


Abstract
The structural basis of ligand affinity can be approached by studying the interactions between a drug and receptor residues; the basis for efficacy is more complex and must involve activation-associated conformational changes. We have used wild-type (WT), a constitutively active mutant (CAM), and a “constitutively inactive” mutant β2 adrenergic receptor (β2AR) to investigate changes in the binding site that accompany binding and activation. The active state (R*) probably involves repositioning of at least some of the agonist-contact residues, thereby optimizing their interactions with agonist and resulting in a higher affinity for agonist. A comparison of the binding affinities of a series of phenethylamine derivatives for WT revealed a remarkable synergism between the various functional groups present in epinephrine. Binding affinity was essentially unchanged with addition of β-OH, N-CH3, or catechol OHs to phenethylamine. In contrast, when each of these same groups was added to the appropriate compound, already containing the other two groups, to make epinephrine, the increase in affinity was quite large (60- to 120-fold). An initial interaction between two or more contacts may stabilize an intermediate conformation of β2AR, R′, either by altering amino acid side chain rotamer conformations or by a more global conformational change involving the repositioning of transmembrane segments. The pattern of these effects was different in the CAM in that fewer interactions were required to observe the synergistic effect, consistent with the hypothesis that the CAM mutation enriches the proportion of receptors in R* or in R′ from which R* is more readily assumed.
The biological activity of an agonist is dependent on both its affinity and its efficacy. The structural basis for affinity can be approached by studying the interaction between a drug and critical contact residues in the receptor. Many of these contact sites for catecholamines have been identified in the β2AR, the best-studied neurotransmitter G-protein-coupled receptor (GPCR). The protonated amine of epinephrine interacts with Asp1133.32 (see Materials and Methods for a description of the indexing of residues) in the third transmembrane segment (TM3) of β2AR (Strader et al., 1988); the catechol OHs interact with Ser2035.42, Ser2045.43, and Ser2075.46 in TM5 (Strader et al., 1989; Liapakis et al., 2000); and the β-OH interacts with Asn2936.55 in TM6 (Wieland et al., 1996). The catecholamine aromatic ring interacts with a cluster of aromatic residues in TM6 (Strader et al., 1994; Cho et al., 1995; Javitch et al., 1998). Based on a model of β2AR, the N-CH3 of epinephrine has been proposed to interact with TM7 (Gouldson et al., 1997); however, this has not yet been experimentally verified.
In contrast to affinity, the structural basis for efficacy is more complex, because the detailed conformational changes associated with receptor activation are not known. Fluorescence spectroscopy (Ghanouni et al., 2001), electron paramagnetic spectroscopy (Resek et al., 1993), and alterations in the accessibility of charged sulfhydryl reagents (Javitch et al., 1997; Rasmussen et al., 1999; Ballesteros et al., 2001; Shi et al., 2002) have been used to infer that activation of GPCRs is associated with movements of their TMs, particularly TM6. Mutagenesis of the residues that contact agonists and/or their modification by fluorescent probes, spin labels, or other sulfhydryl reagents, however, would abolish or dramatically alter their functional interactions with agonist. Thus, it is particularly difficult to determine movements of crucial contact residues, although this information is vital to the elucidation of the mechanism by which agonist interaction with these residues contributes to receptor activation and thus to agonist efficacy.
In the two-state model, receptor exists in an equilibrium between two conformations, the active, R*, and the inactive, R (Leff, 1995). Agonist binds with higher affinity to R* than to R, thereby stabilizing R* and shifting the equilibrium R↔R* to the right. Agonist efficacy is related, therefore, to the ratio between the affinity of a drug for R and R* (affinity shift) and reflects the degree of stabilization of receptor in, and its transition to, R*. The experimental measurement of efficacy, however, is highly system-dependent; in a number of heterologous expression systems, the weak partial agonist dopamine has been shown to display a high intrinsic activity at the β2AR, with ∼90% of the effect of the full agonist epinephrine (Del Carmine et al., 2002). Agonist efficacy has also been approached experimentally by measuring the ratio between the affinities of high- and low-affinity components of agonist binding (Kearn et al., 1999). However, in membranes of COS-7 or HEK 293 cells expressing high levels of wild-type (WT) β2AR, GTP has minimal effects on agonist binding, consistent with the receptor being in excess over G-protein and therefore being predominantly in the low-affinity state (Samama et al., 1993). This makes it difficult to measure this ratio in such a heterologous expression system.
Constitutively active receptor mutants (CAM) are active in the absence of agonist (Samama et al., 1993; Rasmussen et al., 1999; Ballesteros et al., 2001). These receptors seem to more readily assume the R* state and have substantially higher affinity for agonists than does WT receptor. Although signaling by a CAM is dependent on the presence of G-protein, its higher propensity for being in R* and therefore its higher affinity for agonist is observed even in the absence of G-protein (Samama et al., 1993). Thus, a CAM provides a model for the active state of the receptor, and the ratio between the affinities (affinity shift) of agonists for WT and CAM β2AR was shown to correlate with their efficacies, even in an expression system in which the high-affinity component of agonist binding could not be measured (Samama et al., 1993).
As discussed above, many structure-activity relationship studies, by testing the binding to WT β2AR of an assortment of modified adrenergic ligands, have determined the chemical groups that are important for ligand function. In this study, we extend these structure-activity relationship studies by examining the binding of a series of systematically substituted phenethylamine compounds to a series of β2ARs with different proportions in R*, including WT and CAM, to investigate structural changes in the binding site that accompany receptor activation.
Materials and Methods
Numbering of Residues. Residues are numbered according to their positions in the human β2AR sequence. We also index residues relative to the most conserved residue in the transmembrane segment in which it is located (Ballesteros and Weinstein, 1995). By definition, the most conserved residue is assigned the position index “50” (e.g., Pro2115.50) and therefore Val2105.49 and Leu2125.51. This indexing simplifies the identification of aligned residues in different GPCRs.
β2 Plasmids and Site-Directed Mutagenesis. The DNA sequence encoding the WT human β2 adrenergic receptor, epitope-tagged at the amino terminus with the cleavable influenza-hemagglutinin signal sequence followed by the “FLAG” epitope (Kodak IBI, New Haven, CT) and tagged with six histidines at the carboxy terminus (Kobilka, 1995), was a gift from Dr. B. Kobilka (Stanford Brain Research Institute, Palo Alto, CA). This DNA was subcloned into the bicistronic expression vector pcin4 (Rees et al., 1996), a gift from Dr. S. Rees (GlaxoSmithKline, Uxbridge, Middlesex, UK), thereby creating the vector pcin4-SFβ2H. The CAM β2 receptor (Samama et al., 1993) (L266S/K267R/H269K/L272A) was a gift from Dr. R.J. Lefkowitz (Department of Biochemistry, Duke University Medical Center, Durham, NC), and the fragment encoding the CAM mutation was subcloned into pcin4-SFβ2H to create the vector, pcin4-SFCAMβ2H. The mutation of Asp792.50 to Asn (D79N) was generated by the polymerase chain reaction-mediated mutagenesis using Pfu polymerase (Stratagene, La Jolla, CA). The polymerase chain reaction-generated DNA fragment containing the mutation was subcloned into the pcin4-SFβ2H plasmid, and the mutation was confirmed by DNA sequencing.
Cell Culture and Transfection. HEK 293 cells were grown in Dulbecco's modified Eagle's medium/Ham's F-12 medium (1:1) containing 3.15 g/liter glucose and 10% bovine calf serum at 37°C and 5% CO2. Cell transfection with the WT, the CAM, and the D79N β2AR and selection for generation of cells stably expressing the receptors were performed as described previously (Javitch et al., 1997; Liapakis et al., 2000).
Membrane Preparation. Cell suspensions, prepared as described previously (Liapakis et al., 2000), were centrifuged at 1000g for 5 min at 4°C, and the pellets were homogenized in 1 ml of binding buffer (25 mM HEPES, pH 7.4, 5 mM MgCl2, 1 mM EDTA, and 0.006% bovine serum albumin) using an OMNI 1000 Polytron homogenizer at setting 26 to 30 for 10 to 15 s at 4°C. The homogenates were centrifuged at 13,500g for 10 min at 4°C, and the membrane pellets (from a confluent 100-mm dish) were resuspended by homogenization as described above in 1 ml of binding buffer. The membrane suspensions were diluted (typically 1:20-1:40) in binding buffer and used for radioligand binding studies.
[3H]CGP-12177 Binding. Aliquots of diluted membrane suspension (200 μl) were incubated in binding buffer with increasing concentrations of the various agonists tested (obtained from Sigma-Aldrich, St. Louis, MO and Tocris Cookson Inc., Ellisville, MO) in the presence of 0.7 nM [3H]CGP-12177 (Amersham Biosciences Inc., Piscataway, NJ) in competition experiments or with varying concentrations of [3H]CGP-12177 (50-1200 pM) in saturation experiments. The total volume was adjusted to 0.5 ml, and the binding experiments were performed as described previously (Javitch et al., 1997; Liapakis et al., 2000). The amount of membrane used was adjusted to insure that no ligand depletion occurred (i.e., that the specific binding was always equal to or less than 10% of the total concentration of the added radioligand). Specific [3H]CGP-12177 binding was defined as total binding less nonspecific binding in the presence of 1 μM alprenolol (Sigma/RBI, Natick, MA). Data for saturation and competition binding were analyzed by nonlinear regression analysis using Prism 3.0 (GraphPad Software, San Diego, CA). IC50 and Kd values were obtained by fitting the data from the competition studies to a one-site competition model and from the saturation studies to a one-site binding (hyperbola) model, respectively. Ki values were determined using the equation Ki = IC50/(1 + L/Kd) (Cheng and Prusoff, 1973).
cAMP Accumulation Assays. HEK 293 cells stably expressing WT or D79N β2AR were plated onto 96-well cell culture plates (pretreated with poly-l-lysine, 0.1 mg/ml) at a density of 40,000 to 60,000 cells/well. After incubation overnight at 37°C in 5% CO2, the cells were 95 to 100% confluent. The medium was removed, and 100 μl of Opti-MEM (Invitrogen, Carlsbad, CA) with or without (control) 135 μM N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) was added. The cells were incubated for 1 h at 37°C. The medium was removed, and 100 μl of assay buffer (25 mM HEPES, pH 7.4, 2 mM choline, 288 mM sucrose, 0.9 mM CaCl2, 0.5 mM MgCl2, and 1 mM 3-isobutyl-1-methylxanthine) was added. After a 1-h incubation at 37°C, more assay buffer without (basal levels) or with increasing concentrations of agonists was added to a total volume of 200 μl, and the incubation was continued for 10 min at 37°C. At the end of the incubation, the assay buffer was removed. The cells were placed on ice and lysed with 3% trichloroacetic acid. Lysates were incubated on ice for 30 to 60 min and stored at -20°C. After 1 to 5 days, frozen lysates were thawed, centrifuged at 1800g for 10 min at 4°C, and the supernatants were neutralized with 2 N NaOH. Quantification of cAMP in the neutralized supernatants was performed using a competitive binding assay as described previously by Watts et al. (1998) and Nordstedt and Fredholm (1990), with minor modifications. Supernatants were transferred to polypropylene minitubes (20 μl/tube) containing buffer B (100 mM Tris-HCl, pH 7.4, 100 mM NaCl, and 5 mM EDTA) with 1 to 1.1 nM [2,8-3H]-adenosine 3′,5′-cyclic phosphate (Amersham Biosciences Inc.). Subsequently, cAMP-binding protein (∼100 mg of crude bovine adrenal cortex extract in 500 μl of buffer B) was added to each tube. After incubation on ice for 2.5 to 3 h, the mixtures were filtered through GF/B glass fiber filters as described for radioligand binding assays. The amount of cAMP in each sample (one tenth of a well) was determined by comparison with a standard curve of known concentrations of unlabeled cAMP (0.3-100 pmol/tube). EC50 values were obtained by fitting the data to a one-site sigmoidal dose response model using nonlinear regression analysis.
Results
In this study, we tested the functional properties of three β2ARs using a series of different adrenergic ligands. These ligands are phenethylamine and its derivatives that were created by adding one or more of the following functional groups: N-CH3, β-OH, and catechol OHs (Fig. 1). Epinephrine is the fully substituted phenethylamine derivative containing all of the above functional groups.
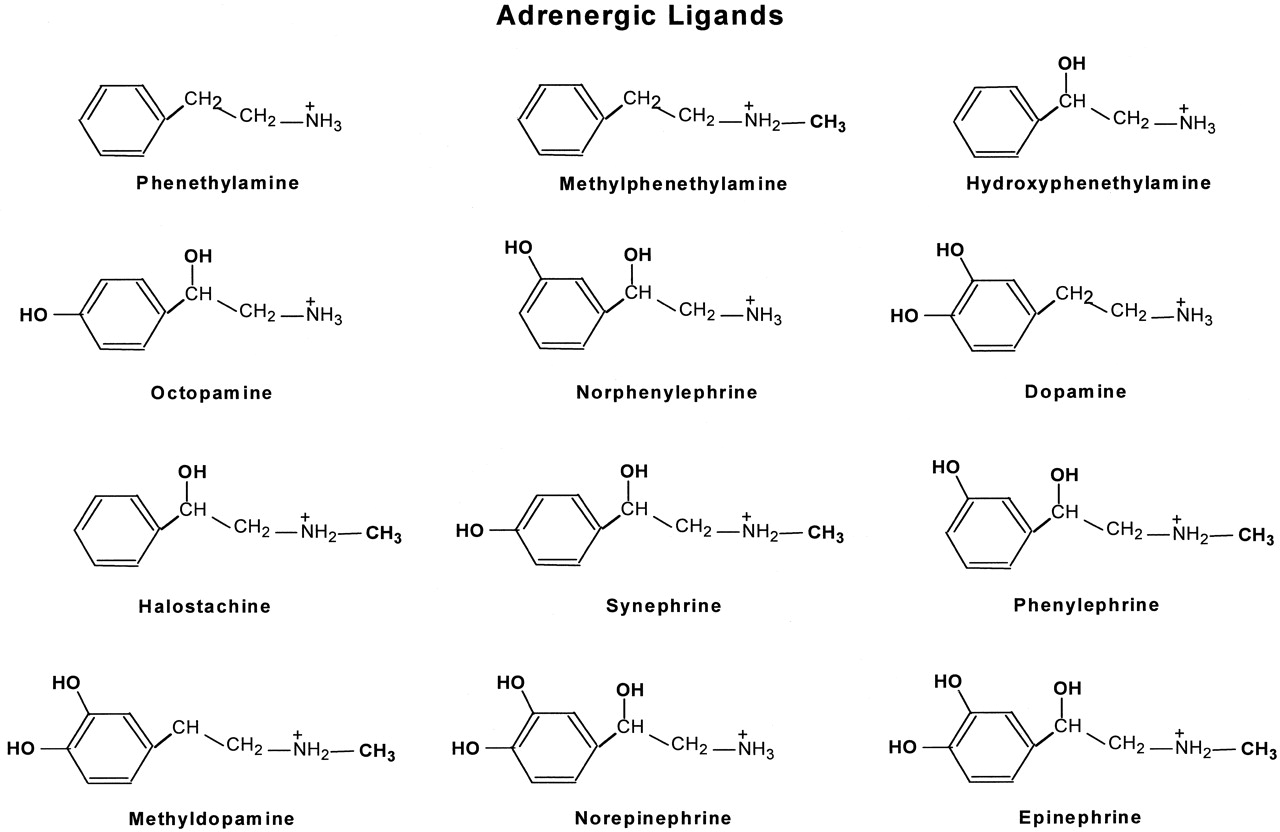
Chemical structures of adrenergic ligands. The adrenergic ligands used in this study are phenethylamine and its derivatives that were created by adding to phenethylamine one or more of the following functional groups: N-CH3, β-OH, and catechol OHs. Epinephrine is the fully substituted phenethylamine derivative containing all of the above functional groups.
Activation of Adenylyl Cyclase by WT β2AR. High expression of the WT β2AR in HEK 293 cells (Bmax = ∼100 fmol/cm2 of culture dish; data not shown) resulted in masking of the partial agonist activity of the drugs tested in this study. Thus, the maximal stimulation of cAMP accumulation by the partial agonists phenylephrine, synephrine, dopamine, and halostachine was similar to that of the full agonist epinephrine (EPI) (data not shown). To overcome this problem, we measured agonist-stimulated cAMP accumulation after inactivating 98 to 99% of the receptors by pretreatment with the alkylating reagent EEDQ (Belleau et al., 1969). In the resulting low receptor reserve environment, we found that phenylephrine, synephrine, halostachine, and dopamine were indeed partial agonists, being 40, 24, 19, and 46% as effective as the full agonist, epinephrine, in maximally stimulating cAMP accumulation in WT receptor (Table 1 and Fig. 2). In contrast, the intrinsic activities of norepinephrine and methyldopamine were similar to that of epinephrine, suggesting that these ligands, which lack the N-CH3 or the β-OH, respectively, fully (or nearly fully) activated the β2AR.
Agonist binding to wild-type and CAM β 2AR and stimulation of cAMP accumulation by wild-type β 2AR
Competition binding assays were performed on HEK 293 membranes stably expressing wild-type or CAM β 2AR, and cAMP accumulation studies were performed in intact HEK 293 cells stably expressing the wild-type β 2AR as described under Materials and Methods. The Ki values were determined according to the method of Cheng and Prusoff using the IC50 values from the competition binding isotherms (Cheng and Prusoff, 1973). The affinity shift is the ratio between the Ki values for WT and CAM β 2AR. Ki/KiPE is the ratio between the Ki values for phenethylamine and each drug. The potencies (EC50 values) of agonists to stimulate cAMP accumulation were determined by nonlinear regression analysis. Values are presented as mean ± S.E. from three to eight independent experiments.

Agonist-induced stimulation of cAMP accumulation by wild-type β2AR. Stimulation of cAMP accumulation by the indicated concentrations of the series of agonists was performed in intact HEK 293 cells stably expressing the wild-type β2AR after treatment with EEDQ, as described under Materials and Methods. The data were fit to a one-site sigmoidal dose-response model by nonlinear regression, and the logEC50 values were calculated and shown in Table 1. Maximal cAMP accumulation is presented as the percentage of the maximal stimulation obtained with 1 mM epinephrine. The values shown in each curve are from a representative experiment performed four to eight times with similar results.
Agonist Binding to WT and CAM β2AR. The affinities of the various agonist derivatives for WT and CAM were determined from competition experiments of [3H]CGP-12177 performed under equilibrium conditions in membranes from HEK 293 cells stably expressing the receptors (Table 1 and Fig. 3, A and B). Phenethylamine, which contains the protonated amine and the aromatic ring, had equal low affinities for the WT and CAM, resulting in an affinity shift of ∼1 (defined as the ratio between their apparent binding affinities for the WT and the CAM) (Table 1). In contrast, epinephrine, which contains the catechol OHs, the β-OH, and the N-CH3 in addition to the protonated amine and the aromatic ring, had an affinity 400- and 16,000-fold higher than phenethylamine in WT and CAM, respectively, thereby resulting in an affinity shift of ∼54 (Table 1).

Competition binding isotherms of adrenergic agonists to wild-type and CAM β2AR. Competition of [3H]CGP-12177-specific binding of the series of agonists was performed, as described under Materials and Methods, on membranes from HEK 293 cells stably expressing wild type (A) or CAM β2AR (B). The data were fit to a one-site competition model by nonlinear regression, and the logIC50 values were calculated. The logKi values determined from the logIC50 values are shown in Table 1. The values shown in each curve are from a representative experiment performed three to eight times with similar results. Of the 12 agonists screened, only those tested for stimulation of cAMP accumulation are shown above.
The maximal ability of the agonists tested to stimulate cAMP accumulation in HEK cells stably expressing WT was highly correlated with their affinity shift (r2 = 0.87, p < 0.005). Thus, the full agonists epinephrine, methyldopamine, and norepinephrine had the highest affinity shifts (54, 68, and 78, respectively), whereas the partial agonists synephrine, phenylephrine, halostachine, and dopamine had affinity shifts of 18, 17, 7, and 10, respectively (Table 1 and Fig. 4).

Chemical structure of agonists and their affinities for wild-type and CAM β2AR. The affinities of agonists for CAM and wild-type β2AR are represented as • and ○, respectively; the connecting line represents the size of the affinity shift. Epinephrine has four functional groups that were substituted in these experiments: two catechol OHs, a N-CH3 group, and a β-OH group. +, the presence of the specified chemical groups for each phenethylamine derivative.
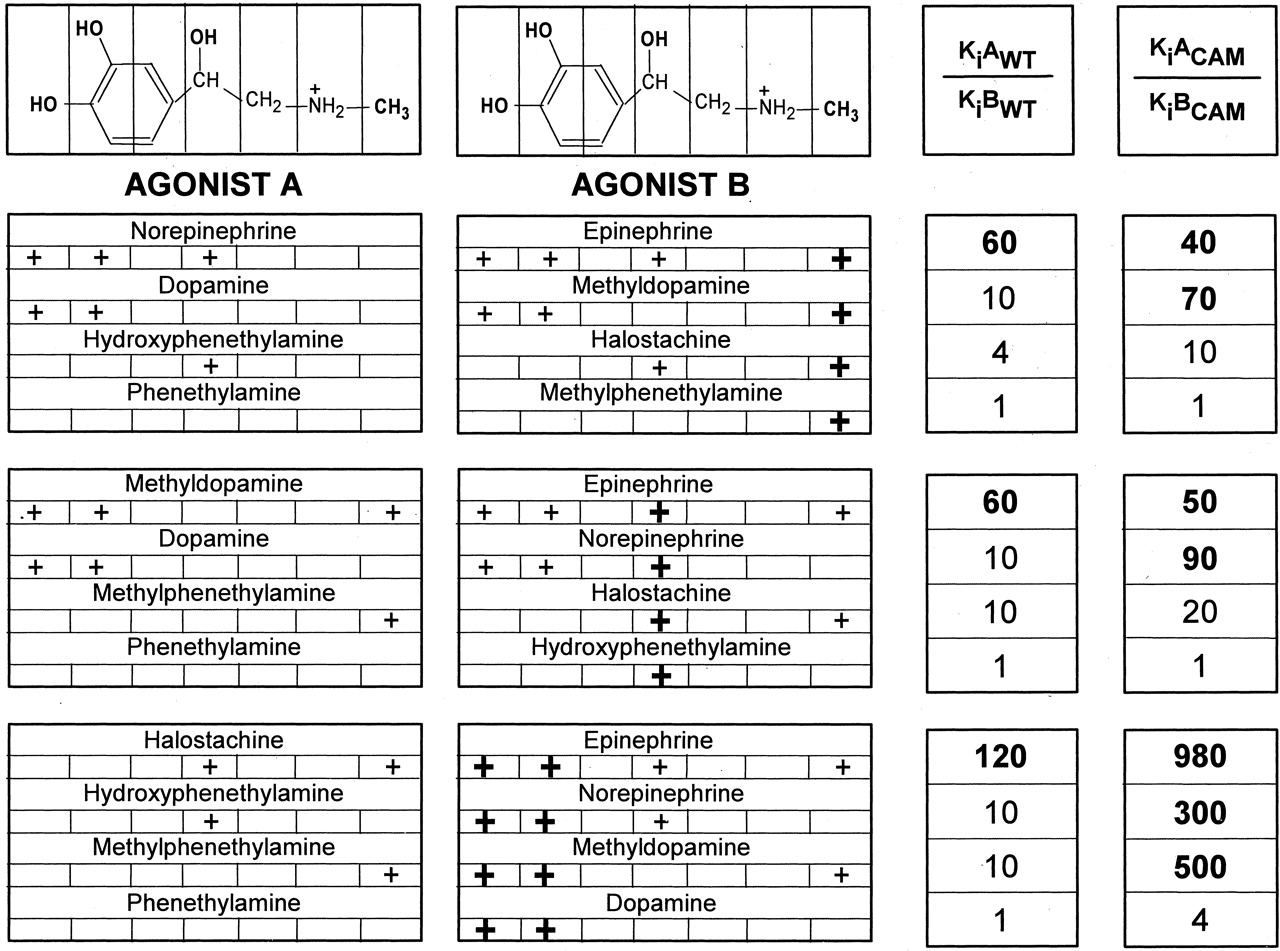
To investigate the interrelationship between the effects of the various functional groups, we expressed the changes in affinity resulting from the addition of each group to the set of substituted phenethylamine compounds. Addition to phenethylamine of either the catechol OHs (dopamine), the β-OH (hydroxyphenethylamine), or the N-CH3 (methylphenethylamine) had no significant effect on affinity for the WT receptor (Table 1 and Fig. 5). Addition of any one of these functional groups to a compound bearing one other group increased their affinities for WT only 4- to 10-fold [considering the catechol OHs as one group and, thus, including adding the N-CH3 to hydroxyphenethylamine or dopamine (Fig. 5), adding the β-OH to methylphenethylamine or dopamine (Fig. 5), or adding the catechol OHs to methylphenethylamine or hydroxyphenethylamine (Fig. 5)]. In marked contrast, addition of the N-CH3 to norepinephrine (Fig. 5), the β-OH to methyldopamine (Fig. 5), or the catechol OHs to halostachine (Fig. 5) (in each case, creating epinephrine) resulted in a synergistic increase of their affinities for WT of 60- to 120-fold.

Affinity change after the addition of N-CH3, β-OH, or catechol OHs into various phenethylamine derivatives. Epinephrine has four functional groups that were substituted in these experiments: two catechol OHs, a N-CH3 group, and a β-OH group. +, the presence of the specified chemical groups for each phenethylamine derivative; the individual group added in each case to agonist A to make agonist B is shown by a bold +. The addition of these functional groups to agonist A changed its affinity for WT and CAM β2AR by KiAWT/KiBWT- and KiACAM/KiBCAM-fold, respectively. KiAWT, KiACAM, KiBWT and KiBCAM are the affinities of agonists A and B for WT and CAM β2AR, shown in Table 1. The KiAWT/KiBWT and KiACAM/KiBCAMA values in bold represent a large synergistic effect on the resulting affinity after addition of various functional groups to agonist A.
Although addition of the catechol OHs to halostachine produced a 120-fold increase in affinity for WT, addition to halostachine of the m-OH alone (thus making phenylephrine) increased the affinity for WT by only 3.5-fold, whereas addition of the p-OH alone (thus making synephrine) had no effect (Table 1). Thus, there was also a dramatic synergistic effect of the simultaneous presence of the catechol OHs, even starting with the presence of the β-OH and the N-CH3. These results are in agreement with a previous study in which the synergism of the catechol OHs on binding affinity was abolished when either the catechol OHs or the -OH group on the side chain at positions 2035.42, 2045.43, or 2075.46 were removed (Liapakis et al., 2000). These results suggest that the synergistic effect of the catechol OHs on the affinity is associated with their interaction with all three OHs at positions Ser2035.42, Ser2045.43, and Ser2075.46 in β2AR.
The synergistic effect of multiple functional groups on affinity was different in the CAM. In contrast to WT, in which the presence of two other functional groups was necessary to enable the full effect of adding the third group, in CAM, a large synergistic effect was achieved by combining the catechol OHs (again considered as one group) with either the N-CH3 or the β-OH. Thus, addition of the N-CH3 to dopamine to make methyldopamine (Fig. 5), β-OH to dopamine to make norepinephrine (Fig. 5), or catechol OHs to methylphenethylamine or hydroxyphenethylamine to make methyldopamine or norepinephrine, respectively (Fig. 5), produced nearly the same increase in affinity as adding these groups to the appropriate compounds to make epinephrine. Despite this enhanced synergism in the CAM, the simultaneous presence of the catechol OHs was still necessary to produce this effect, just as it was necessary in WT. Although addition of the catechol OHs to halostachine produced a 980-fold increase in affinity for CAM (Fig. 5), addition to halostachine of the m-OH alone (thus making phenylephrine) increased the affinity for CAM by only 9-fold, whereas addition of the p-OH alone (thus making synephrine) increased the affinity for CAM by only 2-fold (Table 1).
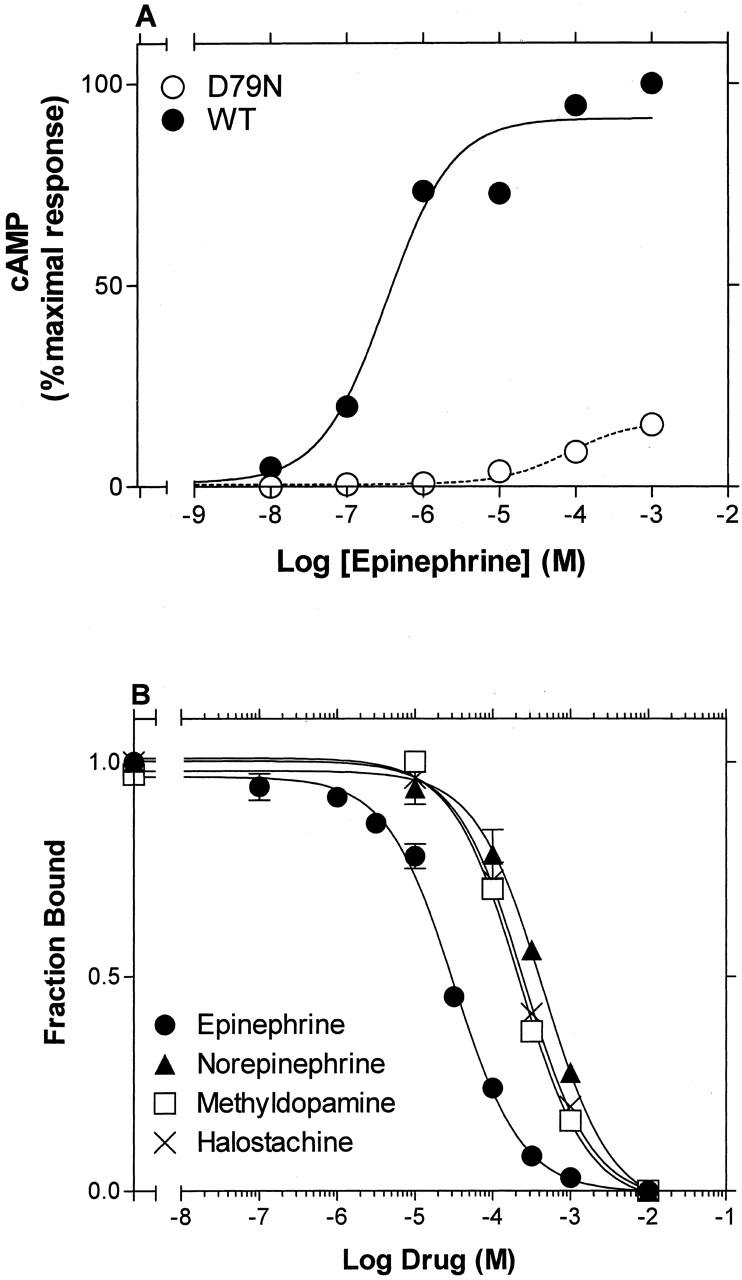
cAMP Accumulation and Ligand Binding Studies on D79N β2AR. The highly conserved Asp2.50 in TM2 has been shown to be important for activation in β2AR (Chung et al., 1988) as well as other GPCRs. Consistent with these reports, mutation of this residue to asparagine, creating D792.50N, dramatically decreased the potency and maximal activity of epinephrine to stimulate cAMP accumulation by 200- and 5-fold, respectively (Fig. 6A).
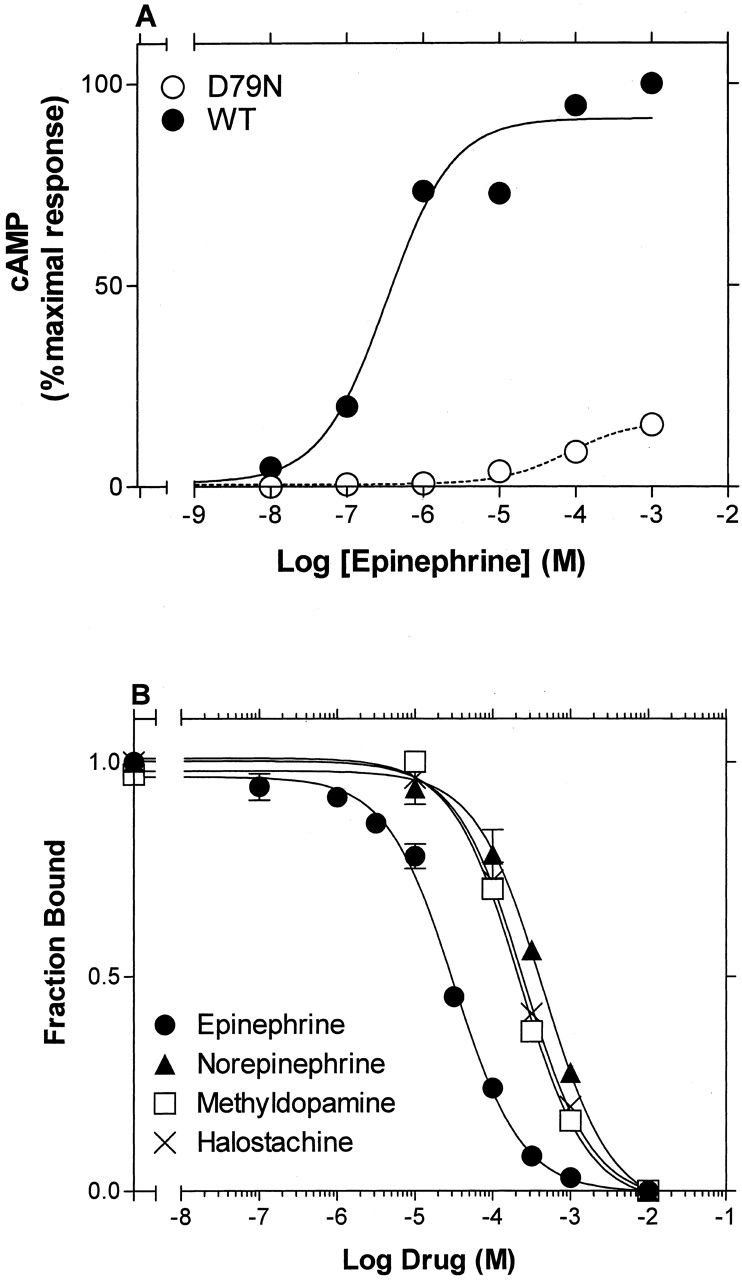
Epinephrine-induced stimulation of cAMP accumulation by wild-type and D792.50N β2AR and ligand binding to D792.50N β2AR. A, stimulation of cAMP accumulation by the indicated concentrations of EPI was performed, as described under Materials and Methods and after treatment with EEDQ, in intact HEK 293 cells stably expressing WT and D792.50N β2AR. The data from three experiments were fit to a one-site sigmoidal dose-response model by nonlinear regression, and the -logEC50 values (mean ± S.E.) were 4.79 ± 0.36 for D792.50N and 7.08 ± 0.33 for WT β2AR. Maximal cAMP accumulation obtained by stimulation of D792.50N with 1 mM EPI is presented as the percentage of the maximal cAMP accumulation obtained by stimulation of WT with 1 mM EPI. The values shown in each curve are from a representative experiment performed three times with similar results. B, competition of [3H]CGP-12177-specific binding by various ligands was performed, as described under Materials and Methods, on membranes from HEK 293 cells stably expressing D792.50N β2AR. The data were fit to a one-site competition model by nonlinear regression, and the logIC50 values were calculated. The -logKi values (mean ± S.E.) determined from the logIC50 values, according to the method of Cheng and Prusoff (1973), were 5.32 ± 0.05 for epinephrine, 4.21 ± 0.06 for norepinephrine, 4.46 ± 0.07 for methyldopamine, and 4.32 ± 0.14 for halostachine. The values shown in each curve are from a representative experiment performed three to four times with similar results.
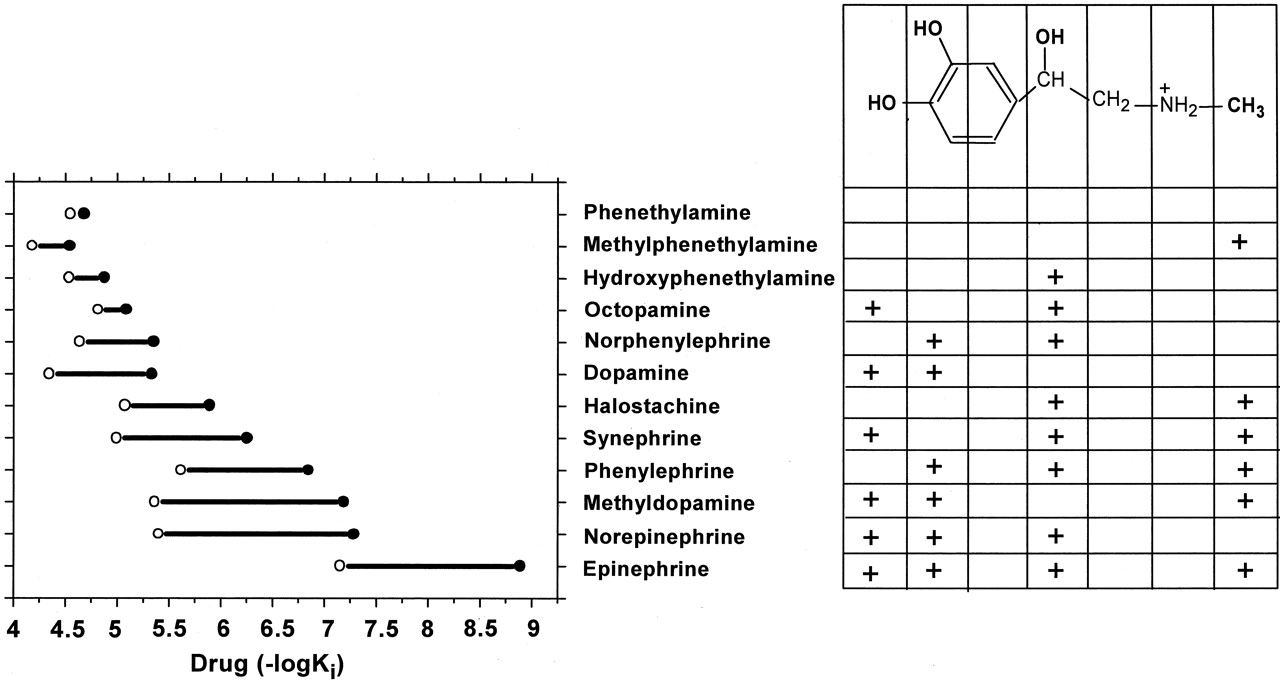
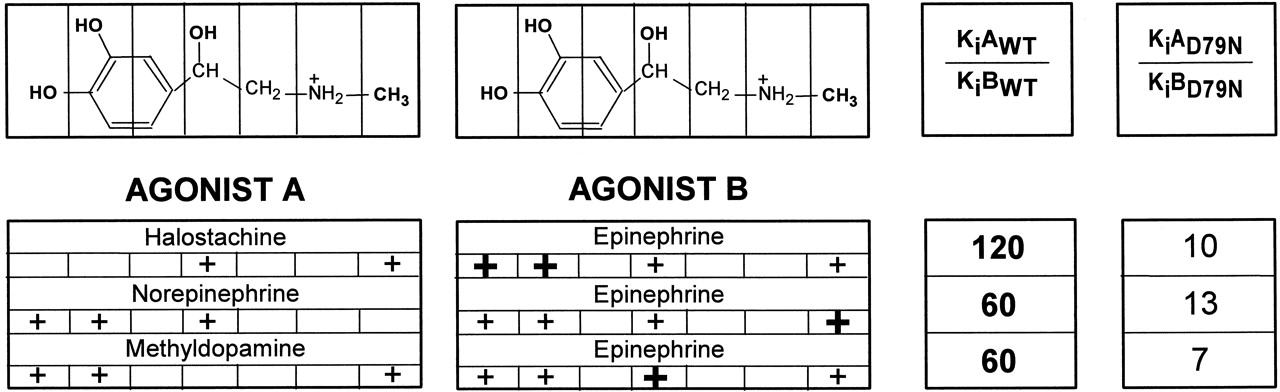
To test whether the impaired ability of D792.50N β2AR to reach the active conformation affected the binding properties of the receptor, we determined the affinities of various agonist derivatives for the D79N mutant in competition experiments with [3H]CGP-12177 (Fig. 6B). It is interesting that the aforementioned synergism of the various functional groups in epinephrine on the binding affinity for WT and CAM was dramatically smaller in D79N: addition of the N-CH3 to norepinephrine, the β-OH to methyldopamine, and the catechol OHs to halostachine, thereby making epinephrine, produced only a 7- to 13-fold increase in affinity for the D792.50N mutant (Fig. 7). Thus, despite the addition of the functional groups to a compound containing two groups, these values were similar to those seen in WT for adding the functional groups to a compound bearing only one group, consistent with impaired synergism and with an impaired ability of D792.50N to reach the active conformation. We refer to such a mutant as a “constitutively inactive” mutant to make the point that relative to WT, its isomerization is shifted toward the inactive state, whereas a CAM is shifted toward the active state.

Affinity change after the addition of the catechol OHs, a β-OH, or a N-CH3 into various phenethylamine derivatives. Epinephrine has four functional groups that were substituted in these experiments: two catechol OHs, a N-CH3 group, and a β-OH group. +, the presence of the specified chemical groups for each phenethylamine derivative; the individual group added in each case to agonist A to make agonist B is shown by a bold +. The addition of these functional groups to agonist A changed its affinity for the WT and D792.50N β2AR by KiAWT/KiBWT- and KiAD79N/KiBD79N-fold, respectively. KiAWT, KiAD79N, KiBWT, and KiBD79N are the affinities of agonists A and B for WT and D792.50N β2AR, shown in Table 1 and Fig. 5B. The KiAWT/KiBWT and KiAD79N/KiBD79N values in bold represent a large synergistic effect on the resulting affinity after addition of various functional groups to agonist A.
Discussion
In contrast to the two-state model (Leff, 1995) in which a receptor isomerizes between an inactive state, R, and an active state, R*, many studies have suggested that β2AR (Ghanouni et al., 2001; Swaminath et al., 2003) and other GPCRs (Noda et al., 1996; Tachibanaki et al., 1997) exist not only in R* and R but in intermediate conformations, R′s, as well. Thus, under the equilibrium conditions of our binding studies, β2AR may exist in a distribution of states, R↔R′s↔R*. Based on the model of Weiss et al. (1996), if a receptor exists in an equilibrium between more than two conformational states, the apparent agonist affinity will represent an average value derived from 1) agonist affinities for each state and 2) the relative proportion at equilibrium of each of the unbound receptor states. Apparent agonist affinity for β2AR should, therefore, represent an average value derived by the affinities for R, R′s, and R* and by the distribution of the unbound receptor among these states. Therefore, a rightward shift of R↔R′s↔R* equilibrium, such as that seen after constitutive activation of β2AR in a CAM, would increase the proportion of receptors in R* and thus the agonist apparent affinity, thereby resulting in an affinity shift compared with WT. In contrast, in a constitutively inactive mutant, such as D792.50N, this equilibrium is shifted leftward, presumably reflecting a decrease in the proportion of receptors in R* and therefore a decreased agonist apparent affinity. Indeed, the apparent affinity of epinephrine for CAM was ∼54-fold higher than that for WT, whereas the affinity of epinephrine for D792.50N was ∼70-fold lower than that for WT, consistent with the notion that CAM isomerizes toward R* to a much larger degree than WT but that WT isomerizes toward R* to a greater extent than D792.50N. This interpretation is consistent with our previous observation that in a rhodopsin homology model, the distance between Asp2.50 and known agonist contact residues is too great for Asp2.50 to be a direct agonist contact (Shi and Javitch, 2002) and that the effect of the mutation of Asp2.50 on agonist binding in β2AR and in other GPCRs is an indirect effect mediated through a change in the distribution of conformational states.
Because R* couples to and activates G-proteins, agonist efficacy must also be related to the degree of receptor transition toward R* and thus to the affinity shift. We found, as was reported previously for a different set of ligands (Samama et al., 1993), that the ratio (affinity shift) between the apparent binding affinities of the agonists tested for CAM and WT was correlated with their maximal abilities to stimulate cAMP accumulation in WT receptor (intrinsic activity). The differential propensities of WT, CAM, and D792.50N β2AR to isomerize toward R* make these receptors excellent tools to investigate, as discussed below, the structural changes in the binding site that accompany receptor activation as well as to discriminate between different possible conformational states of receptor.
The presence of the catechol OHs and either the β-OH or the N-CH3 was absolutely required for full activation of the receptor and for full affinity shift. Thus, despite having much lower affinities than epinephrine, norepinephrine and methyldopamine were full, or nearly full, agonists and had large affinity shifts. These results suggest that the interactions of β2AR with the catechol OHs, the β-OH, and the N-CH3 of epinephrine are optimized in R*, thus stabilizing R* and leading to receptor activation.
Mutagenesis and molecular modeling studies have shown that the contact residues for the catechol OHs are Ser2035.42, Ser2045.43, and Ser2075.46 in TM5 (Strader et al., 1989; Liapakis et al., 2000), whereas the contact residues for the protonated amine and the β-OH are Asp1133.32 in TM3 (Strader et al., 1988) and Asn2936.55 in TM6 (Wieland et al., 1996), respectively. The contact site for the N-CH3 is less clear but may be in TM7 (Gouldson et al., 1997). Thus, the role of epinephrine is to stabilize an activation-associated altered conformational relationship between TM3, TM5, TM6, and possibly TM7. Activation-associated conformational changes, consisting of translations and/or rotations of TM3, TM5, and TM6, have been proposed based on biophysical and biochemical studies in adrenergic and rhodopsin receptors (Farahbakhsh et al., 1993; Farrens et al., 1996; Gether et al., 1997; Javitch et al., 1997; Marjamaki et al., 1999; Shi et al., 2002).
To elucidate further details of the interrelationship of the conformational changes of these various contact residues, we analyzed systematically the energetic contributions of adding each of the functional groups to various substituted phenethylamine derivatives. In WT, the effect of adding each of the groups was 60- to 120-fold greater when added to the otherwise fully substituted compounds norepinephrine, methyldopamine, and halostachine (thus making epinephrine) compared with adding them to phenethylamine itself (Fig. 5). The observed synergistic effect of multiple functional groups on the resulting affinity is associated with the ability of receptor to isomerize toward R*, because the D792.50N mutation, which dramatically impaired activation, resulted in a quite substantial loss of synergism (Fig. 7). In addition, this synergistic effect suggests that an initial interaction between two or more contacts may promote an intermediate conformation of β2AR, R′, that allows more optimal contact with the remaining functional group. In agreement with our data, spectroscopy studies on β2AR have suggested the presence between R and R* of an intermediate conformation, R′, induced by an initial agonist binding (Ghanouni et al., 2001; Swaminath et al., 2003). The existence of intermediate conformations has also been proposed for multiple GPCRs, including rhodopsin (Tachibanaki et al., 1997), AT1-angiotensin (Noda et al., 1996), and M1-muscarinic (Ward et al., 1999).
The pattern of these effects was very different in CAM than in WT in that combination of the catechol OHs with only either the β-OH or the N-CH3 (thus making norepinephrine or methyldopamine, respectively) was sufficient to produce a synergistic effect on the resulting affinity as large as that observed by adding each of the functional groups to the otherwise fully substituted compounds norepinephrine, methyldopamine, and halostachine (thus making epinephrine) (Fig. 5). This synergistic affinity gain from the addition of only two functional groups to produce norepinephrine or methyldopamine is consistent with the CAM mutation enriching the receptor in R′ and/or R*, resulting in synergism with fewer functional groups in our equilibrium binding assays. This enrichment in R′ and/or R* also results in the increased basal activity of CAM.
Because stabilization of a fully active state by agonists requires the presence of either the β-OH or the N-CH3, which interact with residues in TM6 and possibly in TM7, respectively, in combination with the catechol OHs and the protonated amine, which interact with TM5 and TM3 residues, respectively, it is possible that in R′ and/or R*, TM6 and TM7 are conformationally linked and rearrange in concert such that either the interaction of the β-OH with TM6 or the possible interaction of the N-CH3 with TM7 is sufficient for full efficacy. This is consistent with a molecular model of 5-hydroxytryptamine2A receptor in which TM6 and TM7 have been suggested to interact with each other and an activation-associated movement of TM7 affects the motion of TM6 (Luo et al., 1994). In addition, it is possible that the active species of β2AR stabilized by the two functional groups of methyldopamine and norepinephrine might represent different receptor conformations than that stabilized by the three functional groups of epinephrine, despite the fact that all of these active states are able to activate maximal cAMP accumulation. The presence of different active conformations of GPCRs that are stabilized by different agonists has been proposed previously (Perez et al., 1996; Seifert et al., 1999; Strange, 1999; Ghanouni et al., 2001).
Stabilization of fully active species requires the presence of both catechol OHs, since removal from epinephrine of either p-OH or m-OH resulted in partial activation and loss of synergism regardless of the presence of both β-OH and N-CH3. These results are in agreement with previous studies (Strader et al., 1989; Liapakis et al., 2000) and suggest that the interactions between the catechol OHs and the cluster of serines in TM5 play a critical role in the proper positioning of the catechol ring in the binding site crevice and probably the proper positioning of the side chains and/or orientation of the TM5 to achieve the fully active state.
In the dopamine D2 receptor, a cluster of aromatic residues consisting of Phe3826.44, Trp3866.48, Phe3896.51, Phe3906.52, His3936.55 in TM6, Tyr4167.43 in TM7, and Phe1985.47 in TM5 plays an important role in ligand binding and receptor activation (Javitch et al., 1998). Most of these residues are conserved in β2AR, and in this receptor, activation has been inferred to be associated with coordinated rotamer changes of Trp2866.48 and Phe2906.52 in the aromatic cluster along with Cys2856.47 (Shi et al., 2002). This rotamer “toggle switch” may modulate the movement of TM6 about the kink formed by Pro6.50, which is associated with the movement of the cytoplasmic end of TM6 relative to that of TM3, during receptor activation. Based on this model, an agonist could promote the active state by stabilizing Phe6.52 in the t rotamer conformation, thus allowing Trp6.48 to adopt the t conformation, but only in the context of the complete set of interactions described above.
Acknowledgments
We thank Drs. Brian Kobilka, Robert Lefkowitz, and Stephen Rees for gifts of the epitope-tagged β2 receptor DNA, the CAM β2 DNA, and the pcin4 plasmid, respectively. We also thank Myles Akabas, Arthur Karlin, Lei Shi, and Harel Weinstein for valuable discussions.
Footnotes
-
This work was supported in part by National Institute of Mental Health grants MH57324 and MH54137, by the Lebovitz Fund and the Lieber Center, and by the Faculty of Medicine, University of Crete.
-
ABBREVIATIONS: β2AR, β2 adrenergic receptor; EPI, epinephrine; PHE, phenylephrine; SYN, synephrine; HAL, halostachine; TM, transmembrane segment; WT, wild-type/wild type; CAM, constitutively active mutant; GPCR, G-protein-coupled receptor; EEDQ, N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline; HEK, human embryonic kidney; CGP-12177, 4-[3-[(1,1-dimethylethyl)amino]2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one.
- Received October 13, 2003.
- Accepted January 6, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}