


Abstract
The quantitative comparison of the relative potency of agonists is a standard method of receptor and agonist classification. If agonist potency ratios do not correspond in two given tissues, this is used as presumptive data to conclude that the receptors in those two tissues are different. This article presents data to show that a single receptor can demonstrate varying agonist potency ratios in different host cells. These data are described in terms of the production of more than one agonist-selective receptor active state and the interaction of these different active states with multiple G proteins in the membrane to produce cellular response. Stable host human embryonic kidney 293 cells with enhanced quantities of the respective Gα-protein were created. Wild-type and Gα-subunit enriched cells were then transiently transfected with human calcitonin receptor type 2 (hCTR2). Binding did not detect differences in the G protein-enriched cells versus wild-type cells. In contrast, functional studies did show differences between the host cell lines and Gα-subunit enriched cell lines. The relative potency of eight calcitonin agonists was measured in studies of calcium fluorescence in transfected cells containing human calcitonin receptor type 2 by comparing pEC50 (-log molar concentration producing half-maximal response) values. In Gαs-enriched cells, the relative order of potency of the agonists changed. The host-cell dependent differences in potency ratios ranged from 2-fold to more than 46-fold. This finding is not consistent with the idea that all of the agonists produce response in the same manner (i.e., through a common active state of the receptor). These data are consistent with the idea that these different agonists produce arrays of active states that differentially use G proteins. This idea is discussed in terms of the design of stimulus-bias assay systems to detect agonist-selective receptor active states with resulting potential for increased selectivity of agonists.
A classic pharmacologic method of receptor and agonist classification is through the use of agonist potency ratios. Under null conditions, the relative potency of agonists is independent of the host cell for the receptor and depends only on the relative affinity and intrinsic efficacy of the agonists for the receptor type (see Appendix ). Deviations in agonist potency ratios therefore are used as presumptive evidence for differences in receptors. This technique is based on the tacit assumption that the mechanism of response production for the agonists involved is the same (i.e., that they produce the same active state of the receptor that then goes on to activate the stimulus-response mechanisms of the tissue host in a uniform manner). Therefore, differences in agonist potency ratios, in contrast to furnishing evidence for differences in receptor types, may alternatively indicate lack of adherence to this tacit assumption. This article describes the induction of variation in agonist relative potency ratios in different tissue host cells for a single transfected receptor, the hCTR2. This raises the possibility that this effect indicates the production of agonist-specific receptor active states by the different agonists.
Materials and Methods
Molecular Biology and Generation of Stable Cell Lines
Full-length bovine Gαs (short form) cDNA was kindly provided by Dr. Pat Casey at Duke University (Robishaw et al., 1986). The full-length cDNAs for mouse Gαq (Strathmann and Simon, 1990), rat Gαo1 (Jones and Reed, 1987), and rat Gαi1 (Jones and Reed, 1987) were provided by Steve Rees at GlaxoWellcome, UK. TheNco/HindIII fragment of pT7–5/Gαs,BamHI fragment of pSG5/Gαq, HincII fragment of pT7–5/Gαo, and the HincII fragment of pT7–5/Gαi1 were isolated and subcloned into pCIN expression vector (Rees et al., 1996). The full-length calcitonin receptor was isolated and subcloned into expression vector pcDNA3 as described previously (Chen et al., 1997).
The expression vectors containing different G proteins were then transfected into HEK 293 cells using calcium phosphate method (Promega, Madison, WI). On day 3 after transfection, the cells were selected using G418-supplemented media at a concentration of 600 μg/ml. After a 2-week selection, colonies were selected and expanded. Stable lines were checked for expression by immunoblotting.
Gel Electrophoresis and Immunoblotting
HEK 293 membrane preparations, made from stable clones containing various amounts of overexpressed Gα-proteins, were qualitatively evaluated using SDS-polyacrylamide gel electrophoresis according to the procedure of Laemmli (1970). Protein (20 μg) was dissolved in 30 μl of TBS-T buffer (20 mM Tris and 500 mM NaCL, pH 7.5, 0.1% Tween 20) and then incubated with 30 μl of 2× SDS loading buffer containing 5% 2-mercaptoethanol. This mixture was boiled for 5 min and cooled on ice. The denatured protein solution [30 μl (10 μg)] was then loaded into each well of the Novex 10-well, 10% 1.5-mm Tris-glycine gel and run at 120V for 90 to 100 min. The resolved proteins were transferred to Novex nitrocellulose membranes. The nitrocellulose membrane then was incubated in blocking buffer (Megga-Block 1:10 in TBS-T) for 1 h. After washing in TBS-T with 1:100 Megga-Block the membranes were incubated with various Gα specific antibodies (Santa Cruz Biotech, Santa Cruz, CA) for 1 h. After a second washing, the membranes were incubated with a secondary horseradish peroxidase goat anti-rabbit antibody (1:5000) for 1 h. The blot then was developed using the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Piscataway, NJ). Quantitative analysis of the Western blots was done with the BioRad GS-700 Imaging Densitometer (BioRad, Hercules, CA). Bands were measured and analyzed using the BioRad Molecular Analyst software package and data expressed in RDU. Clones were selected based on the increased density of G protein band compared with control.
Transient Transfection of hCTR2
HEK 293 cells enriched with α-subunits Gi, Gs, Go, and Gq were plated at a density of 107cells/225-cm2 flask and grown overnight in DMEM plus 10% fetal calf serum supplemented withl-glutamine (2 mM). Cells were transfected with 40 μg of pcDNA3 vector control (Invitrogen, Carlsbad, CA) or pcDNA3/hCTR2 using the calcium phosphate DNA transfection method (Davis et al., 1986). After a 6-h transfection, media were replaced and cells were grown an additional 48 h when they were collected for assay. HEK 293 cells (wild-type, Gαi-21, Gαs-24, Gαq-15, and Gαo-13 line) were plated to a concentration of 106 cells/100-mm dish. After 24 h, cells were cotransfected with clone 134/pMTR with pRSV/neo at a ratio of 10:1, respectively, according to the calcium phosphate method (Promega).
Binding Studies
Membrane Preparation.
At confluency, cultured cells were harvested by manual scraping of the tissue culture flasks. Cells were pelleted by centrifugation at 2000 rpm for 15 min and then homogenized (three 15-s bursts) in ice-cold HEPES buffer (20 mM HEPES, pH adjusted to 7.4 with NaOH at 23°C). The homogenate was centrifuged at 48,000g for 15 min and washed twice through resuspension with new buffer. After a third centrifugation, the pellet was resuspended in fresh buffer containing 0.2 mM phenylmethylsulfonyl fluoride. Aliquots were frozen at −70°C.
Saturation Binding Curves.
Membranes were equilibrated with either 125I-AC512 (2000 Ci/mmol; Amersham Pharmacia Biotech) or 125I-hCAL (2000 Ci/mmol; Amersham Pharmacia Biotech) in 20 mM HEPES buffer containing 0.5 mg/ml bacitracin, 0.5 mg/ml BSA, and 0.2 mM phenylmethylsulfonyl fluoride (all from Sigma Chemical, St. Louis, MO) for 60 min at 23°C (samples mixed on a Titer Plate Shaker; Lab Line Instruments, Melrose Park, IL). Nonspecific binding was defined as the radioactivity remaining in the presence of 100 nM nonradioactive salmon calcitonin. Incubations were started by addition of membrane in triplicate tubes and binding was terminated by filtration through glass-fiber filters (presoaked 30 min in 0.5% polyethylenimine), with the Skatron semiautomatic cell harvester. Filters were placed in Sarstedt 68.752 51- × 12-mm polypropylene tubes and counted for 1 min in a gamma counter.
Saturation binding data were analyzed with the GlaxoWellcome statistical fitting package RADLIG (GlaxoWellcome Scientific Computing, Plan-les-Ouates, Geneva, Switzerland) to simultaneous equations describing total binding and a linear nonspecific binding curve (to yield a saturable binding curve). Saturation analysis yielded a nonlinear least-squares fit to the logistic equation with a half-maximal fitting parameter (the equilibrium dissociation constant of the ligand/receptor-complex under ideal conditions, denotedK d) and a maximal asymptote (denotedB max, providing an estimate of the maximal number of binding sites in picomoles per milligram of protein). Complex displacement curves were fit to a two-population model, yielding two apparent affinities and relative quantities of the two apparent binding sites (or receptor states). Data with 125I-hCAL indicated complex two-phase binding with a low-capacity, high-affinity binding site and a high-capacity, low-affinity binding site, in keeping with standard agonist binding kinetics
Measurement of Calcium Transient Responses
Stable HEK 293 clones were tested for their ability to mobilize calcium in the FLIPR system. Cells were harvested with 0.05% trypsin (Life Technologies, Gaithersburg, MD) and plated in black 96-well Viewplates (Polyfiltronics, Rockland, ME) at a concentration of 10,000 cells/well in DMEM/F12 phenol red free medium (Life Technologies) containing 5% fetal bovine serum (Life Technologies). Approximately 30 h after cell plating, the media was removed by vacuum and replaced with DMEM/Ham's F12 phenol red-free medium without serum. Cells were kept in serum-free medium approximately 18 h before assay. At the time of assay, cells were washed with 100 μl FLIPR buffer (145 mM NaCl, 5 mM KCl, 0.5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4). Dye was prepared as follows: 2 mM calcium green stock (C 3011; Molecular Probes, Eugene, OR) was prepared in dimethyl sulfoxide. The stock was mixed 50/50 with 20% pluronic acid (P 3000; Molecular Probes) and diluted to 4 μM final concentration in FLIPR buffer. Cells then were loaded with 50 μl of the 4 μM calcium green dye along with 2.5 mM probenecid in 2.5% NaOH and allowed to incubate for at least 1 to 2 h at 37°C. After incubation, the plates were allowed to come to room temperature. Plates were washed in FLIPR buffer twice and 50 μl left in each well. After loading plates into the FLIPR, basal intracellular calcium [Ca2+]i conditions were monitored for 10 s before adding 50 μl of the agonist with an integrated 96-well pipettor. Fluorescence was measured from all 96 wells simultaneously using a charge-coupled device camera. Agonist activity was measured every second for the first 25 s then every 3 s for the next 15 s. Curves were calculated as percentage of 10 μM ionomycin control response.
Results
Stimulus-Biased HEK 293 Cells.
Approximately 100 HEK 293 cell lines were transfected with Gα-subunit cDNA for four different G proteins (Gαi, Gαs, Gαo, and Gαq) and made into stable clones. These were subjected to Western blot analysis for visualization of relative quantities of specific G proteins. As shown in Fig.1, certain clones contained elevated levels of Gαi, Gαs, Gαo, and Gαq protein, respectively. Accordingly, cell lines 21 (for Gαi-enriched cells), 24 (Gαs-enriched), 13 (Gαo-enriched), and 15 (Gαq-enriched) were selected for further study. These stable cells lines then were transiently transfected with hCTR2 cDNA for binding and functional studies.
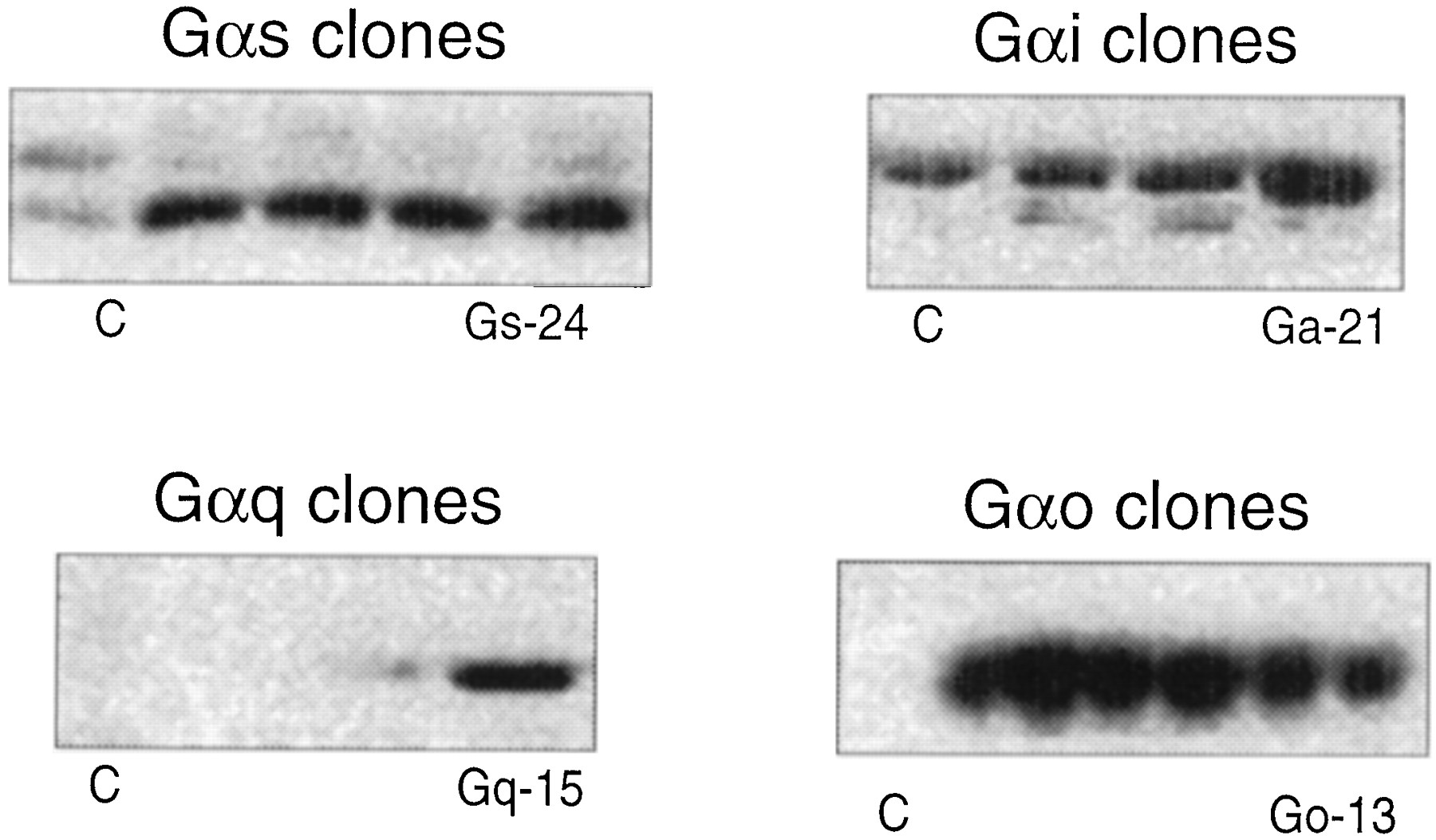
Western blot gels showing relative amounts of Gα-protein in the membrane of various colonies of HEK 293 cells transiently stably transfected with cDNA for Gαi-, Gαo-, Gαs-, and Gαq-. Relative to control, stable colonies 21 (control, 11.26 RDU; Gαi-enriched, 23.47 RDU); 13, (control, 2.06 RDU; Gαo-enriched, 10.41 RDU), 24 (control, 2.05 RDU; Gαs-enriched, 8.01 RDU), and 15 (control, 11.26 RDU; Gαo-enriched, 10.56 RDU) were chosen for receptor cotransfection.
Saturation Binding Studies.
Saturation binding curves were obtained for the agonist 125I-hCAL and the calcitonin receptor antagonist 125I-AC512 (Chen et al., 1997) on membranes prepared from transiently transfected (expressing hCTR2) wild-type HEK 293 cells and Gα-subunit enriched HEK 293 cells. The maximal density of expressed hCTR2 sites as measured with the antagonist radioligand exceeded the number estimated with125I-hCAL (Table 1) consistent with the idea that the G protein limited the production of high-affinity agonist binding state [i.e., the number of receptors exceeded the available G protein (Chen et al., 1997; Kenakin, 1997b)]. Transfection of the four cell lines led to varying levels of receptor expression being highest in the wild-type and Gαi-enriched and lower in Gαs-, Gαο-, and Gαq-enriched cell lines as measured with125I-AC512 binding (Table 1). Little change in the amount of high-affinity binding complex with125I-hCAL was obtained with Gα-subunit enrichment.
Saturation binding of 125I-hCAL and 125I-AC512 to membranes from HEK 293 cells expressing hCTR2
Effect of Receptor Density on Agonist Potency Ratios.
As demonstrated by the different B max values for 125I-AC512 binding, receptor expression of hCTR2 after transient expression was not uniform. This precluded effective comparison among cell types and focused attention on the important aspect of these studies from the standpoint of receptor theory, namely the internal relative profiles of agonists within each cell type. According to the classic models of GPCRs, differences in receptor density should affect absolute potency but not relative potency; this is described more fully in Appendix . The possible effect of calcitonin receptor density differences was examined in functional studies. The relative potency of agonists for hCTR2 under two different transfection conditions in HEK 293 cells was explored in a stable high expression cell line (30.05 ± 3 pmol/mg of protein hCTR2; Chen et al., 1997) and transient expression in wild-type HEK 293 cells. Figure 2A shows dose-response curves for rat calcitonin in the two cell lines. As shown in this figure, the maximal response to was greater in the stable HEK 293 cell line. Figure 2B shows the correlation of the potencies of the agonists (quantified as pEC50, the -log of the EC50, concentration producing half-maximal response) in the two cell lines. Linear regressional analysis (Snedecor and Cochran, 1967; Armitage, 1971) indicated a highly significant regressional coefficient (T = 8.29, d.f. = 6, P < .001). Fig. 2C shows a graphical representation of the pEC50 values in the stable and the transient cell line. This representation shows the uniform phase shift in potency (slightly higher in the stable cell line) for the agonists; the pEC50 values are given in Table2. Although the absolute potencies differed, it is important to note that the relative potency, a parameter dependent only upon the agonists and receptor type(Appendix ), did not.

The relative potency of hCTR2 agonists in a stable HEK 293 cell line containing hCTR2 (30.05 pmol/mg protein) and in a HEK 293 cell line transiently expressing hCTR2. A, dose response curves to rat calcitonin in stable cells (filled circles) and HEK 293 cells transiently transfected with cDNA for hCTR2 (open circles). B, pEC50 values for agonists in transiently transfected cells (abscissae) and stable cells (ordinates). Values shown for eel calcitonin (▴), salmon calcitonin (▵), porcine calcitonin (■), chicken CGRP (⋄), human calcitonin (▪), rat calcitonin (○), rat CGRP (♦), and rat amylin (●). C, change in pEC50 from stable to transient cells (ordinates = pEC50). For B and C, bars represent S.E.M.
Relative potencies of calcitonin receptor agonists in transient wild-type and stably expressing HEK 293 wild-type cells
Stimulus-Biased Assay Systems: Effects of Agonists.
The effects of standard agonists for endogenous receptors in wild-type and Gαs-subunit enriched HEK 293 cells are shown in Fig.3. As can be seen from this figure, carbachol produced 150% maximal ionomycin responses in Gαs-enriched host cells and only 35% maximum response in Gαo-enriched cells (Fig.3A). Similarly, responses to ATP were enhanced in Gαs-enriched host cells and eliminated in Gαo-enriched cells (Fig. 3B). These data indicated that, unlike binding for transfected calcitonin receptors, Gαs-, Gαo-, and Gαq-protein enrichment produced differences in stimulus-response coupling. For both carbachol and ATP, little difference was seen with Gαi-enriched cells compared with wild-type cells.
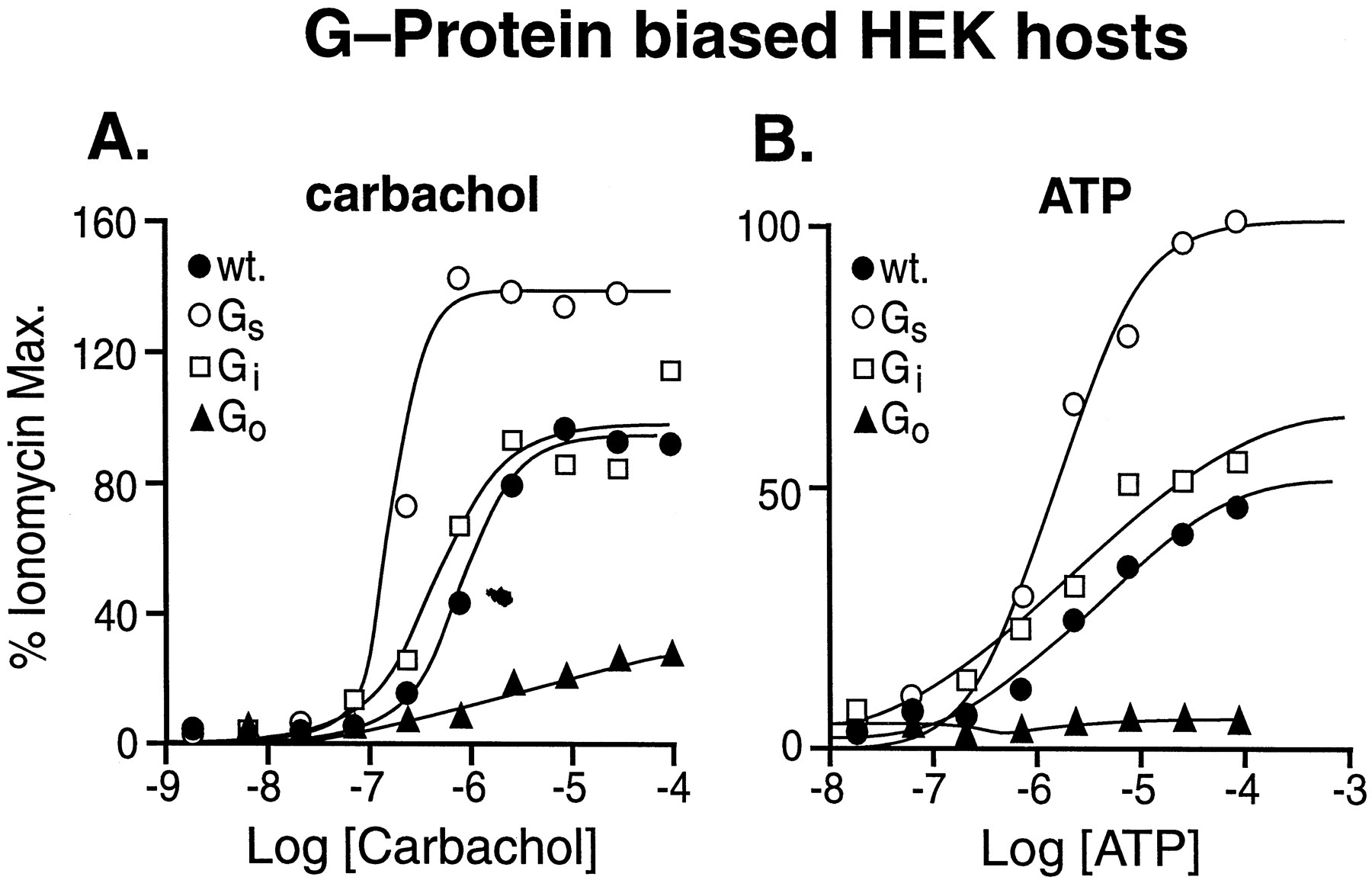
Responses to carbachol (A) and ATP (B) agonists for receptors endogenous to HEK 293 cells in wild-type cells (●), Gαs-enriched cells (○), Gαi-enriched cells (■), and Gαo-enriched cells (▴). No satisfactory dose-response relationships could be obtained with Gαq-enriched cells. Data shown from one of three experiments.
Different patterns of response to calcitonin agonists were observed in cells transiently transfected with hCTR2. Responses to hCAL in Gαo cells were extremely small and difficult to reliably quantify. Responses in Gαq cells were large but erratic and no consistent dose-response curves could be obtained. Accordingly, all further work with hCTR2 were conducted in wild-type and Gαi- and Gαs-enriched cells.
As shown in Fig. 4, the maximal responses, when compared with ionomycin, for amylin (Fig. 4A) and rat calcitonin (Fig. 4B) were enhanced by Gαs enrichment and slightly diminished by Gαi enrichment. Interestingly, the location parameters of the curves to rat calcitonin, but not amylin, also changed with Gα-protein enrichment.
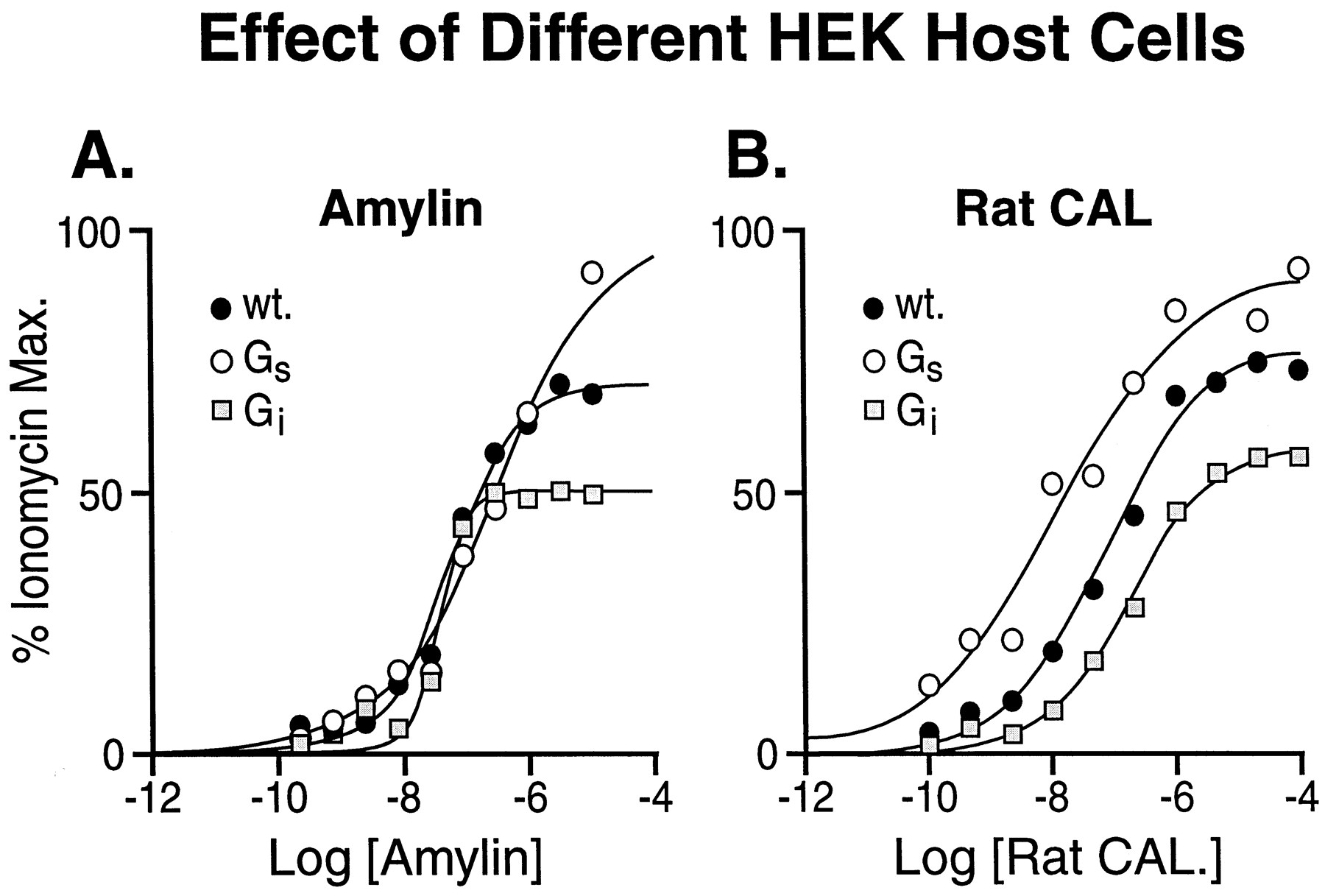
hCTR2-mediated responses to amylin (A) and rat calcitonin (B) in wild-type HEK 293 cells (●), Gαs-enriched cells (○), and Gαi-enriched (■). Data shown from one of three transient transfections.
The relative potency ratios for eight agonists for hCTR2 were compared in wild-type, Gαs-enriched, and Gαi-enriched HEK 293 cells. It should be noted while the maximal responses differed with respect to ionomycin in the different cell types, they did not differ within a given cell type for each of the agonists. Thus, all agonists produced a uniform maximal response (the maximal tissue response) and potency ratios were not complicated by differences in maximal asymptotic response.
Figure 5A shows the correlation between the pEC50 values for the eight agonists in wild-type cells (abscissae) and Gαi-enriched cells (ordinates). Linear regressional analysis of the pEC50 values indicated a highly significant regressional coefficient (T = 13.47, d.f. = 6, P < .001). Fig. 5B shows the individual pEC50 values graphically in wild-type and Gαi-enriched cells. As can be seen from these figures, there was little change in the potencies of the agonists; the pEC50 values are given in Table3.
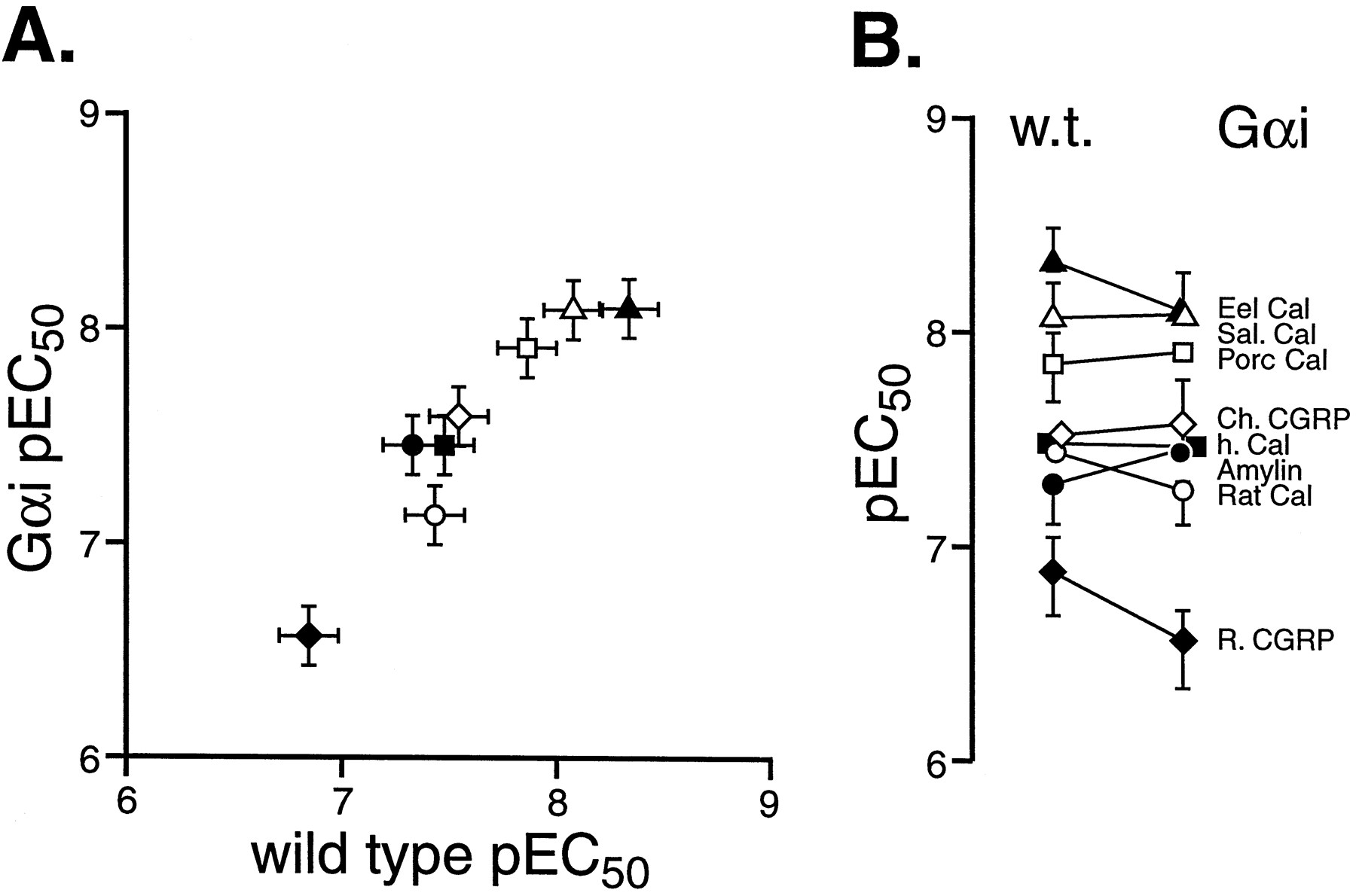
The relative potency of hCTR2 agonists in wild-type transiently transfected HEK 293 cells and Gαi-enriched cells. A, pEC50 values for agonists in wild-type cells (abscissae) and Gαi-enriched cells (ordinates). Values are for eel calcitonin (▴), salmon calcitonin (▵), porcine calcitonin (■), chicken CGRP (⋄), human calcitonin (▪), rat calcitonin (○), rat CGRP (⋄), and rat amylin (●). B, change in pEC50 from stable to transient cells (ordinates = pEC50). For A and B, bars represent S.E.M.
Relative potencies of calcitonin receptor agonists in HEK 293 cells
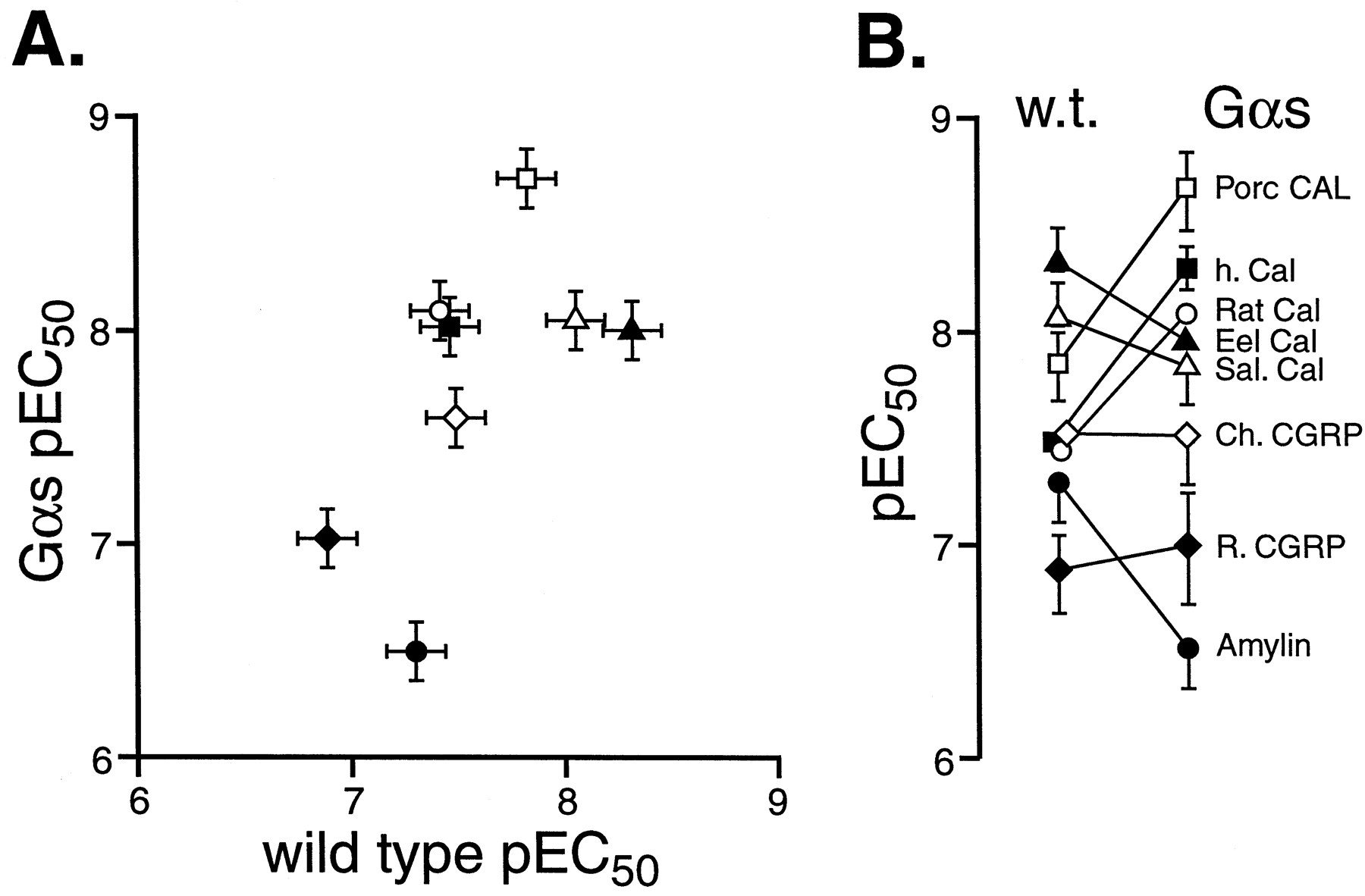
In contrast, Fig. 6A shows that Gαs enrichment produced some striking differences in the relative potencies of agonists. Linear regressional analysis indicated a loss of correlation between wild-type and Gαs-enriched cell pEC50 values (T = 2.17, d.f. = 6, n.s. at 5%). Amylin lost potency in Gαs-enriched cells, some agonists did not change (rat and chicken CGRP, eel and salmon calcitonin) and some selectively increased in potency (human, porcine and rat calcitonin). This resulted in a changed rank order of potency of the agonists in Gαs-enriched cells (over wild-type)—see Fig. 6B. The pEC50 values are shown in Table 3.
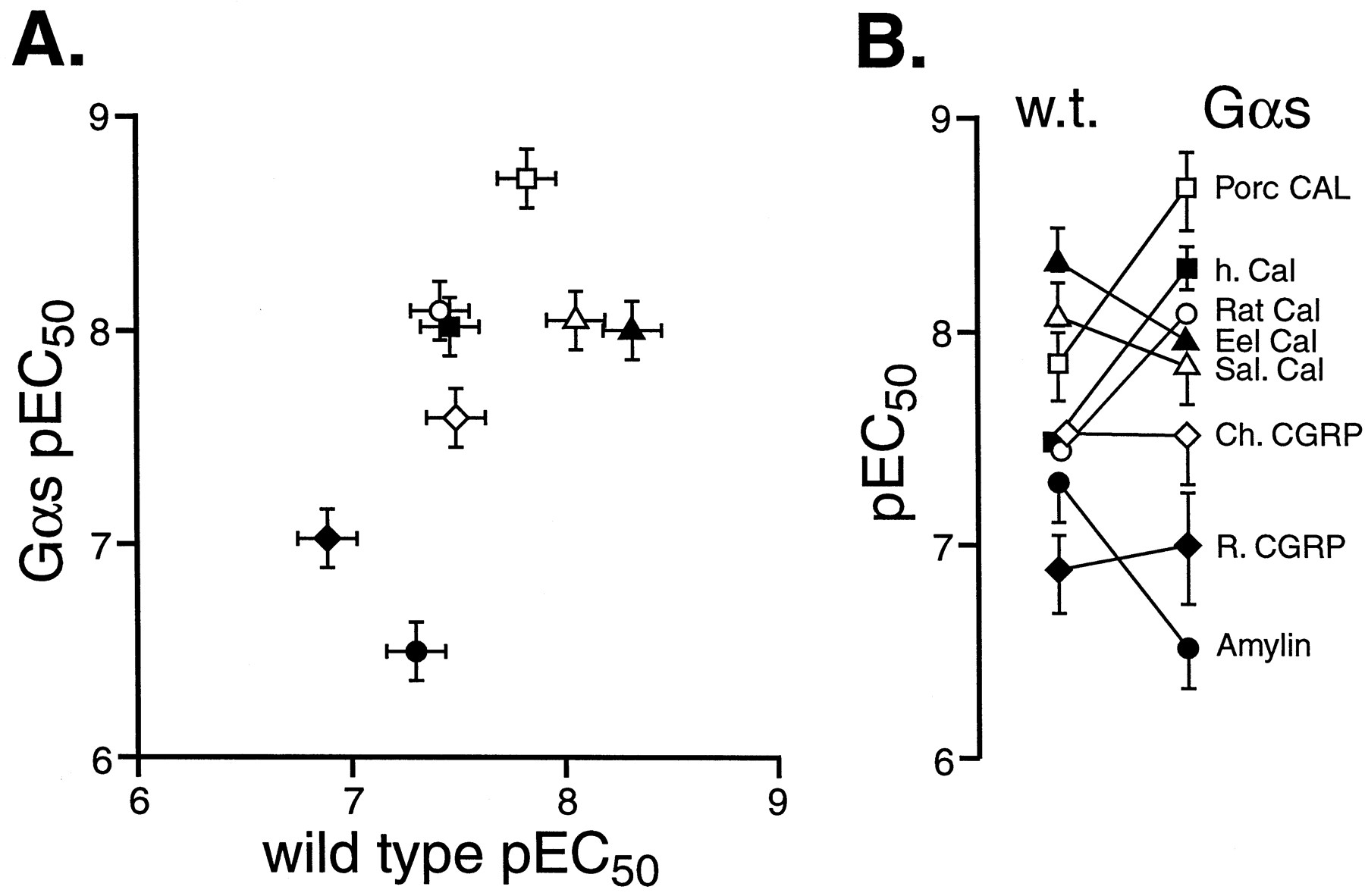
The relative potency of hCTR2 agonists in wild-type transiently transfected HEK 293 cells and Gαs-enriched cells. A, pEC50 values for agonists in wild-type cells (abscissae) and Gαs-enriched (ordinates). Values are for eel calcitonin (▴), salmon calcitonin (▵), porcine calcitonin (■), chicken CGRP (⋄), human calcitonin (▪), rat calcitonin (○), rat CGRP (⋄), and rat amylin (●). B, change in pEC50 from stable to transient cells (ordinates = pEC50). For A and B, bars represent S.E.M.
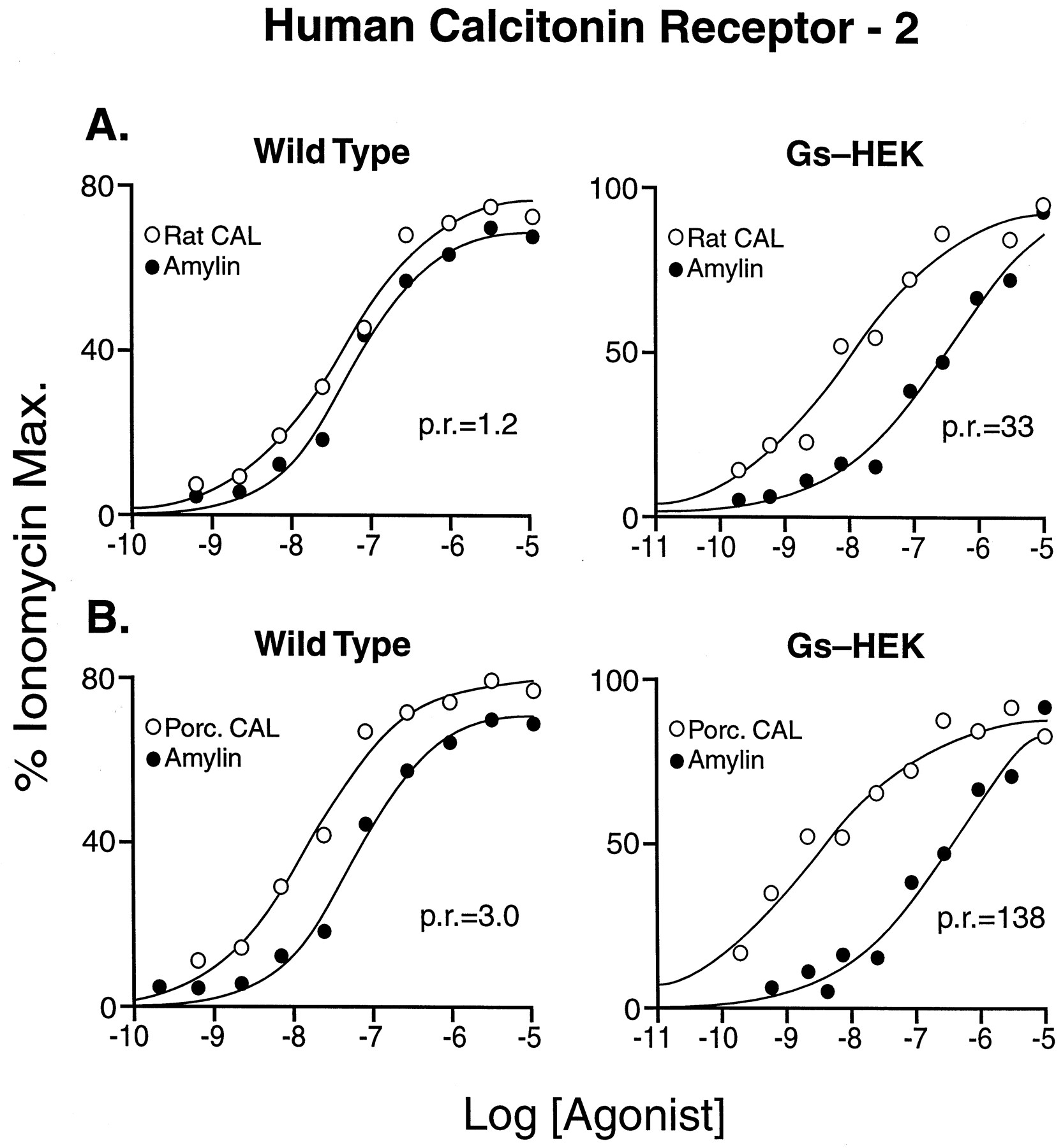
Figure 7 shows two examples of differing relative potencies in wild-type and Gαs-enriched cells. Thus, although the relative potency of rat calcitonin and rat amylin was 1.2 in wild-type cells, it increased by a factor of 27 (to 33) in Gαs-enriched host cells (Fig. 7A). Similarly, the potency ratio of porcine calcitonin and rat amylin changed by a factor of 46, from 3.0 to 138 (Fig. 7B).
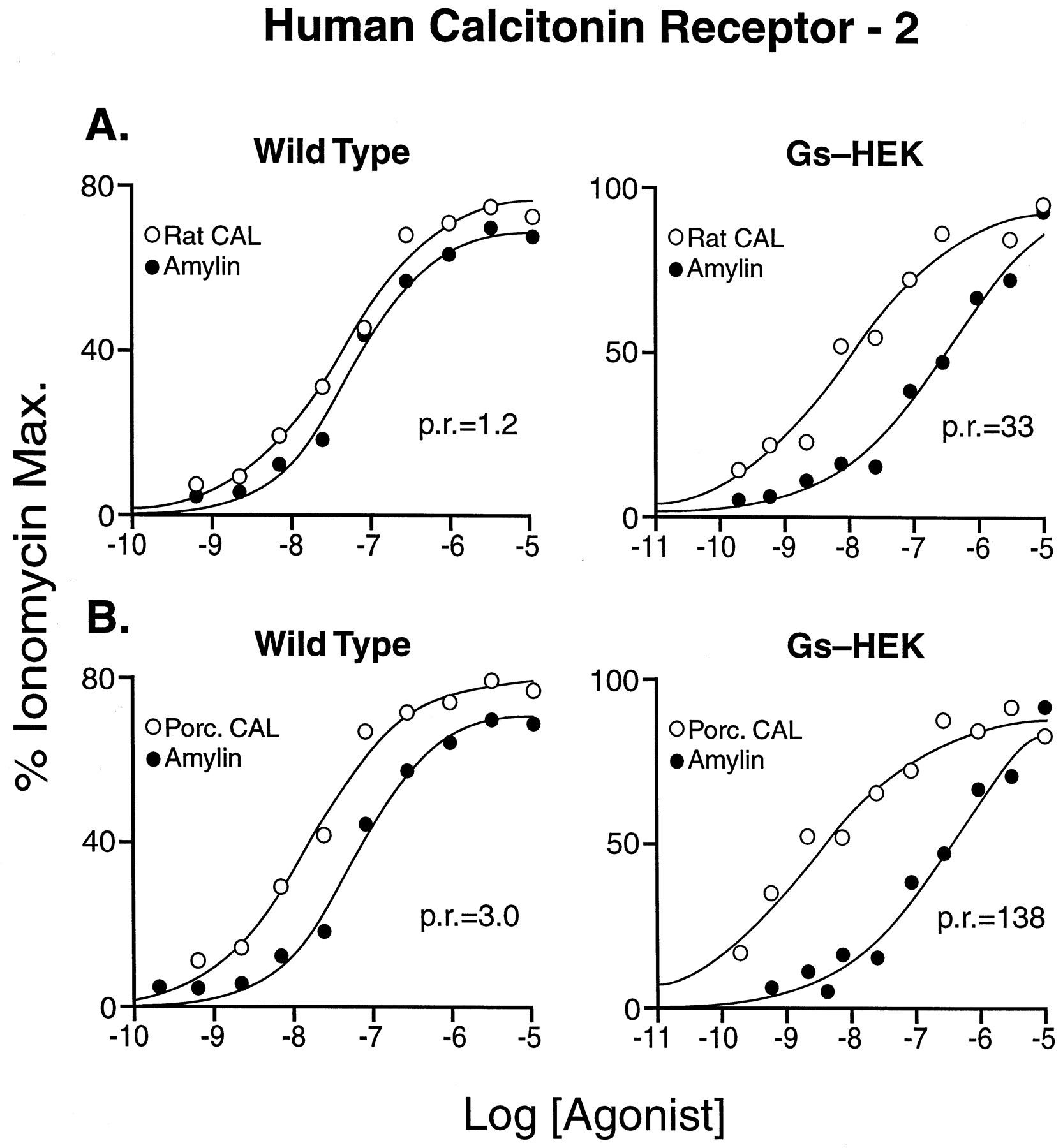
Dose-response curves for amylin (●) and rat calcitonin (○) (A) and amylin (●) and porcine calcitonin (○) (B) in wild-type HEK 293 cells (left) and Gαs-enriched HEK 293 cells (right). p.r., potency ratios for the agonists.
Discussion
The observation of varying relative potencies of agonists for the same receptor in different host systems is highly unusual. If this were observed in different natural systems, it would be taken as presumptive evidence for differences in the receptors mediating the responses in those systems. The fact that this was observed for a single receptor in different host cells is particularly interesting in that such behavior cannot be accommodated by the tacit assumption made in classical receptor occupancy theory, namely that all agonists form a single active receptor state that then initiates cellular signaling.
In this study, the measurement of selective G protein enhancement was operational. Although the relative increase in the G protein content of the stimulus-biased cells was measured (relative to wild-type control), the resulting data are not relevant to the actual quantity of G protein accessible to the transfected receptor and available for receptor coupling and served only as a method of choosing cells for receptor transfection. The change in the responses to ligands for endogenous receptors (ATP and carbachol) and transfected hCTR2 was more relevant in that a bias, in terms of G protein coupling to these receptors, was demonstrated. This operational observation of G protein enrichment was used as the basis for further study of relative agonist potencies.
The fact that the amount of high-affinity agonist complex was so much lower than the total receptor number (as measured with the antagonist radioligand) indicated that the amount of G protein was limiting. Gα-subunit enrichment theoretically could have resulted in differences in the amount of high-affinity binding of an agonist radioligand. In keeping with these predictions, coexpression of secretin receptors with Gαs has been shown to lead to an increase in the number of high-affinity binding sites for125I-secretin from 1.8% to 15% (Ishihara et al., 1991). However, for pleiotropic receptors that interact with more than a single G protein (such as hCTR2; Horne et al., 1994), the degree to which high-affinity binding would be enhanced depends upon the relative stoichiometries of the G proteins involved (i.e., if a relatively minor G protein is enhanced in the presence of a high concentration of the major interactant G protein for that receptor, then enrichment of a secondary protein may not be detectable as an increase in the number of high affinity binding sites). The relationship of the B max for an agonist observed in saturation binding and G protein is given from eq. 7 ofAppendix as αL (1 + γ2[G2]/β2K2) [Rtotal]/(1 + αL (1 + γ1[G1]/β1K1+ γ2[G2]/β2K2)). It can be seen that enrichment of one of the G proteins would have little effect on B max if the other G protein binding the receptor was in a relatively greater concentration or if the affinity of the receptor active state (denoted by γ) was lower for the enriched G protein. Thus there is a limitation of mass equivalence in the measurement of Gα-subunit protein in binding studies.
The limitation in binding studies of mass equivalence is not present in functional studies. When cell function is used as a measure of receptor stimulus, there can be disconnections between the amount of high-affinity complex and resulting amount of stimulus (and resulting response) produced by that complex. In general, stimulus-response mechanisms within cells greatly amplify the result of receptor/G protein interaction therefore G protein enrichment, insufficient to result in changes in high-affinity binding, may still produce observable effects on function. In light of the data with Gαs-enrichment, this seems to be what occurred in these studies. The implications of this finding are worth considering.
Within a given cell type, if all agonists produce a uniform active receptor state, then the relative potency of these agonists will not vary (see Appendix ). The data obtained in Gαs-enriched cells cannot be accommodated by this hypothesis, leading to the possibility that some of the agonists in this study produce at least two different receptor active states (see below). There are increasing data in the literature, from a range of experimental approaches, to suggest that this occurs with other receptors. Experimental data support the idea that agonist specific receptor activation for PACAP 1 receptor (pituitary adenylate cyclase activating polypeptide receptor type 1; Spengler et al., 1993), Drosophila tyramine receptors (Robb et al., 1994), and β2-adrenoceptors (Chidiac et al., 1994). Agonist specific receptor activation has directly been proposed for dopamine D2s (Wiens et al., 1998), 5-HT2c (Berg et al., 1998), 5-HT3 (Van Hooft and Vijverberg,1996), β2-adrenoceptors (Krumins and Barber, 1997;Seifert et al., 1999), cannabinoid CB1 receptors (Bonhaus et al., 1998; Glass and Northup, 1999) and μ-opioid receptors (Keith et al., 1996; Blake et al., 1997; Yu et al., 1997).
Agonist-specific receptor active states have been hypothesized on the basis of a number of experimental approaches. These include observation of protean agonism [reversal of efficacy from positive to negative in quiescent versus constitutively active receptor systems (Kenakin, 1997c)], differences in agonist-induced receptor internalization, kinetic rates of activation, differences in rates of hydrolysis of G protein-induced nucleotide hydrolysis, and reversal of the relative potency of agonists for different stimulus-response pathways. Many of these approaches uncover agonist-selective receptor states, but it is still a theoretical hypothesis that these states are involved in signaling and will result in stimulus-trafficking. The present approach directly demonstrates that the agonists tested produce differing response patterns which may translate to different therapeutic profiles. On the other hand, complex differential G protein coupling would be expected to be dependent upon receptor/G protein stoichiometry and would thus be quite system dependent. Under these circumstances, it would not be expected that a given stimulus-biased assay such as this one would be universally useful for the detection of stimulus trafficking. With this in mind, it would be prudent to use as many techniques as possible to test ligand agonism because some may work better than others for given receptors and agonists.
The ternary complex model for GPCRs, either in the form of the extended ternary complex model (Samama et al., 1993), or the cubic ternary complex model (Weiss et al., 1996a,b,c) commonly are described as “two-state” models referring to the two spontaneously occurring active and inactive states of the receptor. The concept that agonists produce unique active receptor states may seem to be in conflict with these classic models. However, it should be noted that these models really are “multistate” models when describing activation of receptors by ligands. This is because the presence of the ligand opens the possibility of a modified affinity of the activated receptor for G proteins through thermodynamic constants α (extended ternary complex model) or γ and δ (cubic ternary complex model); see Table 4 and Fig.8. Thus, the existing models of GPCR function accommodate agonist-specific receptor active conformations.
Parameters for the ternary complex models
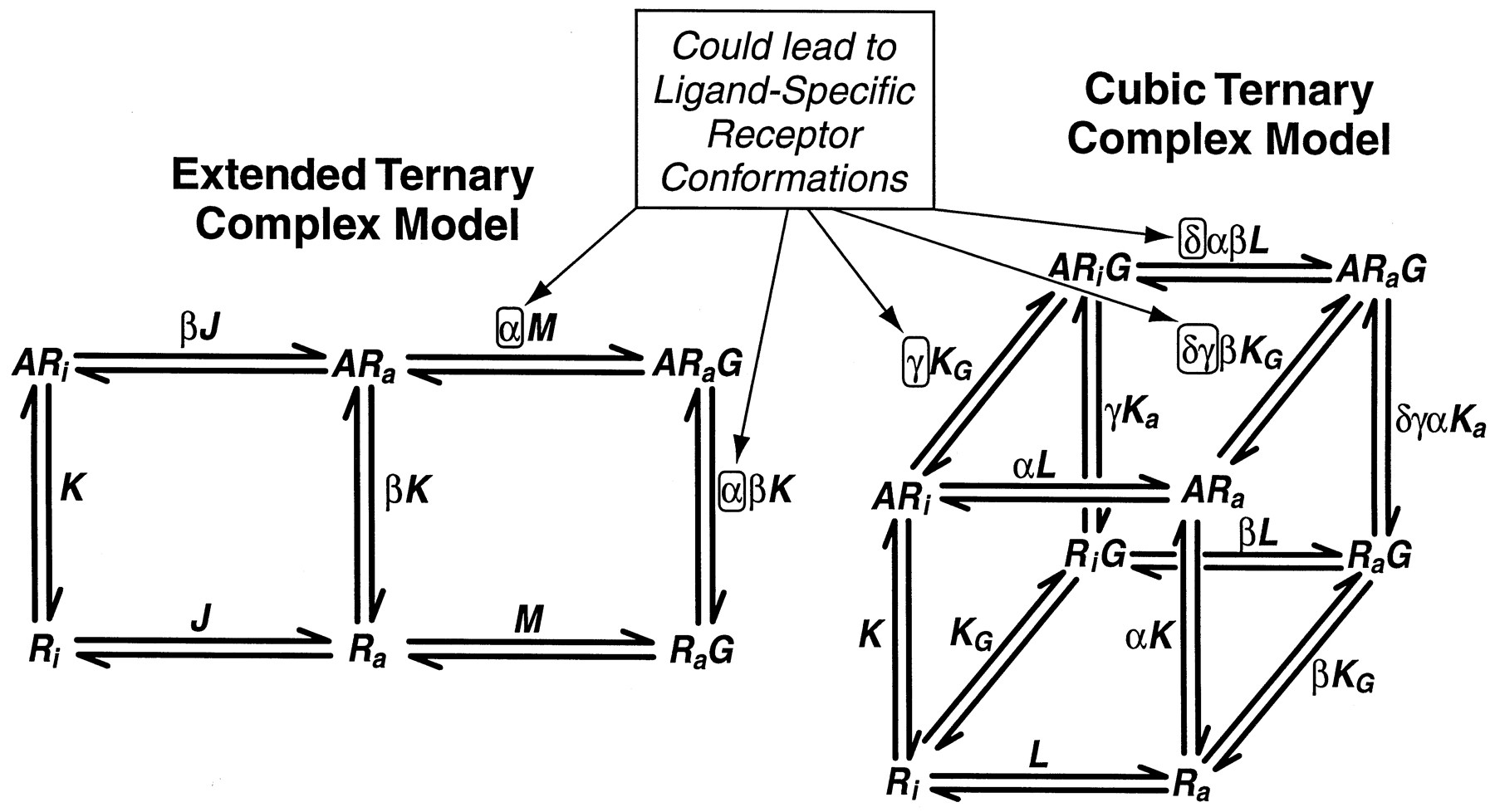
Two models of GPCR activation. The extended ternary complex model (Samama et al., 1993) allows interaction of only the active state receptor with G protein. The cubic ternary complex model (Weiss et al., 1996a,b,c) allows interaction of both the active and inactive state with G protein. The binding of ligand allows changes in the receptor affinity for G protein through the factors α, γ, and δ. This allows for agonist-specific receptor active states within the framework of these models.
The formation of agonist-specific receptor active states becomes important from a cellular signaling point of view when multiple G proteins are involved. It is known that many G protein coupled receptors are pleiotropic with respect to the G proteins with which they interact (Kenakin, 1996). As discussed previously, human calcitonin receptors are among those known to activate Gs, Gi, and Gq (Horne et al.,1994). This could be important for signaling because studies have shown that different regions of the cytosolic loops of seven-transmembrane receptors activate different G proteins (Ikezu et al., 1992; Wade et al., 1999). Under these circumstances, it would not be expected that different overall receptor conformations would expose these different G protein-interacting sequences to signaling mechanisms in an identical manner. These ideas, taken in conjunction, open the theoretical possibility that different active receptor conformations selectively activate different G proteins to direct stimulus to different biochemical pathways in cells. When this occurs, it would be predicted that the relative potencies of agonists producing different active states will vary according to the relative amounts of G protein available for stimulus-response coupling. This is discussed further inAppendix .
Stimulus-biased assay systems, such as the Gα-subunit-enriched HEK cells used in this study, are not meant to reflect natural physiology but rather are designed to furnish unique information about agonists. In drug discovery programs designed to find agonists, there usually are two possible targets: a complete mimic of the physiologically endogenous agonist or a mimic of a subset of agonism produced by the physiological agonist. Previously, the latter profile has been achieved solely by finding agonists for receptor subtypes or restriction of pharmacokinetics. The production of selective receptor active states theoretically offers another level of selective agonism. Specifically, if the spectrum of signal transduction initiated by a given agonist could be reduced, then a subset of physiological responses could be produced. If the pleiotropic nature of the endogenous receptor signaling is associated with a concomitant plethora of physiological responses (some of which are not desired in the therapeutic field), then reducing these may lead to a better mating of replacement agonist therapy for pathophysiological disorders. From this standpoint, agonist-selective receptor active states could represent the next effective level of agonist selectivity (Kenakin, 1997a).
Presently the relevance of agonist-specific cellular signaling to therapeutic targeting of synthetic agonists is not clear. The extent to which this idea can be capitalized upon therapeutically is unclear. At the least, however, it allows a method of classifying agonists by measures beyond those based simply on strength of agonism. This latter idea assumes that all agonists produce a uniform receptor active state that goes on to produce physiological response on the basis of stoichiometry and nothing more. The idea of agonist specific receptor active states extends that concept to include the “quality” of efficacy as well as the “quantity” of efficacy in describing the activity of agonists. The use of stimulus-biased host cells for surrogate expression of receptors may be a useful tool in this regard.
Acknowledgments
We thank Susan Armour for excellent technical assistance and Donna McGhee for expert preparation of the manuscript. We also thank Deborah Jones-Hertzog for statistical advice.
Effect of Receptor Density on Agonist Potency Ratios
Extended Ternary Complex Model.
This model (Samama et al., 1993) defines agonism with the following equation:
For two agonists a and b, the relative potency is defined as:
Cubic Ternary Complex Model.
This model (Weiss et al., 1996a,b,c) defines agonism with the following equation:
For two agonists a and b, the relative potency is defined as:
Effect of Receptor Density on Relative Potency
It can be seen from the two expressions for relative potency, in either the extended ternary complex model or the cubic ternary complex model, that the effects of receptor density cancel and molecular constants reflecting affinity and intrinsic efficacy control relative potency. Under these circumstances, the relative potency of two agonists (providing they both produce full agonist response) is a unique identifier of the agonist and receptor type. The guideline used in this process dictates that differences in the relative potency of full agonists denotes differences in the receptors' mediating response. However, it can also be seen that differences in potency ratios can be brought about by differences in α or γ that reflect changes in the affinity of the ligand for the active (over the inactive) receptor state and differences in the affinity of the receptor for G protein when ligand is bound. This latter factor could be relevant if the ligand forms a different receptor state.
Relative Potency Ratios for Promiscuous Receptor/G Protein Interactions
The Ternary Complex Model [for this particular example, the extended ternary complex model by Samama et al. (1993) is used] can be expanded to include the interaction of the active State receptor (Ra) with two G proteins (denoted G1 and G2). Under these circumstances, the equation denoting response to an agonist (as defined by the production of a ternary complex with the agonist and active-state receptor with either G1 or G2) as:
Footnotes
- Received May 17, 2000.
- Accepted August 21, 2000.
-
Send reprint requests to: Terry Kenakin, Ph.D., Department of Receptor Biochemistry, Glaxo Wellcome Research and Development, 5 Moore Drive, Research Triangle Park, NC 27709. E-mail:tpk1348{at}glaxo.com
Abbreviations
- hCTR2 human calcitonin receptor type 2
- HEK, human embryonic kidney
- TBS-T
- Tris-buffered saline/Tween-20
- RDU
- relative density units
- AC512
- [Arg18,Asn30, Tyr32]9–32 salmon calcitonin
- hCAL
- human calcitonin
- FLIPR
- fluorometric imaging plate reader
- DMEM
- Dulbecco's modified Eagle's medium
- GPCR
- G protein-coupled receptor
- CGRP
- calcitonin gene-related peptide
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}