see Appendix). Thus, the relative potency of agonists A and B in a multieffector system is given by:  given:
given: 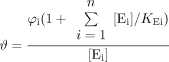 where ϑ is a tissue constant for all agonists in any given tissue. This term relates to the particular cellular milieu of coupling mechanisms available to the receptor. The ratio of maxima of the agonists for any pathway is given by:
where ϑ is a tissue constant for all agonists in any given tissue. This term relates to the particular cellular milieu of coupling mechanisms available to the receptor. The ratio of maxima of the agonists for any pathway is given by:  It can be seen from an examination of eqs. 5 and 7, for any given pathway i, if the ratio is measured to be >1, this can occur only if τAi > τBi. Therefore, a reversal of the relative potency or maximal responses can occur only if the relative efficacies for that pathway reverse (i.e., τAi < τBi). Because τ = [Rtot]/KEi and [Rtot] is constant for both agonists, such a reversal can occur only if the equilibrium dissociation constant of the agonist-occupied receptor changes. If this is observed, it would indicate a change in the nature of the agonist-activated receptor (i.e., a different receptor active state). It should also be noted that cell-based selectivity would depend only on values of ϑ for cells and is not controllable through the structure of the agonist.
It can be seen from an examination of eqs. 5 and 7, for any given pathway i, if the ratio is measured to be >1, this can occur only if τAi > τBi. Therefore, a reversal of the relative potency or maximal responses can occur only if the relative efficacies for that pathway reverse (i.e., τAi < τBi). Because τ = [Rtot]/KEi and [Rtot] is constant for both agonists, such a reversal can occur only if the equilibrium dissociation constant of the agonist-occupied receptor changes. If this is observed, it would indicate a change in the nature of the agonist-activated receptor (i.e., a different receptor active state). It should also be noted that cell-based selectivity would depend only on values of ϑ for cells and is not controllable through the structure of the agonist.
Should Functional Selectivity Be an Expected Event?
Seven-transmembrane receptors are allosteric proteins and are therefore capable of adopting different conformations. An important outcome of this behavior is that the changes in conformation can be global in nature; i.e., changes in numerous regions of the protein may occur simultaneously. Thus, a modulator may stabilize one or more pre-existing but possibly rare conformations of the receptor, and those may show altered positions of amino acids in numerous locations. It is useful to think about this type of effect in terms of receptor active states; a starting point for this is to consider the nature of a seven-transmembrane receptor active state.
A receptor active state interacts with a membrane component to elicit a change in cellular behavior. Thus, a change in the receptor conformation presumably opens the door to the binding of the receptor to an effector protein such as a G-protein or β-arrestin. For example, an 11-amino acid peptide sequence from the C-terminal region of the third intracellular loop of the β-adrenoceptor (Thr284–Thr291) has been shown to have the unique ability to initiate Gs-mediated adenylate cyclase activation in turkey erythrocytes (Münch et al., 1991). This suggests that conformations that expose this region of the receptor will cause cytoplasmic signaling. The corollary to this idea is that conformations of the receptor that prevent exposure of this region to Gs-protein will be inactive and not signal. This predicts the existence of numerous “active” and “inactive” conformations [referred to as an “ensemble” (Kenakin, 2002b)]. The existence of multiple states is supported by point mutation studies carried out on the α1B-adrenoceptor in which it was found that amino acid substitution at position 293 of the receptor produces a constitutively active receptor state. It is noteworthy that substitution of 20 different amino acids in this location resulted in 20 different levels of constitutive activity, indicating 20 different conformations capable of signaling (Kjelsberg et al.,1992) and a low level of fidelity with respect to the conformational requirements for activation.
Seven-transmembrane receptors can also demonstrate the allosteric trait of probe dependence. For example, the CXCR4 receptor antagonist AMD3100 and antibody P140 block chemotaxis produced by the natural CXCR4 agonist stromal-derived factor 1-α. However, these antagonists have no effects at all on the response to the stromal-derived factor 1-α peptide fragments [Ala-Ser-Leu-Trp] and [Arg-Ser-Val-Met] (Sachpatzidis et al., 2003). Such probe dependence would be predicted to be amplified in systems in which different regions of the receptor mediate the affinities (and efficacies) of the probes. Thus, cells in which different regions of the receptor interact with different G-proteins (i.e., see Ikezu et al., 1992) define sensitive systems to detect differences in receptor conformation. The basis for this expectation is the notion that different tertiary protein conformations would not be expected to produce identical movements of these different intracellular loops and that these differences would be detected by cytosolic interactants with the receptor (see Fig. 5). In fact, this has been shown to be the case. For example, the CB1 cannabinoid ligand desacetyllevonantradol, a positive agonist for Gi1 and Gi2, is an inverse agonist for Gi3. Likewise, (R)-methanandamide is an inverse agonist for Gi1 and Gi2 and a positive agonist for Gi3 (Mukhopadhyay and Howlett, 2005). A logical interpretation of these data is to postulate that the receptor conformations stabilized by these ligands produce different changes in the various regions interacting with these G-proteins to produce heterogeneous effects (i.e., classical allosteric probe dependence).
Binding profiles for antibodies also can be sensitive indicators of tertiary conformations of specific regions of receptors. For example, it has been shown that allosteric modulators of the chemokine receptor CCR5 produce different binding profiles for various antibodies to the receptor (Kenakin, 2007). These data are consistent with the notion that a modulator can produce different conformational effects in various regions of receptors. If these regions interact with cellular signaling mechanisms, this could translate into differences in receptor signaling.
Biased agonism and receptor-based functional selectivity was first defined in systems in which 7Transmembrane receptors interacted with multiple G-proteins in a pleiotropic manner (Kenakin, 1995a; Lawler et al., 1999). However, a new paradigm for 7Transmembrane receptor signaling has been defined in the form of G-protein-independent, β-arrestin-mediated signaling (Lefkowitz, 2004, 2006; Terrillon and Bouvier, 2004; Lefkowitz and Shenoy, 2005; Luttrell, 2005); this sets the stage for further multiple receptor region allosteric dependence. Thus, although β-adrenoceptor blocking agents such as atenolol and bisoprolol are inverse agonists for Gs-protein- and β-arrestin-mediated extracellular signal-regulated kinase activation, others, such as ICI118,551 and propranolol, are inverse agonists for Gs-protein and positive agonists for the extracellular signal-regulated kinase pathway (Azzi et al., 2003; Baker et al., 2003; Galandrin and Bouvier, 2006). Likewise, agonists for the chemokine CCR5 receptor RANTES and AOP-RANTES both produce CCR5-mediated calcium response (Proudfoot et al., 1999) and both induce receptor phosphorylation. However, aminooxypentane-RANTES functions as a “superagonist” of phosphorylation, producing 300% maximal effect of RANTES (Oppermann et al., 1999), which indicates differential effects on the sites on CCR5 responsible for calcium signaling and GRK binding.
Site-directed mutagenesis studies suggest that the interaction of receptors with β-arrestin are complex, involving a large number of surface charges, and that elements of arrestin are differentially engaged by various functional forms of the receptor (Charest et al., 2005; Hanson and Gurevich, 2006). This idea, when coupled to the fact that receptors have been shown directly to adopt different conformations in response to ligand-binding (Gether et al., 1995; Ghanouni et al., 2001; Palanche et al., 2001, Yao et al., 2006; Swaminath et al., 2004), suggests that not all ligands that cause engagement with β-arrestin will do so in a uniform manner. Furthermore, because the β-arrestin/receptor complex can internalize and function as a signaling scaffold for mitogen-activated protein kinases (receptosomes), it is as yet unclear whether the changes in the conformation of β-arrestin in this process (Xiao et al., 2004) are sensitive to the type of ligand bound to the receptor.

Schematic diagram depicting levels of conformational aberration produced in different areas of the receptor upon stabilization of receptor conformations by different agonists. Arrows depict various regions of interaction of the receptor with cytosolic interactants such as different G-proteins and β-arrestin. It might be surmised that dissimilar conformations affect these various regions to varying degrees causing respective differences in effect for diverse coupling mechanisms.
Heterogeneous probe dependence would be expected to increase with increasing numbers of receptor probes; in this case, “probe” refers to the cellular interactants coupling to the receptor to initiate cellular response. The list of such probes is increasing, ranging from different G-proteins [the thyrotropin receptor has been shown to interact with all four G-protein families (Laugwitz et al., 1996)] to β-arrestin(s), GRKs, receptor activity-modifying proteins, PDZ proteins, and numerous other membrane-bound and cytosolic interactants (Bockaert and Pin, 1999; Brady and Limbird, 2002; Bockaert et al., 2004; Gavarini et al., 2006). Each of these interactions is defined by a distinct affinity equilibrium constant. Therefore, multiple receptor conformations would not be expected to produce uniform multiple relative propensities to activate different signaling pathways; i.e., different conformations would be expected to produce functional selectivity.
It is premature to conclude whether the theoretical prediction of widespread functional selectivity will be the exception or the rule with different agonists. This should become clearer with the accumulation of more data with new synthetic agonists. This is analogous to the situation encountered with the discovery of inverse agonism for the opioid receptor ligand ICI17864. Although this seemed to be an exception when first observed (in apparent disagreement with the theoretical prediction that it should be the rule), the subsequent widespread testing of antagonists in constitutively active systems confirmed that inverse agonism is the rule, not the exception. One estimate showed that approximately 85% of orthosteric antagonists were inverse agonists. This is in agreement with theoretical prediction indicating that identical affinities for different receptor conformations would not be expected (Kenakin, 2004). It will be interesting to determine whether the ability to run high-throughput screens to detect synthetic agonists in functional screening mode (as opposed to binding mode) will subsequently increase the number of agonists (both orthosteric and allosteric) available to study selective pathway stimulation. In addition, just as the availability of constitutively active systems enabled studies to address the prevalence of inverse agonism, the availability of assays that independently measure various aspects of receptor function (i.e., G-protein, β-arrestin interaction, internalization, and phosphorylation) will uncover selectivity in molecules previously thought to uniformly mimic natural agonists.
Conclusions
The identification of receptor-based functional selectivity is a useful endeavor in terms of using the chemical structure of the ligand as a control point to induce selective cellular function. From this standpoint, it is important to identify true receptor-based selectivity and differentiate it from general selectivity that can be obtained courtesy of the wiring in any given cell type. Therefore, true reversal of potency ratio or maximal response should be the hallmark for differentiating these functional selectivities, and accurate nomenclature of the result should follow to avoid confusion in the literature.
Appendix
The model consists a single receptor bound by a ligand A that can interact with various elements in the cell membrane after activation according to the operational model of agonism (Black and Leff, 1983). The amount of ligand-bound receptor is given by mass action.  where Ka is the equilibrium association constant of the ligand-receptor complex. The subsequent complex with an interactant Ei is given by:
where Ka is the equilibrium association constant of the ligand-receptor complex. The subsequent complex with an interactant Ei is given by: 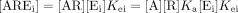 where Kei is the equilibrium association constant of the ternary AREi complex.
where Kei is the equilibrium association constant of the ternary AREi complex.
The receptor conservation equation for all of the receptor species for all membrane interactants is given by:  It can be shown that the fraction of receptor bound to any one reactant is given by ρAEi = [AREi]/[Rtot]. For ρAE1 = [ARE1]/[Rtot] for a system with n receptor interactants:
It can be shown that the fraction of receptor bound to any one reactant is given by ρAEi = [AREi]/[Rtot]. For ρAE1 = [ARE1]/[Rtot] for a system with n receptor interactants:  where KA, KE1 and KEi are equilibrium dissociation constants (1/KA,1/KE1, and 1/KEi, respectively).
where KA, KE1 and KEi are equilibrium dissociation constants (1/KA,1/KE1, and 1/KEi, respectively).
The stimulus-response pathway(s) producing response from the activation of each pathway controlled by the interactants is modeled by a simple logistic input-output forcing function of the form: 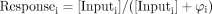 where φi is a parameter describing the efficiency of the coupling process (relationship between [AREi] and the rest of the stimulus response mechanism of the cell). The input for this function is the number of receptors bound by the ligand and coupled to the particular process given by ρAEi [Rtot]. Substituting this into eq. 11 yields:
where φi is a parameter describing the efficiency of the coupling process (relationship between [AREi] and the rest of the stimulus response mechanism of the cell). The input for this function is the number of receptors bound by the ligand and coupled to the particular process given by ρAEi [Rtot]. Substituting this into eq. 11 yields: 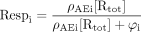 Substituting for ρAEi yields:
Substituting for ρAEi yields:  For an agonist A, substituting τAi for [Rtot]/KEi yields:
For an agonist A, substituting τAi for [Rtot]/KEi yields:  It is useful to define the following cell specific term:
It is useful to define the following cell specific term:  which causes eq. 15 to be rewritten as:
which causes eq. 15 to be rewritten as:  It can be seen from this equation that the observed potency of agonist A for the response pathway is given by:
It can be seen from this equation that the observed potency of agonist A for the response pathway is given by: 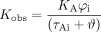 For two agonists A and B, the potency ratio is
For two agonists A and B, the potency ratio is  Likewise, the maximal response from eq. 17 is:
Likewise, the maximal response from eq. 17 is: 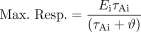 The relative maximal responses to agonists A and B are then:
The relative maximal responses to agonists A and B are then: 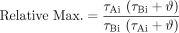
Footnotes
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.107.040352.
-
ABBREVIATIONS: HEK, human embryonic kidney; PACAP, pituitary adenylyl cyclase-activating protein; GRK, G protein-coupled receptor kinase; CCR5, chemokine receptor; ICI118,551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol; RANTES, regulated on activation normal T cell expressed and secreted; PDZ, postsynaptic density 95/disc-large/zona occludens; ICI174,864, 2[N,N′-diallyl-Tyr1,Aib2,3]Leu5-enkephalin; LP1805, N,N-(2-methylnaphthyl-benzyl)-2-aminoacetonitrile; AMD3100, 1,1′-[1,4-phenylenebis(methylene)]bis-1,4,8,11-tetraazacyclotetradecane octahydrochloride; UK-14304, 5-bromo-N-(4,5-dihydro-1H-imidazol-2-yl)-6-quinoxalinamine.
- Received July 29, 2007.
- Accepted September 27, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}