


Abstract
β-Blockers have beneficial effects in heart failure, although the underlying mechanism is unknown. β2-Adrenoceptors, however, are proportionally higher in the failing human heart. This study shows several clinically used β-blockers are agonists at the human β2-adrenoceptor. Although these agonist effects were small at the cAMP level, they were substantial at the level of cAMP response element (CRE)-mediated gene transcription. Some of the effects of “β-blockers” seen in heart failure may be related to the β2-agonist actions of these compounds. CRE-gene transcription responses to β2-agonists, forskolin, and cAMP-analogs were sensitive to p42/44-mitogen-activated protein (MAP) kinase pathway inhibitors. p42/44-MAP kinase activation was also shown directly by western blotting and enzyme-linked immunosorbent assay techniques. N-[2-(4-bromocinnamylamino)ethyl]-5-isoquinoline (H89; a protein kinase A inhibitor) stimulated cAMP accumulation and CRE gene transcription via the β2-adrenoceptor at concentrations at which protein kinase A was inhibited, providing evidence for an alternative pathway. Propranolol, however, produced paradoxical effects; it reduced basal cAMP accumulation (via β2-mediated inverse agonism) but stimulated β2-mediated CRE gene transcription. This cannot be explained by a sequential pathway from Gs-adenylyl cyclase-cAMP to CRE binding protein phosphorylation. Both responses to propranolol were insensitive to pertussis toxin, thus excluding Gi-protein involvement. Propranolol CRE gene transcription responses were attenuated by p42/44-MAP kinase inhibitors and propranolol was also found to directly stimulate the p42/44-MAP kinase pathway. Studies of inositol phosphate accumulation and of protein kinase C or Rho kinase inhibitors on CRE-gene transcription provided no evidence for Gq/11 or G12/13 involvement. These data suggest that propranolol can simultaneously act as an inverse agonist through a Gs-coupled mechanism while stimulating the p42/44-MAP kinase pathway through an alternative G-protein-independent mechanism.
β-Blockers are beneficial in the management of hypertension and ischemic heart disease because of antagonism of the inotropic and chronotropic actions of endogenous catecholamines at cardiac β1-adrenoceptors (Heidenreich et al., 1999; Morgan et al., 2001). Recently, clinical observations have found that β-blockers have a beneficial effect on survival in heart failure, although the mechanism is unknown (CIBIS-II, 1999; MERIT-HF, 1999; Eichhorn and Bristow, 2001). Moreover, it may not be a simple class effect because improvements in heart failure were not seen with all β-blockers (BEST, 2001; Port and Bristow, 2001; Nicholas et al., 1990). Recently another study found that the beneficial antihypertrophic effects of propranolol, seen in a rat model of heart failure, were independent of its β-antagonist effect (Marano et al., 2002). Therefore, it seems that the beneficial effects of β-blockers cannot be simply attributed to long-term antagonism of endogenous catecholamine responses. Furthermore, although β1-adrenoceptors are the predominant subtype in healthy human hearts (82%), in heart failure the proportion of β1-adrenoceptors falls such that the β2-adrenoceptor comprises 36% of the total β-adrenoceptor population (Port and Bristow, 2001). Thus, the beneficial actions of β-blockers may actually occur via the β2-adrenoceptor.
The β2-adrenoceptor, like the β1-adrenoceptor, is a G-protein coupled receptor that couples to Gαs-proteins and hence, via adenylyl cyclase, stimulates an increase in intracellular cAMP (Kobilka, 1992). This activates protein kinase A (PKA), leading to phosphorylation of protein targets, including the cyclic AMP response element binding protein (CREB). The β2-adrenoceptor has also been shown to stimulate the MAP kinase pathway (Daaka et al., 1997). It has been suggested that this involves coupling to Gαi-proteins as a consequence of PKA-mediated phosphorylation of the β2-adrenoceptor and a switch from Gαs- to Gi-coupling (Daaka et al., 1997; Lefkowitz et al., 2002). However, others have shown that Gαi switching is not an essential prerequisite for MAP kinase activation (Schmitt and Stork, 2000; Friedman et al., 2002). This is currently an area of debate (Lefkowitz et al., 2002). Initially, MAP kinase activation was also thought to require internalization of the receptor (Daaka et al., 1998) however, several alternative mechanisms have been reported more recently (Friedman et al., 2002). For, example, β2-adrenoceptor-mediated cAMP stimulates Epacs, which in turn activate Rap-1, B-Raf, and hence the MAP kinase cascade (de Rooij et al., 2000). Alternatively, PKA itself has been shown to directly activate Rap-1 (Schmitt and Stork, 2000).
The β-blocker CGP 12177 has been shown to stimulate β1-adrenoceptor responses in both cellular studies (Konkar et al., 2000; Baker et al., 2003) and in whole-animal studies (Kaumann et al., 2001). However, further work has suggested the existence of two different active sites of the human β1-adrenoceptor: 1) where classic agonists (catecholamines) and β-antagonists act and 2) where CGP 12177 is an agonist and relatively resistant to inhibition by β-adrenoceptor antagonists (e.g., propranolol, CGP 20712A; Konkar et al., 2000). Furthermore, several clinically used β-blockers have since been shown to be agonists of both β1-adrenoceptor-mediated cAMP accumulation and downstream CRE-mediated gene transcription, some via the catecholamine site, others through the secondary site, and at least two compounds through both sites (Baker et al., 2003). The precise nature of the “sites” remains unknown and could represent separate binding sites, dimerization, or association with particular scaffold proteins.
We have recently reported that CGP 12177 is a potent agonist of CRE-mediated gene transcription and produced a small stimulation of cAMP accumulation in CHO cells expressing the human β2-adrenoceptor (Baker et al., 2002). Several other β-blockers have been reported to have weak agonist actions via the β2-adrenoceptor at the level of cAMP accumulation (e.g., pindolol; Jasper et al., 1990; Chidiac et al., 1994), although many have been shown to have inverse agonist activity; that is, they decrease basal cAMP accumulation and in effect “turn off” β2-adrenoceptor signaling. This inverse activity has been reported both at the level of cAMP accumulation (Chidiac et al., 1994; Azzi et al., 2001) and cardiac contractility in transgenic mice (Bond et al., 1995). The present study was therefore undertaken to: a) evaluate the functional effect of a range of β-blockers on CRE-mediated gene transcription via the human β2-adrenoceptor; b) examine whether there is any evidence for a secondary site analogous to that of the human β1-adrenoceptor, and c) explore the relationship between the effects of these compounds on cAMP accumulation, CRE-mediated gene transcription, and MAP kinase pathways.
Materials and Methods
Materials
Cell culture reagents were from Sigma Chemical (Poole, Dorset, UK), except for fetal calf serum, which was from PAA Laboratories (Teddington, Middlesex, UK). [3H]Adenine, [3H]CGP 12177, [myo-3H]inositol, [14C]cAMP, and enhanced chemiluminescence Western blotting detection reagents were obtained from Amersham Biosciences (Buckinghamshire, UK); CGP 12177 and U0126 were from Tocris Cookson (Avonmouth, Bristol, UK), and scintillation fluid Cocktail Plus and Microscint 20 were from PerkinElmer Life and Analytical Sciences (Pangbourne, Berkshire, UK). The cAMP analogs (8-bromo-cAMP and CPT-cAMP), pertussis toxin (PTX), SB 203580, RO 318220, Gö 6976, Gö 6983 and Y 27632 were from Calbiochem (Nottingham, UK). The FACE ERK1/2 ELISA kits were from Active Motif Europe (Rixensart, Belgium). The primary antibodies phospho-ERK and total ERK were from PerkinElmer Life and Analytical Sciences (Hitchin, Hertfordshire, UK). The secondary antibody (peroxidase-conjugated goat anti-mouse immunoglobulins) was from DAKO Cytomation (Ely, Cambridgeshire, UK). Bupranolol was a gift from Prof. Sian Harding (Imperial College, London, UK). Sigma Chemical supplied all other reagents.
Cell Culture
CHO cells stably expressing the human β2-adrenoceptor and a reporter gene, secreted placental alkaline phosphatase (SPAP), under the transcriptional control of a six-CRE promoter (Baker et al., 2002), were used. CHO cells stably transfected with the human adenosine A1 receptor and the CRE-SPAP reporter were used as a positive control cell line for PTX experiments. Untransfected cells (CHO-K1) and those stably transfected with the CRE-SPAP reporter alone (CHO-SPAP) were used as controls where stated. All CHO cell lines were grown at 37°C in Dulbecco's modified Eagle's medium/nutrient mix F12 (DMEM/F12) containing 10% fetal calf serum and 2 mM l-glutamine in a humidified 5% CO2/95% air atmosphere.
CRE-Mediated Gene Transcription (SPAP)
Cells were grown to confluence in 24-well plates then serum-starved for 24 h before experimentation in DMEM/F12 containing 2 mM l-glutamine (serum-free media). Where used, PTX (100 ng/ml) and PDBu (1 μM) were added to the serum-free media and thus had 24 h of incubation before experimentation. On the day of experimentation, the media was removed and replaced with 1 ml of fresh serum-free media. Where used, antagonists were added to this media and incubated for 30 min at 37°C in a humidified atmosphere of 5% CO2/95% air. Intracellular inhibitors were incubated for 1 h at 37°C. Agonists (in 10 μl, each condition in triplicate) were then added and incubated for 5 h in the same 5% CO2 atmosphere. Media and drugs were then removed and replaced with 300 μl of fresh serum-free media and incubated for a further hour. Samples of media (20 μl) from each well were then transferred to 96-well plates and CRE-dependant SPAP reporter activity was quantified as described previously (Baker et al., 2002).
[3H]Cyclic AMP Accumulation
Cells were grown to confluence in 24-well plates. Where PTX was used, the cells were grown to confluence in 24-well plates and the media replaced with serum-free media containing PTX (100 ng/ml) for 24 h before experimentation (i.e., identical to PTX use in the SPAP assay). Confluent cells were prelabeled with [3H]adenine (4 μCi/ml) for 2 h at 37°C in 1 ml/well Hanks' balanced salt solution containing 20 mM HEPES, pH 7.4 (HBH). The [3H]adenine was removed, each well was washed twice with 1 ml of HBH and then incubated for 30 min with 1 ml of HBH containing the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 1 mM). Agonists in 10 μl were then added and the cells were incubated for a further hour before the reaction was terminated by the addition of 50 μl of concentrated HCl to each well. Cyclic [3H]AMP was separated from other [3H]adenine nucleotides by sequential Dowex and alumina chromatography, and each column was corrected for efficiency by comparison with [14C]cAMP recovery as described previously (Baker et al., 2002).
MAP Kinase Activation
Western Blotting. Cells were grown to confluence in six-well plates, washed, then incubated in 2 ml of serum-free media for 24 h. Cells were stimulated for 10 min at 37°C by the addition of agonist in 200 μl of serum-free media. The reaction was terminated by washing the cells with ice-cold phosphate-buffered saline (PBS) then adding 200 μl of ice-cold lysis buffer [20 mM Tris/HCl, pH 7.4, 1 mM EGTA, 0.5% (v/v) Triton X-100, 1 mM NaF, 1 mM dithiothreitol, 70 mM β-glycerophosphate, 10 μl of protease inhibitor cocktail (Sigma)/7 ml of buffer). Cells were then scraped from the wells and centrifuged in 0.5-ml Eppendorf tubes for 5 min at 12,000 rpm at 4°C. Supernatant was collected and sampled for protein content by the method of Lowry et al. (1951). Supernatant (70 μl) was added to 70 μl of SDS/polyacrylamide gel electrophoresis Laemmli sample buffer (Sigma), boiled at 95°C for 5 min, and then subjected to Western blot analysis. Protein samples (15 μg) were separated by SDS/polyacrylamide gel electrophoresis (11.25% acrylamide gel) using the mini protein-II system (Bio-Rad, Hemel Hempstead, UK). After transfer of the proteins to a nitrocellulose membrane, the membranes were blocked for 1 h in antibody blocking buffer [5% (w/v) low-fat dried milk in wash buffer (25 mM Tris, 125 mM NaCl, and 0.1% Tween 20, pH 7.6)] at room temperature. Blots were then incubated with either anti-phospho-ERK 42/44 or anti-total ERK 42/44 overnight in antibody blocking buffer at 4°C. The blots were washed for an hour (37°C) in wash buffer and then incubated with the secondary horseradish peroxidase-conjugated goat anti-mouse antibody for 1 h at 37°C in antibody blocking buffer. The secondary antibody was then removed and the membranes were washed for 1 h in wash buffer (37°C) before developing the blots using the enhanced chemiluminescence detection system (Amersham Biosciences).
ELISA. Cells were grown to confluence in 96-well plates, then washed and incubated in 200 μl of serum-free media for 24 h. Cells were stimulated for 10 min (37°C) by the addition of 20 μl of agonist in serum-free media and the reaction terminated by removal of the media and addition of 100 μl of 4% formaldehyde in PBS. ERK-42/44 phospho MAP kinase was then detected using the FACE ERK-1/2 ELISA kit (Active Motif) as per the manufacturer's instructions.
[3H]CGP 12177 Whole-Cell Binding
Cells were grown to confluence in 96-well view plates. The media was removed and 200 μl of serum-free media containing H89 and 0.3 nM [3H]CGP 12177 was added to each well. The plates were then incubated for an hour at 37°C in a 5% CO2 atmosphere. Nonspecific binding was determined using 100 nM ICI 118551. The media and drugs were removed, the cells were washed twice with 200 μl of PBS, 200 μl of Microscint 20 (PerkinElmer Life and Analytical Sciences) was added to each well, and the plates were counted on a TopCount liquid scintillation counter (PerkinElmer Life and Analytical Sciences).
[3H]Inositol Phosphate Accumulation
Cells were grown to confluence in 24-well plates in 0.5 ml of DMEM/F12 media containing [myo-3H]inositol (592 kBq/ml), 2 mM glutamine, and 10% fetal calf serum. After being washed twice with 1 ml/well serum-free media, cells were incubated for 30 min in 1 ml of serum-free media containing 20 mM LiCl (37°C, 5% CO2). Agonists (in 10 μl) were then added and the incubation continued for 1 h. Reactions were terminated by aspiration of all incubation medium and drugs followed by the addition of 1 ml of cold (-20°C) methanol/0.12 M HCl (1:1, v/v) to each well. The cells were left at -20°C for at least 2 h before isolating total [3H]inositol phosphates as described previously (Megson et al., 2001). Total [3H]inositol phosphate levels were determined by liquid scintillation counting.
Data Analysis. A maximal isoprenaline concentration was included in each separate experiment for both [3H]cAMP accumulation and SPAP gene transcription to allow agonist responses to be expressed as a percentage of the isoprenaline maximum. Agonist concentration-response curves were fitted to a four-parameter logistic equation through computer-assisted nonlinear regression using the program Prism 2 as described previously (Baker et al., 2002).
All antagonist dissociation constants were assessed at fixed antagonist concentrations (assuming competitive antagonism) by observing the shift in the agonist concentration-response curve using the equation DR = 1 + [A]/KD, where DR is the dose-ratio of the concentrations of agonist required to produce an identical response in the presence and absence of antagonist, [A] is the concentration of antagonist, and KD is the antagonist dissociation constant. All data are presented as mean ± S.E.M. from triplicate determinations. The n in the text refers to the number of separate experiments.
Results
Agonist and Inverse Agonist Responses of β-Blockers. ICI 118551, previously reported to be a selective inverse agonist at the β2-adrenoceptor (Bond et al.,. 1995; Azzi et al., 2001), decreased the basal production of [3H]cAMP to 36.1 ± 4.4% of untreated levels (log IC50, -9.08 ± 0.18, n = 8; Fig. 1) thus confirming its inverse agonist activity in these cells at the level of cAMP accumulation. As would be expected for a cAMP-mediated downstream event, an inverse agonist response was also seen at the level of CRE-mediated gene transcription (log IC50, -9.62 ± 0.13, reduction to 85.6 ± 2.7% of untreated basal, n = 9; Fig. 1). Atenolol behaved in a similar manner (Table 1). A range of other β-blockers (bisoprolol, bupranolol, metoprolol, sotalol, and timolol) all had inverse agonist activity at the level of [3H]cAMP accumulation but no response was detectable for gene transcription (Table 1).

[3H]cAMP accumulation and CRE-mediated SPAP production in CHO-β2 cells in response to ICI 118551. Bars represent basal responses and responses to 10 μM isoprenaline. Data points are triplicate mean ± S.E.M. from a single experiment representative of 7 (cAMP) and 8 (SPAP) other separate experiments.
Responses of a range of β-blockers at the level of cAMP accumulation and CRE-mediated gene transcription (SPAP)
Values represent mean ± S.E.M. of n determinations
Labetolol (log EC50, -8.08 ± 0.02, n = 12; Fig. 2) stimulated an increase in [3H]cAMP accumulation that was 4.49 ± 0.47-fold over basal (8.50 ± 0.62% of the maximal response elicited by 10 μM isoprenaline). An agonist response was also seen at the level of CRE-mediated gene transcription (log EC50, -8.76 ± 0.04, 3.71 ± 0.27-fold over basal, 66.92 ± 5.23% of maximum isoprenaline, n = 14; Fig. 2). These agonist responses (for both [3H]cAMP accumulation and SPAP gene transcription) were antagonized by ICI 118551 to yield log KD values of -9.59 ± 0.03 (n = 5) and -9.84 ± 0.07 (n = 8), respectively (Fig. 2, Table 2). These values are similar to the log IC50 values for ICI 118551 above.

[3H]cAMP accumulation and CRE-SPAP production in response to labetolol in the presence and absence of ICI 118551. Bars represent basal responses and responses to 10 μM isoprenaline or ICI 118551 (30 nM for SPAP; 10 nM for cAMP) alone. Data points are mean ± S.E.M. of triplicate determination, and this single experiment is representative of four (cAMP) and seven (SPAP) other separate experiments.
log Kd values for ICI 118551 and (±)-propranolol as antagonists of “β -blocker”-stimulated cAMP accumulation and CRE-mediated gene transcription
Pindolol, acebutolol, alprenolol, and carvedilol also stimulated agonist responses at the level of both cAMP accumulation and CRE-gene transcription (Fig. 3, Table 1). The gene transcription responses to pindolol, acebutolol, and alprenolol were also antagonized by ICI 118551 to yield similar KD values (Fig. 3, Table 2). The selective β1-adrenoceptor antagonist CGP 20712A also stimulated an agonist response at the level of CRE-gene transcription at concentrations compatible with its binding to β2-adrenoceptors although no [3H]cAMP response was observed (Table 1).

CRE-SPAP production in response to alprenolol (a) and pindolol (b) in the presence and absence of 30 nM ICI 118551. Data points (mean ± S.E.M. of triplicate determination) are from a single experiment that is representative of three (a) and five (b) separate experiments. Bars show basal SPAP production and responses to 10 μM isoprenaline or 30 nM ICI 118551 alone.
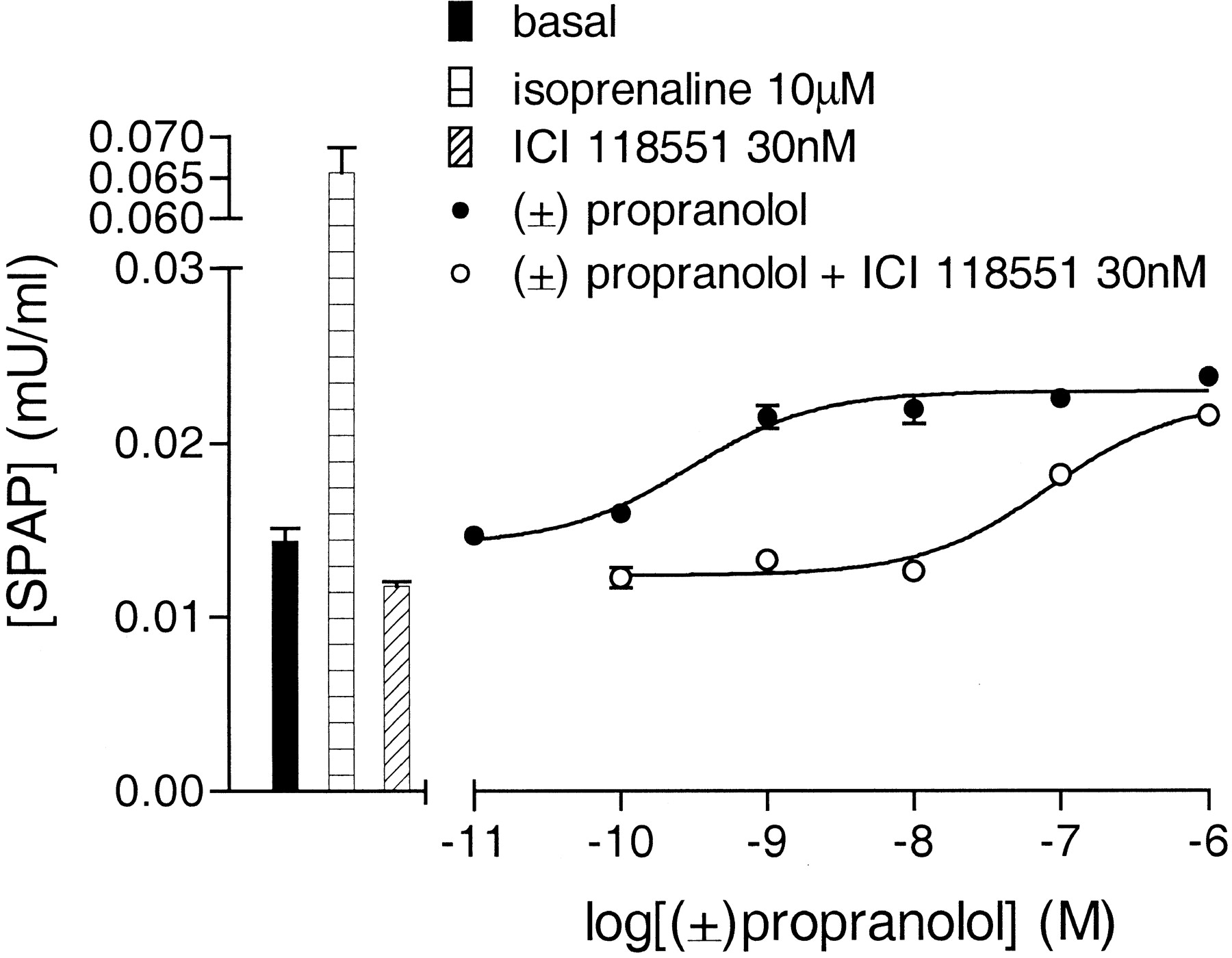
Agonist and Inverse Agonist Effects of Propranolol. Propranolol, previously reported to be an inverse agonist (Chidiac et al., 1994; Azzi et al., 2001), acted as an inverse agonist at the level of [3H]cAMP accumulation by reducing the basal accumulation of cAMP to 54.24 ± 7.32% of the untreated value (log IC50, -9.17 ± 0.08, basal, n = 7; Table 1). However surprisingly, propranolol stimulated an increase in SPAP production that was 14.2 ± 0.9% of the isoprenaline maximal (log EC50, -9.4 ± 0.05, 1.55 ± 0.03 fold over basal, n = 25; Table 1). Because propranolol exists as two stereo-isomers, experiments with the separate stereoisomers were conducted. The S(-)-propranolol isomer was found to be more potent than the R(+) isomer in both functional assays. However, both isomers were inverse agonists at the level of cAMP accumulation but agonists at CRE-mediated gene transcription (Fig. 4, Table 1) with a S(-) to R(+) potency ratio of 77.98 ± 12.08, n = 3, and 67.71 ± 3.00, n = 3, for the cAMP and gene transcription responses, respectively. This is similar to the findings of Howe and Shanks (1966), who found S(-)-propranolol to be 50-fold more potent than R(+)-propranolol in humans.

[3H]cAMP production (a) and CRE-SPAP production (b) in CHO-β2 cells in response to the stereoisomers of propranolol: R(+)- and S(-)-propranolol. Bars show basal levels of [3H]cAMP accumulation and SPAP production and that in response to 10 μM isoprenaline. Each experiment is representative of three separate experiments.
To confirm that propranolol was acting via the human β2-adrenoceptor, first its agonist effect on gene transcription response was antagonized by ICI 118551. Because the inverse agonist nature of ICI 118551 is relatively large compared with the racemic propranolol agonist response, an apparent KD value was calculated for ICI 118551 by comparing the EC50 values of the two curves. This yielded an apparent log KD value of -9.88 ± 0.11 (n = 3; Fig. 5), again similar to that above. Racemic propranolol was also able to antagonize the more efficacious agonist responses of labetalol and alprenolol (Table 2). Thus the propranolol IC50 value as an inverse agonist of [3H]cAMP accumulation, the propranolol EC50 value as an agonist of CRE-mediated gene transcription, and the propranolol KD values as an antagonist, were all very similar, as would be expected if all these interactions were occurring via the same receptor site (Tables 1 and 2).

CRE-SPAP production in response to (±)-propranolol in the absence and presence of 30 nM ICI 118551. Bars represent basal SPAP production and responses to 10 μM isoprenaline and 30 nM ICI 118551 alone. This single experiment is representative of three separate experiments. Given the degree of inverse agonism of ICI 118551 relative to the propranolol stimulation, an apparent KD value for ICI 118551 was calculated by comparing the EC50 values of the two curves.
Lack of Responses in Native CHO-K1 Cells. Further confirmation that all of these responses were occurring via the transfected human β2-adrenoceptor came from experiments with native CHO cells and those transfected with only the CRE-SPAP reporter. In both of these cells lines, the direct adenylyl cyclase activator forskolin stimulated an increase in [3H]cAMP accumulation, but there was no response to any of the other β-blockers (n = ≥3 for each drug in each assay up to concentrations of 100 μM). A similar lack of responses was seen at the level of cAMP accumulation and CRE-SPAP production in CHO-SPAP cells expressing the reporter only and not the receptor. This confirms that the cAMP and SPAP responses to the β-blockers, including propranolol, described above, are dependent upon the presence of the transfected human β2-adrenoceptor.
Effects of MAP Kinase Inhibitors. In view of the data obtained with propranolol, we investigated the potential contribution from other signaling pathways in the CRE-mediated reporter gene response. The potential involvement of the p42/44 MAP kinase pathway was investigated by using the intracellular inhibitors PD 98059 (50 μM), U0126 (10-50 μM; both known to be MEK inhibitors; English and Cobb, 2002), and RO 318220 [10 μM; known to be a protein kinase C (PKC) and Rsk-2 inhibitor; Alessi, 1997]. U0126 and RO 318220 decreased basal CRE-SPAP response to 54.8 ± 6.7% (n = 20) and 74.1 ± 1.9% (n = 19) of untreated basal (Fig. 6). The isoprenaline CRE-SPAP response (log EC50, -8.30 ± 0.07, 5.9 ± 0.2-fold over basal, n = 46) was inhibited to varying degrees by all of the MAP kinase p42-p44 inhibitors (Fig. 6, a-c; Table 3). The less efficacious β2-agonists that, unlike isoprenaline, do not stimulate internalization of the β2-adrenoceptor (Baker et al., 2002) induced responses that were also inhibited by these p42/44 MAP kinase inhibitors (Table 3). To ensure that all of these MAP kinase inhibitors were not affecting the secretory part of the pathway, cells were lysed and intracellular SPAP measured. This yielded identical results.

CRE-SPAP production in response to isoprenaline (a-c) and forskolin (d) in the absence and presence of several MAP kinase pathway inhibitors: 50 μM PD 98059, 50 μM U0126, and 10 μM RO 318220. Bars represent basal SPAP production in the presence of the inhibitor alone (a-c) and in response to 10 μM isoprenaline alone (d). These separate experiments are representative of 12 (a), 6 (b), 10 (c), and 3 (d) separate experiments.
SPAP gene transcription responses to β2-agonists, partial agonists, and β -blockers after preincubation with inhibitors (50 μM U0126, 50 μM PD 98059, or 10 μM RO 318220)
The percentage of maximum isoprenaline response is expressed as a percentage of the response to 10 μM isoprenaline measured in the same experiment. The remaining columns indicate the percentage of that ligand's maximum response (in the absence of inhibitor).
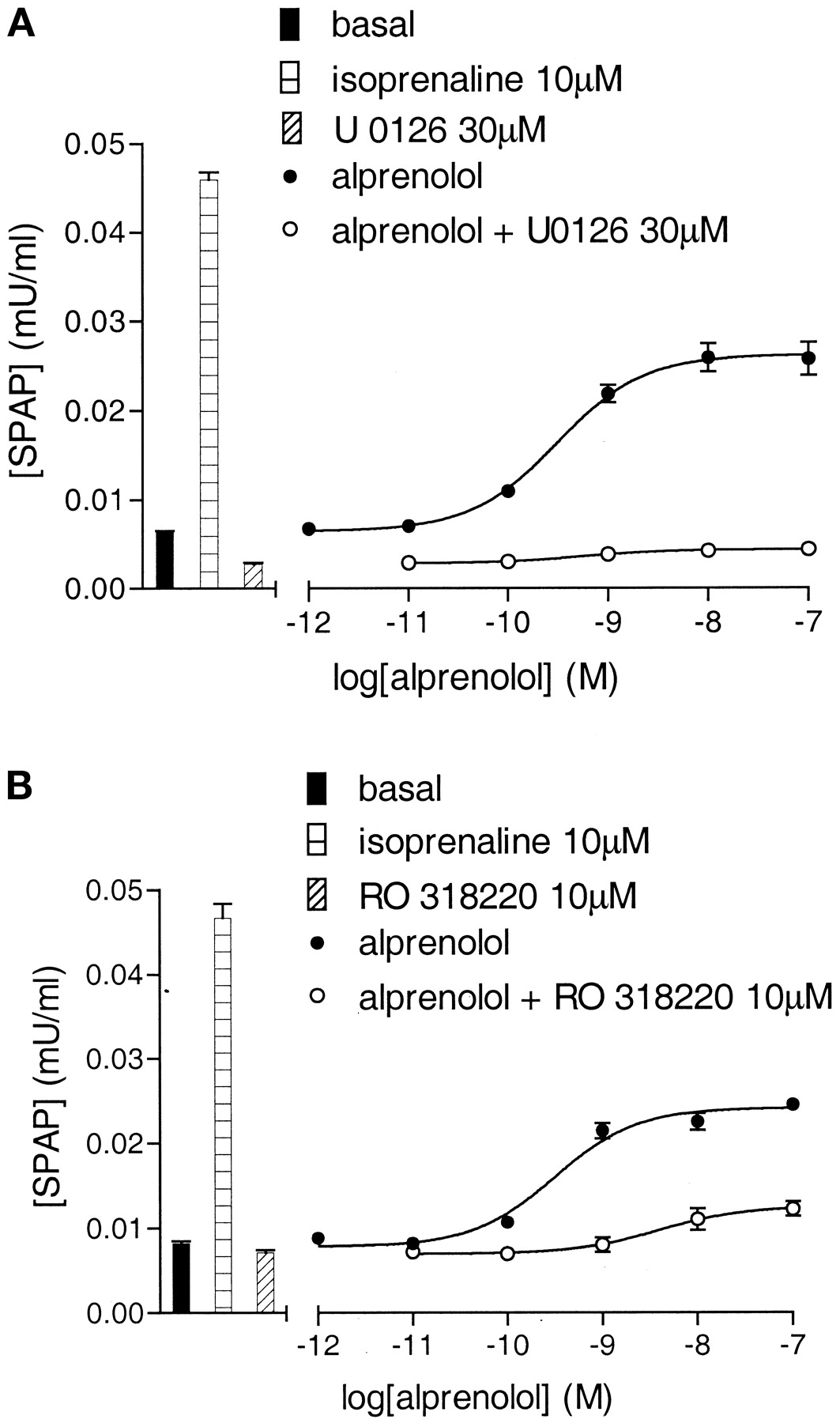
U0126 (30 μM) and RO 318220 (10 μM) inhibited the agonist CRE-gene transcription responses to both alprenolol and propranolol (Fig. 7; Table 3). In contrast, the P38 MAP kinase inhibitor SB 203580 (3 μM) produced no marked inhibitory effects on the maximum agonist gene transcription responses to isoprenaline (99.8 ± 4.4% of control; n = 3), salbutamol (95.0 ± 4.2% of control, n = 3), alprenolol (97.3 ± 3.5%; n = 3), or propranolol (90.1 ± 10.3; n = 3; also see Fig. 12c).
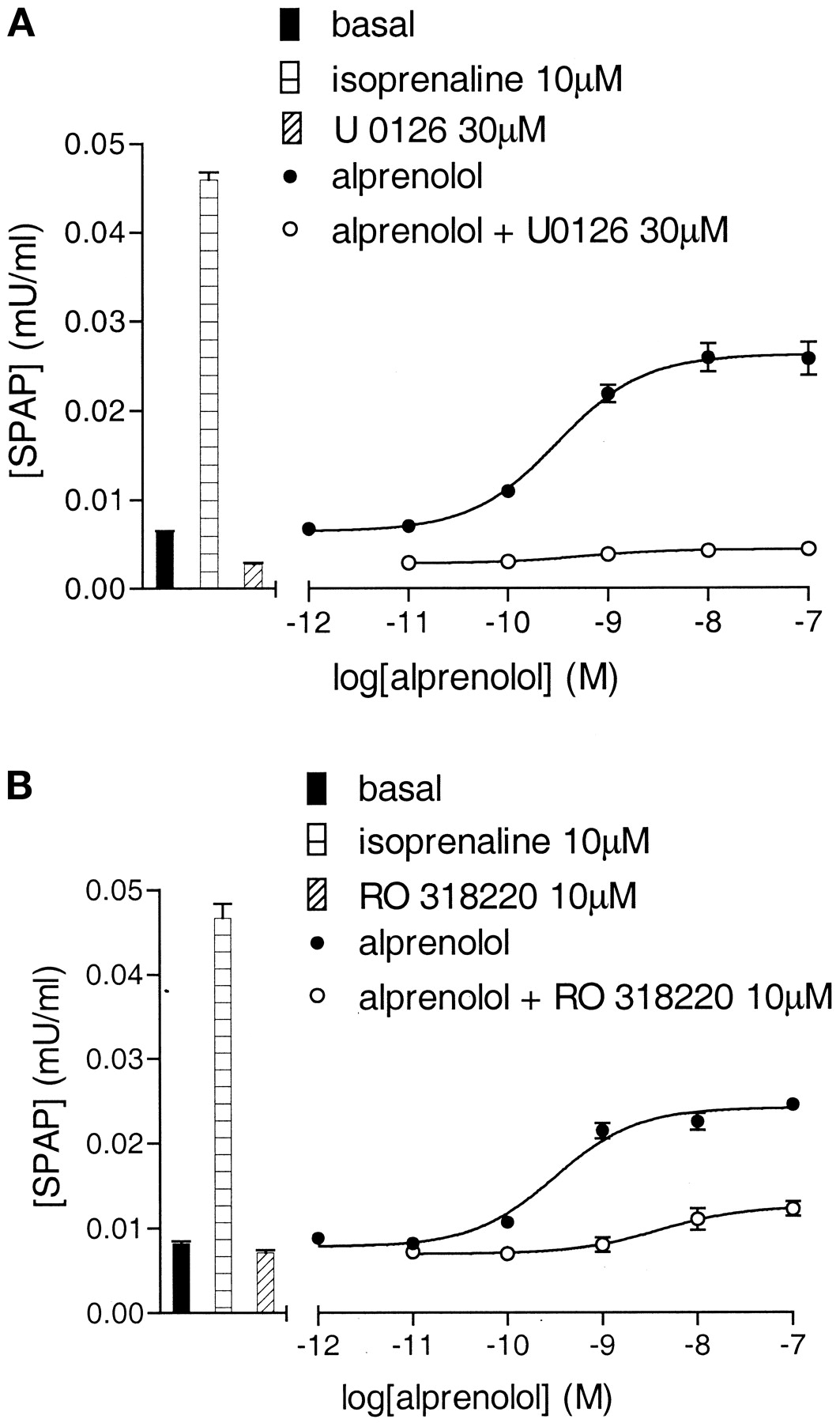
CRE-SPAP production in response to alprenolol in the absence and presence of 30 μM U0126 (a) and the absence and presence of 10 μM RO 318220 (b). Bars show basal SPAP production and that in response to 10 μM isoprenaline, 30 μM U0126, or 10 μM RO 318220 alone. Data points are from single experiments and are representative of five (a) and three (b) other separate experiments.

a, CRE-SPAP production in response to (±)-propranolol in the absence and presence of 3 μM Gö 6983, 3 μM Gö 6976, and after 24-h pretreatment with 1 μM PDBu. Bars represent basal SPAP production and responses to 10 μM isoprenaline, 3 μMGö 6983, 3 μMGö 6976 alone, and after 24-h treatment with 1 μM PDBu. Data are mean ± S.E.M. of triplicate determinations in a single experiment that is representative of three separate experiments. b, [3H]inositol phosphate accumulation in response to 10 μM isoprenaline, 10 μM (±)-propranolol, and 100 μM UTP. Data are mean ± S.E.M. of six determinations in a single experiment. Similar data were obtained in four other experiments. c, CRESPAP production in response to (±)-propranolol in the absence and presence of 3 μM SB 203580 and 3 μM Y 27632. Bars represent basal SPAP production and responses to 10 μM isoprenaline, 3 μM SB 203580, and 3 μM Y 27632 alone. Data are mean ± S.E.M. of triplicate determinations in a single experiment that was representative of three separate experiments.
Effects of Forskolin and cAMP Analogs. The involvement of cAMP and PKA in generating the CRE-SPAP responses was investigated using the direct adenylyl stimulator forskolin, two membrane-permeant analogs of cAMP, the phosphodiesterase inhibitor IBMX, and the PKA inhibitor H89. Forskolin caused an increase in both [3H]cAMP accumulation (log EC50, -6.33 ± 0.09, 71.3 ± 14.6% isoprenaline maximum response, n = 3) and CRE-mediated gene transcription (log EC50, -6.32 ± 0.05, 91.3 ± 2.9% of the isoprenaline maximum response, n = 7). To determine whether the MAP kinase involvement was up- or downstream of cAMP, the CRE-SPAP forskolin responses were examined in the presence of the MAP kinase inhibitors. The forskolin CRESPAP response was also inhibited by U0126 and RO 318220 (Fig. 6d, Table 3), suggesting that the MAP kinase contribution occurs downstream of cAMP. CPT-cAMP stimulated an increase in CRE-mediated gene transcription (log EC50, -4.02 ± 0.06; 85.2 ± 4.8% of isoprenaline maximum, n = 3; Fig. 8). This response was inhibited by 30 μM U0126 to 24.9 ± 1.5% of the control response (Fig. 8). Similar data were obtained with 8-bromo-cAMP (1 mM stimulated a response 76.9 ± 3.4% of the isoprenaline maximum and this was inhibited by 30 μM U0126 to 36.9 ± 1.2% of the control response; Fig. 8, n = 3). Elevation of intracellular cAMP with 3 mM of the phosphodiesterase inhibitor IBMX also stimulated a CRE-mediated gene transcription response (63.7 ± 4.2% of the isoprenaline maximum; n = 3), and this was similarly sensitive to 30 μM U0126 (23.7 ± 1.5% control response, Fig. 8). This confirms the forskolin findings above and suggests the MAP kinase involvement is downstream of cAMP.
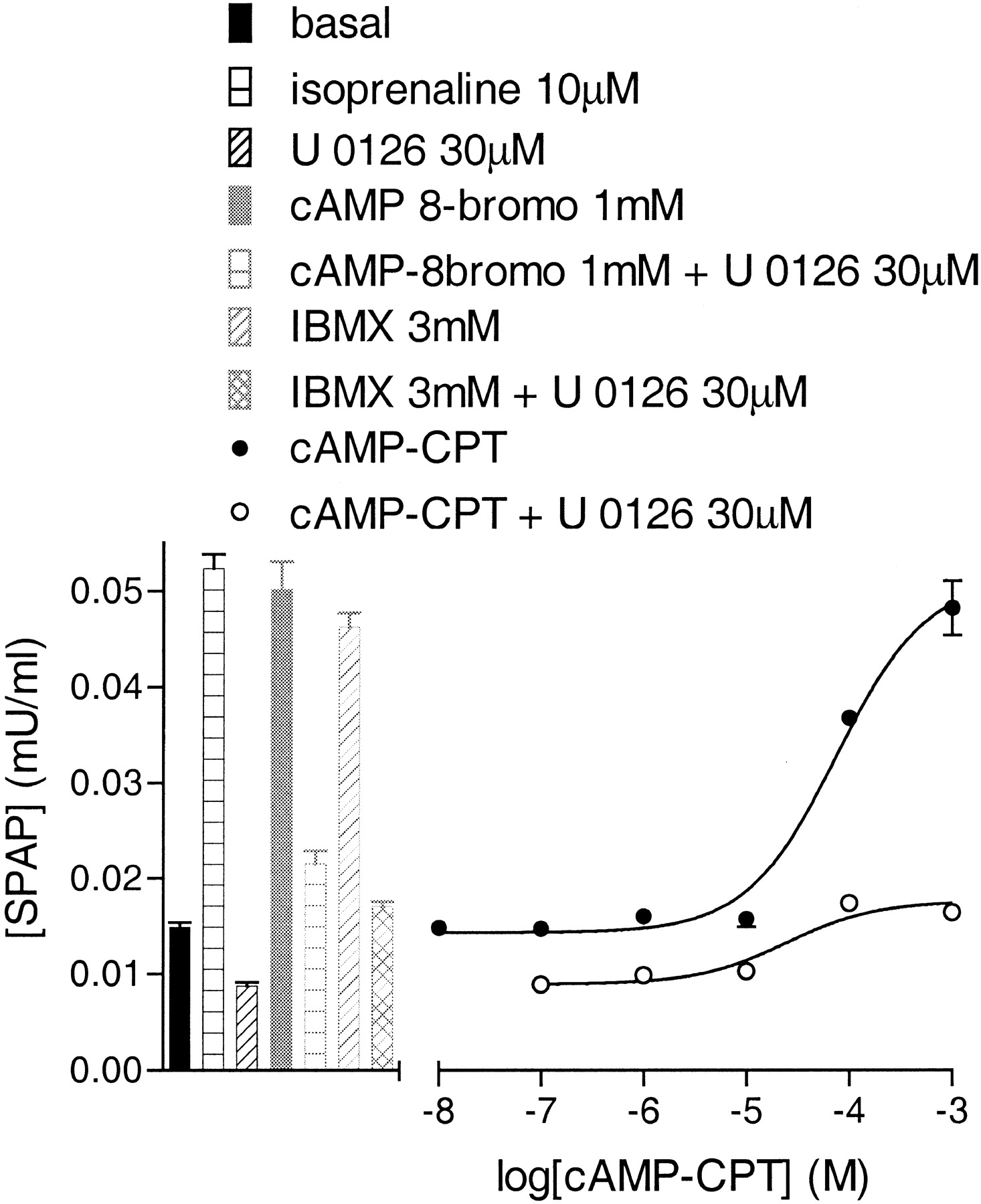
CRE-SPAP production in response to 8-bromo-cAMP (1 mM), IBMX (3 mM), or CPT-cAMP in the presence and absence of 30 μM U0126. Bars show basal SPAP production and responses to 10 μM isoprenaline or 30 μM U0126 alone. Also shown as bars are the responses to 8-bromo-cAMP (1 mM) or IBMX (3 mM) in the presence and absence of 30 μM U 0126. Data points are from a single experiment and are representative of three separate experiments.
Direct Evidence of p42/44 MAP Kinase Activation. To directly confirm that isoprenaline, alprenolol, propranolol, and forskolin were indeed able to activate the p42/44 (ERK-1/2) MAP kinase pathway, two different methods were used to detect the active phosphorylated MAP kinase. Western blot analysis showed that all four agents were able to increase the levels of phospho-ERK over basal after 10 min of stimulation (n = 3; Fig. 9a). Similarly, quantitative ELISA confirmed that all 4 agents were able to elicit a significant stimulation over basal (p < 0.001; one-way ANOVA with post hoc Neuman-Keuls analysis, n = 4; Fig. 9b) of p42/44 phospho-ERK MAP kinase. Of particular note is the fact that propranolol, which decreases cAMP, also produced a significant increase in p42/44 MAP kinase activation.

a, Western blot showing detection of the activated phospho-p42/44 MAP kinase and total p42/44 MAP kinase under basal conditions and in response to 10% serum, 10 μM isoprenaline, 10 μM alprenolol, 10 μM (±)-propranolol, and 100 μM forskolin. This single experiment is representative of three separate experiments. b, ELISA showing detection and quantification of phospho-p42/44 MAP kinase under basal conditions and in response to 10% serum, 10 μM isoprenaline, 10 μM alprenolol, 10 μM (±)-propranolol, and 100 μM forskolin. Data points are means ± S.E.M. from a single experiment that is representative of four separate experiments. *, p < 0.001, one-way ANOVA with post hoc Neuman-Keuls analysis. Two-way ANOVA of the combined data from all four experiments confirmed significant effects of isoprenaline, alprenolol, propranolol, and forskolin (p < 0.01). Serum was included in only three experiments and was significant in each case (p < 0.001; one-way ANOVA).
Effects of the PKA Inhibitor H89. H89 has been previously shown to be a β-adrenoceptor antagonist (Penn et al., 1999). In whole CHO-β2 cell binding, H89 inhibited the specific binding of the radioligand [3H]CGP 12177 to give a log KD value of -6.21 ± 0.03 (n = 4). H89 stimulated an increase in [3H]cAMP accumulation (log EC50, -5.66 ± 0.06, 5.03 ± 0.8% of the isoprenaline maximum, n = 4; Fig. 10). This cAMP response was inhibited by ICI 118551 (log KD, -8.83 ± 0.07, n = 3; Fig. 10). H89 also stimulated an increase in CRE-SPAP production (log EC50, -6.51 ± 0.05, 28.34 ± 1.51% of the maximum isoprenaline response, n = 4). This H89 response was also inhibited by ICI 118551 (n = 4; Fig. 10). No H89 responses were seen in control cells containing the reporter construct, but not the receptor (n = 4). Thus at concentrations required to inhibit PKA (10 μM; Penn et al., 1999), H89 caused substantial agonism of β2-CRE-mediated gene transcription. The PKA-inhibitory effect of H89 on CRE-gene transcription however was also seen. 10 μM H89 substantially reduced the forskolin-stimulated CRE-SPAP response to 51.08 ± 2.77% of control forskolin (n = 4, Fig. 11a), suggesting that part of the CRE-SPAP response is PKA-sensitive. In experiments with isoprenaline as the agonist, H89 behaved as a partial agonist, shifting the isoprenaline curve to the right and reducing the maximum response (to 58.1 ± 1.2% of control, n = 4; Fig. 11b) in keeping with both its partial β-agonist- and PKA-inhibitory effects. These data therefore confirm the presence of both PKA-dependent and -independent pathways downstream from cAMP in the generation of CRE-gene transcription responses.

[3H]cAMP accumulation and CRE-SPAP gene transcription in response to H89 in the presence and absence of ICI 118551. Bars represent basal responses and responses to 10 μM isoprenaline or ICI 118551 (30 nM SPAP; 10 nM cAMP) alone. Data are mean ± S.E.M. from a single experiment representative of three (cAMP) and four (SPAP) separate experiments.
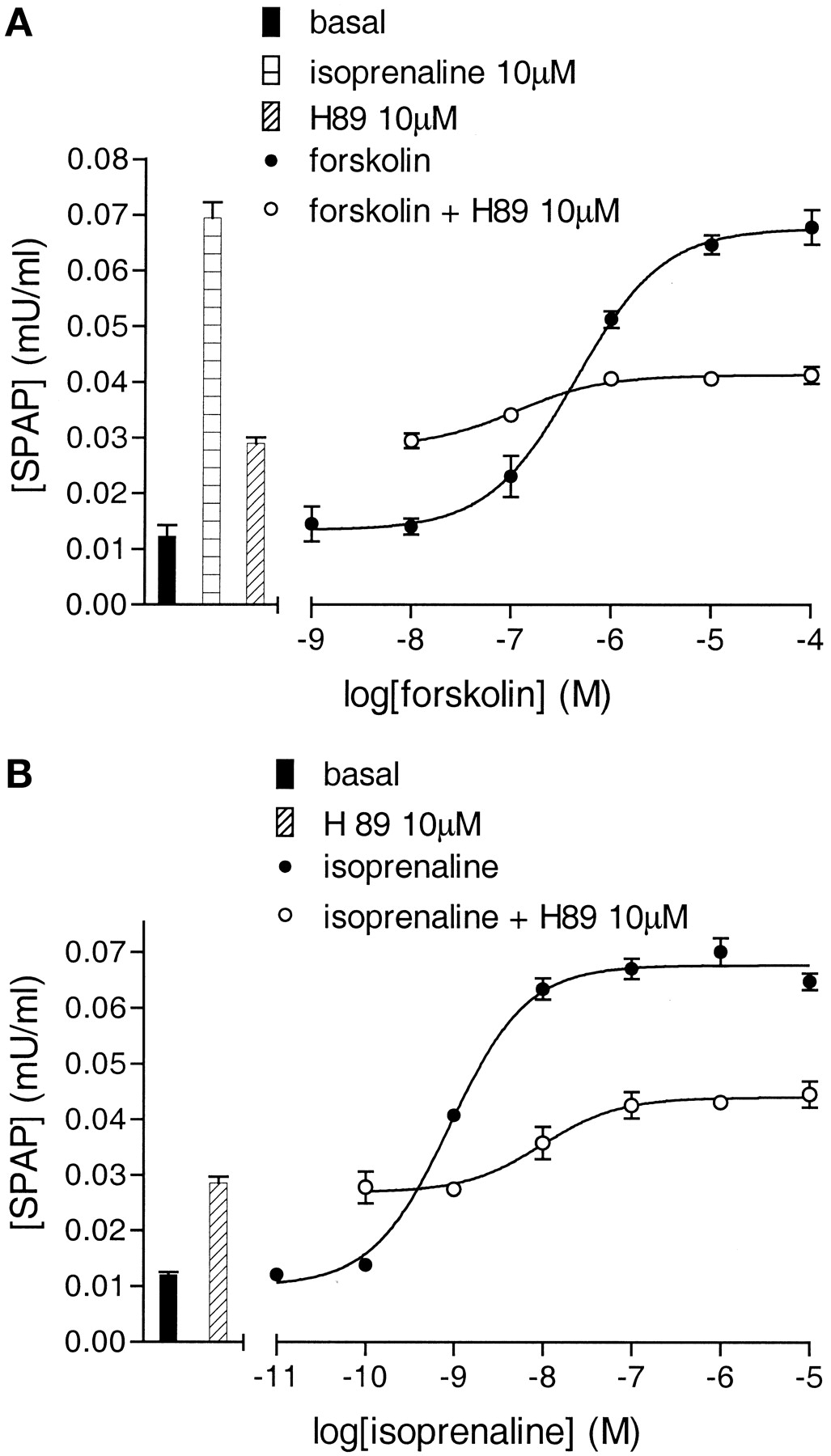
CRE-SPAP production in response to forskolin (a) and isoprenaline (b) in the presence and absence of 10 μM H89. Bars represent basal SPAP production and responses to 10 μM isoprenaline and 10 μM H89 alone. Data points are from a single experiment that is representative of four separate experiments in each case
Lack of Gi-Mediated Involvement—The Effects of PTX Pretreatment. Work from others has suggested that the human β2-adrenoceptor can switch coupling from Gs to Gi proteins (Daaka et al., 1997). Phosphorylation of the receptor by PKA has been suggested as the mechanism by which Gs-coupling is terminated and Gi-coupling initiated (Daaka et al., 1997). The ability of the β2-adrenoceptor to couple to Gs and Gi might therefore provide a potential mechanism for the different propranolol effects seen above. In addition, some of the “inverse agonism” seen with other β-blockers could potentially be explained by a Gi-mediated inhibition of cAMP accumulation rather than true inverse agonism at the receptor-Gs complex. We therefore investigated the contribution of Gi-protein coupling in the generation of CRE-gene transcription responses. The CRE-gene transcription response to the efficacious agonist isoprenaline was not effected by 24 h of preincubation with PTX (Table 4) and neither were the responses to the less efficacious agonists salbutamol and salmeterol (Table 4). Similarly, no effect was seen with the β2-partial agonist, CGP 12177. Thus, no evidence for a switch to Gi-coupling was seen with a range of agonist efficacies in these cells. Unsurprisingly, therefore, no effect of PTX was seen in the CRE-SPAP agonist responses of the β-blockers; importantly, however, no effect of PTX was seen on the propranolol CRE-SPAP agonist response either. Because the inverse agonist CRE-SPAP response to ICI 118551 was only small, a low concentration of forskolin (100 nM) was added to enhance adenylyl cyclase activity and, if anything, to make any contribution from Gi-proteins more obvious. There was however, still no change in the ICI 118551 response seen after PTX treatment (Table 4). We therefore found no evidence of Gi-protein involvement in any of the CRE-gene transcription responses.
cAMP and CRE-SPAP gene transcription responses to β 2-agonists, partial agonists, and β -blockers after pre-incubation with PTX (100 ng/ml for 24 h)
No CRE-SPAP responses to timolol, bisoprolol, metoprolol, or sotalol were seen in either the absence or after incubation with PTX.
In view of the clear inverse agonism seen at the level of cAMP accumulation, the effect of PTX at this level was also examined to ensure that there was no contribution from Gi-mediated effects either. Once again, no PTX effects were seen for any of the responses, including ICI 118551 and the inverse agonist [3H]cAMP accumulation response of propranolol (Table 4).
To ensure that these batches of PTX at this concentration were capable of inactivating Gi-proteins, parallel experiments with CHO-A1-SPAP cells were performed. Here, stimulation of the Gi-coupled adenosine A1-receptor by the agonist N6-cyclopentyl adenosine caused a decrease in cAMP accumulation and also in downstream SPAP production and this was ablated by 24 h of preincubation with PTX (100 ng/ml) in both assays (n = 4, data not shown).
Lack of Involvement of Gq/11- and G12/13-Protein-Mediated Pathways. To investigate whether propranolol can signal to CREB via Gq/11-protein-dependent stimulation of phospholipase C and subsequent activation of the MAP kinase cascade via PKC, experiments were performed in the presence of two different PKC inhibitors. Neither Gö 6976 (3 μM) or Gö 6986 (3 μM) produced any significant attenuation of the CRE-mediated SPAP response to propranolol (Fig. 12a). These two PKC inhibitors were also without marked effect on the responses to isoprenaline (112.4 ± 3.1% and 78.5 ± 7.6% of control for 3 μM Gö 6983 and 3 μM Gö 6976, respectively; n = 3) and alprenolol (108.8 ± 5.7% and 97.4 ± 8.3%; n = 3). Similarly, down-regulation of PKC isoforms α, δ, and ϵ (Megson et al., 2001) with 24-h treatment with 1 μM PDBu produced no significant attenuation of the responses to propranolol (Fig. 12a), isoprenaline (103.9 ± 11.7% of control; n = 3), and alprenolol (122.7 ± 8.7%; n = 3). Finally, no significant stimulation of [3H]inositol phosphate accumulation was produced by 10 μM propranolol (104.3 ± 3.0% basal, n = 5) or 10 μM isoprenaline (93.0 ± 2.4% basal, n = 5) in these cells (Fig. 12b). UTP (100 μM), however, stimulated a marked stimulation of inositol phosphate accumulation (246.0 ± 8.2% basal, n = 5) by activating endogenous P2y2 receptors.
Finally, the potential role of G12/13-protein involvement was assessed. The small molecular weight GTPase Rho has been shown to be the major effector of the G12/13 family of heterotrimeric G-proteins (Gohla et al., 1999). However, the Rho kinase inhibitor Y 27632 (3-10 μM; Klages et al., 1999) had no significant effect on responses to propranolol (Fig. 12c). Similarly no effects was seen with alprenolol (115.1 ± 15.1% control, n = 3, 10 μM Y 27632) and isoprenaline (115.4 ± 10.6% control, n = 3, 10 μM Y 27632) as agonists.
Discussion
β-Blockers have recently been used in the management of heart failure (CIBIS-II, 1999; MERIT-F, 1999; Eichhorn and Bristow, 2001) where simple catecholamine blockade seems not to be the sole explanation for the beneficial effects seen. β-Blockers have even been proposed as a potential future treatment in chronic airway disease in a manner analogous to their use in heart failure (Bond, 2001). Here, we have shown that many clinically used β-blockers have small agonist actions at the level of cAMP accumulation, whereas others are inverse agonists. However, at the level of gene transcription, many of these β-blockers produce substantial agonist responses that may underlie some of their clinical effects.
All of the β-blocker agonist responses seem to contain a single-component response and each response was antagonized by the selective β2-ligand ICI 118551 to yield comparable KD values. This is markedly different from the human β1-adrenoceptor, where β-blockers reveal two distinct sites through which different compounds elicit agonist responses (Konkar et al., 2000; Baker et al., 2003). The data obtained here for β2-adrenoceptor antagonism by ICI 118551 provides no evidence that clinically used β-blockers can discriminate an analogous second site on the human β2-adrenoceptor.
Propranolol decreased cAMP accumulation while stimulating a CRE-mediated gene transcription response. The β2-inverse agonist response of propranolol at the level of cAMP has been reported previously (Chidiac et al., 1994; Azzi et al., 2001). The agonist gene transcription response to propranolol, however, was also β2-adrenoceptor-mediated because it was inhibited by ICI 118551. Propranolol itself also antagonized the agonist responses to more efficacious β-blockers, and the responses were not present in cells lacking the receptor. These propranolol-induced responses show that the relationship between cAMP and CRE-mediated gene transcription cannot be a simple linear cascade.
We therefore examined the potential contribution of the MAP kinase cascade in generating the CRE-SPAP response. Whereas SB 203580, a p38 MAP kinase inhibitor, had no effect, PD 98059, U0126, and the Rsk-2 inhibitor RO 318220 all reduced the CRE-SPAP responses to isoprenaline and a range of other β2-agonists of differing efficacy. Although U 0126 and PD 98059 inhibit the activation of both MEK1 and MEK2 and interact with a common site on the enzyme, U0126 has a much higher affinity than PD 98059 for MEK1/2 (Favata et al., 1998; Davies et al., 2000), and this probably explains the differing degrees of inhibition. However, both of these MEK1/2 inhibitors have been reported to have little activity on other kinases including PKA (Favata et al., 1998; Davies et al., 2000). A substantial part of the CRE gene transcription response to β-adrenoceptor stimulation, therefore, actually occurs via the p42/44 MAP kinase pathway (Fig. 13). Forskolin, a direct adenylyl cyclase activator, cAMP analogs, and IBMX also stimulated responses that were sensitive to these inhibitors, suggesting that the p42/44 MAP kinase pathway involvement is downstream of cAMP (Fig. 13).

Schematic representation of the signaling cascades from the β2-adrenoceptor to CRE gene transcription. The pathway activated by propranolol from the β2-adrenoceptor to MEK-1 is likely to involve a heterotrimeric G-protein-independent mechanism. However, the exact point at which this pathway feeds into the cascade above the level of MEK-1 remains to be established.
However, the propranolol-induced agonist gene transcription response was also sensitive to U0126. This suggests that a further separate pathway exists from receptor to CRE-gene transcription that is independent of cAMP. Direct confirmation for the activation by propranolol (and also isoprenaline, alprenolol, and forskolin) of the p42/44 MAP kinase pathway was provided by both Western blotting and ELISA analysis of the production of active phospho-p42/44 MAP kinase. This increase in phospho-p42/44 MAP kinase seen in response to propranolol shows conclusively that this activation can occur independently of an increase in cAMP, because propranolol itself actively decreased cAMP levels.
H89, a well known PKA inhibitor, has also been reported to be a β-antagonist (Penn et al., 1999). H89 stimulated both cAMP accumulation and CRE-mediated gene transcription, and these H89-agonist responses were blocked by ICI 118551, confirming that they were β2-adrenoceptor-mediated. Thus, at concentrations at which H89 should be blocking PKA (Penn et al., 1999), there was substantial downstream H89-induced, β2-mediated CRE gene transcription. This suggests the existence of an alternative PKA-independent pathway to CRE gene transcription, although incomplete H89-induced PKA inhibition cannot be excluded. H89 also reduced the maximum forskolin and isoprenaline-stimulated gene transcription responses, presumably by the inhibition of PKA. Thus, the H89 data suggest the existence of two pathways downstream from cAMP: one involving a cAMP-dependent, PKA-independent activation of the MAP kinase pathway and another involving PKA activation. In the latter case, the link between PKA and CREB phosphorylation could be mediated either directly or via the p42/44-MAP kinase pathway (Fig. 13). These data do not explain, however, the agonist effect of propranolol on CRE-mediated gene transcription and therefore suggest that propranolol must be recruiting the p42/44-MAP kinase pathway in a non-Gs-protein-dependent manner.
The inverse agonist nature of several β-blockers was clearly seen at the level of cAMP accumulation, but these inverse agonist responses were relatively difficult to ascertain at the level of CRE-mediated gene transcription. If the cascade is linear, then the relatively large decreases in cAMP should be translated into clearly visible decreases at the level of CRE-mediated gene transcription. The discrepancy in the extent of the response between the two levels suggests that the relationship between cAMP production and gene transcription is not a simple linear cascade. It also raises the question of whether some of the decreases in basal cAMP accumulation are occurring via another G-protein activation rather than true inverse agonism. Furthermore, an explanation for the paradoxical responses to propranolol could be that propranolol caused the β2-adrenoceptor to couple to more than one G-protein or switch coupling from Gs to another G-protein. This has previously been reported at the β2-adrenoceptor with evidence of a switch occurring from Gs to Gi coupling after activation of PKA (Daaka et al., 1997). Alternatively, propranolol-induced Gi protein coupling alone could be an explanation in that the Gαi could decrease cAMP, whereas the βγ-subunit stimulates a secondary non-cAMP-mediated cascade.
Preincubation with pertussis toxin (which ADP-ribosylates Gαi and interferes with receptor activation) had no effect on the CRE gene transcription responses to a range of β2-agonists of differing efficacy and thus failed to demonstrate any Gi-involvement in the 5-h gene transcription responses. PTX also had no effect on the inverse agonist responses at the level of either cAMP accumulation or CRE-gene transcription, suggesting that they are true Gs-induced inverse agonist responses and not a consequence of Gi/o-protein coupling. Furthermore, neither the cAMP inverse agonist or CRE-SPAP stimulation response to propranolol was altered by preincubation with PTX, clearly demonstrating that there is no Gi/o-protein involvement in the generation of either of these propranolol-elicited responses.
The role of the other G-proteins was then investigated. Gq/11 involvement is unlikely, because propranolol (or isoprenaline) was unable to stimulate inositol phospholipid hydrolysis in these cells. In addition, preincubation with two PKC inhibitors, Gö 69076 and Gö 69083, was without significant effect on isoprenaline-, propranolol-, and alprenolol-stimulated CRE-SPAP responses. Similarly, down-regulation of PKC isoforms α, δ, and ϵ (Megson et al., 2001) with 24-h treatment with PDBu was without significant effect. Because these different methods did not detect any Gq/11-PKC involvement, this also confirms that the reduction of the responses seen with RO 318220 above was indeed caused by inhibition of Rsk-2 (Alessi, 1997) and not PKC activity. Finally, the G12/13 family of heterotrimeric G-proteins activate the small molecular weight GTPase Rho (Gohla et al., 1999). However, the Rho kinase inhibitor Y 27632 (Klages et al., 1999) had no significant effect on responses to propranolol, alprenolol, or isoprenaline. It seems possible, therefore, that propranolol (and possibly other β-blockers) may activate other inputs to the secondary cAMP-independent MAP kinase pathway by a mechanism that does not involve heterotrimeric G-proteins but is still β2-adrenoceptor-mediated.
There is increasing evidence that G-protein-coupled receptors (including the β2-adrenoceptor) can elicit functional responses independently of G-protein interactions (Hall et al., 1998; Luttrell et al., 1999; Brzostowski and Kimmel, 2001; Fan et al., 2001; Seta et al., 2002). It is quite possible, therefore, that different agonist-directed conformations of the β2-adrenoceptor exist that can activate separate signaling cascades (i.e., G-protein-dependent and -independent pathways) with consequent differences in observed agonist efficacy for compounds such as propranolol. Thus, a molecule may induce a conformational change in the receptor such that the G-protein-coupling region of the receptor leads to inverse agonism, but a different region (e.g., the C-terminal tail) elicits an agonist response via a different signaling protein. This would be equivalent to a certain extent to the concept of agonist trafficking to different G-proteins proposed by Kenakin (1995). However, in classic agonist trafficking, the interaction with different heterotrimeric G-proteins (e.g., Gs and Gi) is usually considered to be mutually exclusive, whereas if other signaling proteins (non-G-proteins) are involved that target different sites on the receptor, this may not be the case. In this situation (agonist-directed domain signaling), agonism of one pathway and inverse agonism of another could occur simultaneously. Because propranolol is able to act as an agonist of the cAMP-independent MAP kinase-gene transcription response but as an inverse agonist on the cAMP response, its ability to elicit opposing responses via the same receptor to two different signaling cascades (i.e., cAMP-dependent and cAMP-independent pathways) can be clearly demonstrated. However, it is possible that those β-agonists (including many clinically used β-blockers) that stimulate both cAMP accumulation and MAP kinase activation may also be able to stimulate MAP kinase independently of cAMP. The relative extent to which these β-agonists stimulate MAP kinase activation via these cAMP-dependent and -independent pathways however remains to be established.
Acknowledgments
We thank Anne Webber for providing the CHO-A1-SPAP cell line.
Footnotes
-
J.G.B. holds a Wellcome Trust Clinical Training Fellowship.
-
ABBREVIATIONS: PKA, protein kinase A; CRE, cAMP response element-binding protein; MAP, mitogen-activated protein; CGP-12177, 4-[3-[(1,1-dimethylethyl)amino]2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one; CGP 20712A, [2-(3-carbamoyl-4-hydroxyphenoxy)-ethyl-amino]-3-[4-(1-methyl-4-trifluormethyl-2-imidazolyl)-phenoxy]-2-propanolmethanesulfonate; CRE, cAMP response element; CHO, Chinese hamster ovary; U0126, 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophynyltio)butadiene; PTX, pertussis toxin; SB 203580, 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole;; RO 318220, 3-[1-[3-(amidinothio)propyl-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide; Gö 6976, 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole; Gö 6983, 2-[1-(3-dimethylaminopropyl)-5-methyloxyindol-3-yl]-3-(1H-indol-3-yl)maleimide; Y 27632, trans-4[(1R)-1aminoethyl-N-4-puridinylcyclohexanecarboxamide; ERK, extracellular signal-regulated kinase; ELISA, enzyme-linked immunosorbent assay; SPAP, secreted placental alkaline phosphatase; DMEM/F12, Dulbecco's modified Eagle's medium/nutrient mixture F12; PDBu, phorbol-12,13-dibutyrate; HBH, Hanks buffered saline solution/HEPES; IBMX, 3-isobutyl-1-methylxanthine; PBS, phosphate-buffered saline; ICI 118551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)-amino]-2-butanol; PKC, protein kinase C; ANOVA, analysis of variance; MEK, mitogen-activated protein kinase kinase; CPT-cAMP, 8-(4-chlorophenylthio)adenosine-3′,5′-cyclic monophosphate; 8-bromo-cAMP, 8-bromoadenosine-3′,5′-cyclic monophosphate.
- Received January 31, 2003.
- Accepted August 28, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}