


Abstract
Artemisinin drugs are of utmost importance in the treatment of malaria, because they represent the sole class of therapeutically used antimalarial drugs to which malaria parasites have not yet developed resistance. The major disadvantage of these medicines is the comparatively high recrudescence rate, which has been attributed to the remarkable decrease of artemisinin plasma concentrations during multiple dosing. Autoinduction of CYP2B6-mediated metabolism has been implicated as the underlying mechanism. So far, the molecular mechanism of induction by artemisinin has not been resolved. Because the xenosensors pregnane X receptor (PXR) and constitutive androstane receptor (CAR) have been shown to mediate induction of drug-metabolizing enzymes and drug transporters, we investigated the hypothesis that artemisinin induces cytochrome P450 expression by activating PXR and/or CAR. By combining in vitro transfection methods and quantitative analyses of gene expression in cell lines and primary human hepatocytes, we here show that artemisinin drugs activate human PXR as well as human and mouse CAR and induce the expression of CYP2B6, CYP3A4, and MDR1 in primary human hepatocytes and in the human intestinal cell line LS174T. Furthermore, we demonstrate that artemisinin acts as a ligand of both nuclear receptors, because it modulates the interaction of the receptors with coregulators. In conclusion, activation of PXR and CAR and especially the resulting induction of CYP3A4 and MDR1 demonstrate that artemisinin has a higher risk of potential drug interactions than anticipated previously.
Malaria, a parasitic infection by protozoans, is one of the most threatening human infections worldwide. Among the four human malaria parasites, Plasmodium falciparum causes the majority of all severe cases and deaths. Because of the extensive use of monotherapy in the past, this parasite has rapidly developed resistance to several commonly used and cost-effective antimalarial drugs, including chloroquine, sulfadoxine-pyrimethamine, and mefloquine, in many malaria-affected areas (White, 2004). Spread of resistant parasites is regarded to be a major contributor to the global resurgence of malaria in the last decades. Today, artemisinin drugs represent the only therapeutically used antimalarial drug class to which resistance has not yet been developed. Therefore, these drugs are increasingly used in the treatment of drug-resistant falciparum malaria, especially in combination therapy with a second antimalarial drug (White, 2004).
Artemisinin, a sesquiterpene lactone endoperoxide, was isolated in 1972 by Chinese researchers from the leaves of the herb Artemisia annua, which has been used in traditional Chinese medicine for the treatment of fever for centuries (Klayman, 1985). Since then, artemisinin and some of its semisynthetic derivatives proved to be safe and effective antimalarial drugs. However, monotherapy with artemisinin drugs results in comparatively high recrudescence rates, which have been attributed to the decrease of artemisinin plasma concentrations to 20 to 30% of the initial values during the standard treatment of patients and healthy volunteers for 5 to 7 days with daily dosing. Thus, it has been argued that artemisinin drugs induce their own elimination, most likely by induction of first pass extraction, because artemisinin half-lives do not change concomitantly with plasma levels (Ashton et al., 1998). Artemisinin induces the enzymatic activity of cytochrome P450 (P450) enzymes CYP2C19 and CYP2B6 in vivo (Svensson et al., 1998; Mihara et al., 1999; Simonsson et al., 2003). The latter enzyme is primarily involved in the elimination of artemisinin in human liver microsomes in vitro (Svensson and Ashton, 1999), and it has been suggested that induction of CYP2B6 enzymatic activity would explain the autoinduction of artemisinin elimination (Simonsson et al., 2003).
Induction of drug-metabolizing P450 enzyme activity by artemisinin may also result in drug-drug interactions in combination therapy, because most antimalarial drugs used in combination with artemisinin are substrates of P450 enzymes (Giao and de Vries, 2001). Therefore, it is of interest to resolve the molecular mechanism of induction by artemisinin, which has not yet been elucidated. Several hypothetical mechanisms may account for induction. Artemisinin may induce the expression of genes by induction of gene transcription, processing or stabilization of mRNA, translation, or enzyme stabilization. Among these different mechanisms of induction, regulation of gene transcription, mRNA stabilization, and substrate/ligand stabilization of the enzyme have been shown to be most important for the regulation of P450 expression (Porter and Coon, 1991).
In the past few years, the basal molecular mechanism of xenobiotic induction of P450s of the subfamilies 2B, 2C, and 3A has been resolved. The nuclear receptors pregnane X receptor (PXR; NR1I2) and constitutive androstane receptor (CAR; NR1I3) mediate the induction of these drug-metabolizing P450 enzymes and also of drug transporters of the ABC family by many structurally diverse xenobiotics. PXR and CAR act by induction of the transcription of their target genes, which is mediated by specific binding sites in promoter and enhancer regions of these genes to which PXR/RXR and CAR/RXR heterodimers bind (for review, see Honkakoski et al., 2003).
In this study, we have investigated the hypothesis that artemisinin drugs induce P450 enzymes at the transcriptional level by activating PXR and/or CAR. We here report that artemisinin and its clinically used derivatives artemether and arteether activate human PXR as well as mouse and human CAR. Most likely, artemisinin acts as an agonist of these nuclear receptors. Treatment of intestinal LS174T cells and primary human hepatocytes with increasing concentrations of artemisinin led to the specific induction of CYP2B6, CYP2C19, CYP3A4, and MDR1 mRNA expression. These findings provide new insights into potential drug interactions of artemisinin.
Materials and Methods
Chemicals. Artemisinin, artemether, arteether, and dihydroartemisinin were generous gifts from Dafra Pharma (Oud-Turnhout, Belgium). 5α-Androst-16-en-3α-ol (androstenol) and digoxin were purchased from Sigma-Aldrich (Taufkirchen, Germany). CITCO, TCPOBOP, rifampin, and verapamil were obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA), CN Biosciences (Beeston, UK), Merck (Darmstadt, Germany), and Abbott (Ludwigshafen, Germany), respectively. [3H]Digoxin and [3H]inulin were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). [14C]Artemisinin was synthesized as described previously (Avery et al., 1996). PSC-833 was kindly provided by Novartis (Basel, Switzerland).
Plasmids. The expression plasmids encoding human PXR and CAR have been described previously (Geick et al., 2001; Burk et al., 2002). Expression plasmids for human VDR and TRα1 were constructed by cloning the open reading frames of the respective receptors, amplified by PCR out of Caco-2 TC7 or human small intestine cDNA using appropriate primers, into expression vector pcDNA3 (Invitrogen, Carlsbad, CA). The respective 5′ upstream primers introduced optimized Kozak consensus sequences. Expression plasmids for human GRα, HNF4α1, TRβ1, and mouse PXR were constructed by subcloning the cDNA inserts of pRShGR (Giguere et al., 1986), pMT2-human HNF4α (kindly provided by I. Talianidis, Institute of Molecular Biology and Biotechnology, Heraklion, Greece), peA101 (American Type Culture Collection, Manassas, VA), and pSG5-mouse PXR (kindly provided by S. Kliewer, University of Texas Southwestern Medical Center, Dallas, TX) into pcDNA3. The mouse CAR expression plasmid pCR3-mCAR was kindly provided by M. Negishi (National Institute of Environmental Health Sciences, Research Triangle Park, NC).
The direct repeat (DR)3 reporter gene plasmid [pGL3(DR3)3Tk] has been described previously (Hustert et al., 2001). The DR4 reporter gene plasmid contains a dimer of the DR4(I) motif of the MDR1 –7.8 kb enhancer (Geick et al., 2001). Enhancer/promoter reporter gene plasmids of CYP3A4 [pGL3-CYP3A4(–7830Δ7208-364)] and CYP2B6 (pB-1.6k/PB/XREM) have been described previously (Hustert et al., 2001; Wang et al., 2003). pB-1.6k/PB/XREM was kindly provided by E. L. LeCluyse (School of Pharmacy, University of North Carolina, Chapel Hill, NC). The enhancer/promoter plasmid of MDR1 [p-7971(Δ7012-227)MDR] was constructed from BglII/PvuII-digested p-7975(Δ7012-1804) (Geick et al., 2001). Digestion with these enzymes removed the sequence between –1803 and –227 of the MDR1 promoter. Construction of the HNF4α-dependent reporter gene, containing a dimer of the HNF4α response element of the human PXR promoter cloned in front of the Tk promoter (–105 to +51) and of the GR-dependent reporter gene, containing four copies of the GR response element of the tyrosine aminotransferase gene promoter cloned in front of Tk, will be described elsewhere. The GAL4-dependent reporter gene construct pGL3-G5 has been described previously (Arnold et al., 2004).
The sequence encoding the RID of human SMRT (GenBank accession no. U37146, amino acids 1109–1330) was amplified by PCR from Caco-2 TC7 cDNA using appropriate primers and cloned into vector pM (BD Biosciences Clontech, Palo Alto, CA), resulting in an expression plasmid encoding the fusion protein of GAL4-DNA binding domain (DBD) and SMRT-RID. Sequences encoding the ligand binding domains (LBDs), or parts of them, of human PXR (amino acids 108–434/132–188/189–434) and of human CAR (amino acids 105–348/105–150/151–348) were amplified by PCR using appropriate primers and cloned into vectors pVP16 or pM (BD Biosciences Clontech), as indicated. The resulting plasmids encode fusion proteins of the VP16 activation domain (AD) or GAL4-DBD and the respective regions of human PXR or human CAR. Expression plasmids encoding fusion proteins of the GAL4-DBD and RIDs of steroid receptor coactivator 1, transcriptional intermediary factor 2, activator of thyroid hormone and retinoic acid receptors, and DRIP205, respectively, have been described previously (Arnold et al., 2004). The identities of all cloned DNA fragments were verified by sequencing.
Cell Culture. The human colon adenocarcinoma cell lines LS174T and Caco-2 were obtained from American Type Culture Collection and cultivated as described previously (Geick et al., 2001). The origin and culture of HepG2 and COS1 cells have been described previously (Arnold et al., 2004).
Transient Transfections and Mammalian Two-Hybrid and Reporter Gene Assays. Transient transfections of LS174T and COS1 cells and mammalian two-hybrid assays for the analysis of CAR interactions in COS1 cells were performed as described previously (Burk et al., 2002; Arnold et al., 2004). Two-hybrid assays for the analysis of PXR interactions in HepG2 cells were performed essentially as described for COS1 (Arnold et al., 2004). In brief, HepG2 cells were plated at a density of 1.5 × 105 cells/well 1 day before transfection. The cells were transfected with 150 ng of the reporter gene plasmid pGL3-G5, 10 ng each of expression plasmids encoding GAL4 or VP16 fusion proteins, and 20 ng of β-galactosidase reference plasmid pCMVβ per well. Luciferase and β-galactosidase activities were analyzed as described previously (Burk et al., 2002). To identify statistical significant differences, statistical tests were performed as indicated with the mean values of at least three independent experiments done in triplicates using InStat version 3.05 for Windows 95 (GraphPad Software Inc., San Diego, CA).
Primary Human Hepatocytes. Human liver tissue was obtained from liver resections of patients with primary or secondary liver tumors. The collection of tissue was done according to the Declaration of Helsinki and the institutional guidelines of the Charité, Humboldt University, Berlin, Germany, and with the informed written consent of each patient. Hepatocytes were isolated and cultured as described previously (Yuan et al., 2004). After 2 days of culture, hepatocytes were maintained for 24 h in Hepato-STIM cell culture medium supplemented with 0.01 μg/ml epidermal growth factor (BD Biosciences Discovery Labware, Bedford, MA), 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Thereafter, cells were treated for 48 h with the indicated chemicals that were added to the culture medium as 1000× stocks in Me2SO or with an equivalent amount (0.1%) of Me2SO only.
Isolation of Total RNA and Real-Time PCR. Total RNA and first strand cDNA were prepared as described previously (Burk et al., 2002). PCR reactions were set up with cDNA corresponding to 25 pg (18S rRNA) or 50 ng (all other assays) of total RNA and the TaqMan Universal PCR Mastermix (Applied Biosystems, Foster City, CA). Expression levels of CYP2B6, CYP2C19, CYP3A4, MDR1, PXR, CAR, and 18S rRNA were quantified by TaqMan real-time quantitative PCR using the 7500 Real-Time PCR system (Applied Biosystems). The experiments were performed according to a standard protocol for the 7500 Real-Time PCR system. Assays were done in triplicate. Oligonucleotide primers and probes were designed with the PrimerExpress software version 1.5 (Applied Biosystems). Primers were used at a final concentration of 400 nM (CYP2B6 and CAR), 900/300 nM (CYP2C19), or 900 nM (MDR1) and probes at 400 nM (CYP2B6), 250 nM (CYP2C19), 240 nM (MDR1), or 200 nM (CAR). The probes were labeled at 5′ with the reporter dye 6-carboxyfluorescein (FAM) and at 3′ with a minor groove binder and nonfluorescent quencher (MGB/NFQ) (Applied Biosystems). Oligonucleotides used for CYP2B6 were as follows: primers 5′-GCT GAA CTT GTT CTA CCA GAC TTT TTC-3′ (exon 4) and 5′-GAA AGT ATT TCA AGA AGC CAG AGA AGA G-3′ (exon 5), probe FAM-5′-TGT ATT CGG CCA GCT GT-3′-MGB/NFQ (exon 4/5). The specificity of the assay was determined using CYP2B6 and CYP2B7 cDNA plasmids. Serial dilutions of the linearized CYP2B6 cDNA plasmid were used to create the calibration curve. Oligonucleotides used for CYP2C19 were as follows: primers 5′-GAC TTT ATT GAT TGC TTC CTG ATC AA-3′ (exon 5) and 5′-GCA GTG ATT ACC AAG TTT TCA ATA GTG-3′ (exon 6), probe FAM-5′-ATG GAG AAG GAA AAG CAA-3′-MGB/NFQ (exon 5/6). The specificity of the assay was determined using CYP2C8, CYP2C9, CYP2C18, and CYP2C19 cDNA plasmids. Serial dilutions of the linearized CYP2C19 cDNA plasmid were used to create the calibration curve. Oligonucleotides used for MDR1 were as follows: primers 5′-CTG GTG TTT GGA GAA ATG ACA GAT A-3′ (exon 4) and 5′-TGG TCA TGT CTT CCT CCA GAT TC-3′ (exon 5), probe FAM-5′-TCA AAC ATC ACT AAT AGA AG-3′-MGB/NFQ (exon 4/5). To create the calibration curve of the MDR1 assay, serial dilutions of linearized plasmid pMDR V15′ were used. Oligonucleotides used for CAR were as follows: primers 5′-AGG TTG CAG AAG TGC TTA GAT GCT-3′ (exon 3) and 5′-TGC TCG CCG CAA TGC-3′ (exon 4), probe FAM-5′-TGA GGA AAG ACA TGA TAC TGT-3′-MGB/NFQ (exon 3/4). The CYP3A4 and PXR assays were performed as described previously (Wolbold et al., 2003). All expression levels were normalized with respect to the expression of 18S rRNA, as determined using the TaqMan ribosomal RNA control reagents (Applied Biosystems) and serial dilutions of Caco-2 cDNA for the calibration curve.
Relative quantification was calculated with the RQ study software module of the 7500 System SDS software version 1.2.3 (Applied Biosystems), which uses the comparative threshold cycle (ΔΔCT) method. 18S rRNA was used as the endogenous control.
Vectorial Transport Studies. Transport was studied using 7-day postconfluent cultures of Caco-2 cells grown as polarized monolayers on semiporous filter inserts in 12-well Costar Transwell plates (Corning Glassworks, Corning, NY). Confluent Caco-2 cells undergo enterocytic differentiation and express MDR1/P-glycoprotein (Pgp) in their apical membrane, allowing the study of vectorial (basal-to-apical) transcellular transport. Before the start of the experiment, culture medium in the apical and basal compartments was replaced by serum-free OptiMEM medium (Invitrogen), and transepithelial electrical resistance was measured to analyze the integrity of the monolayers (>200 Ohm). Transport experiments with digoxin (5 μM) and artemisinin (0.52, 5.25, and 10.25 μM) were performed as described previously (Pauli-Magnus et al., 2000) with radiolabeled [3H]digoxin or [14C]artemisinin as tracers. In addition, involvement of Pgp was demonstrated, using 1 μM Pgp inhibitor PSC-833 in parallel experiments. Substrate experiments were performed at least in duplicates. Inhibition of digoxin transport by artemisinin was studied by adding increasing concentrations ranging from 10 to 200 μM to both the basal and the apical compartment. IC50 value for inhibition by artemisinin was calculated using the net basal-to-apical transport rate at 4 h (with Prism 4.01; GraphPad Software Inc.). Control experiments indicated that translocation of inulin was not significantly altered in the presence of artemisinin, indicating that integrity of the monolayers was not affected by artemisinin. Statistical differences in digoxin/artemisinin net transport in the absence and presence of PSC-833 were tested for significance using unpaired t test (Instat 3.05; GraphPad Software Inc.). A P value <0.05 was considered to be statistically significant.
Pgp Inhibition Studies by ATPase Assays. Purified membrane vesicles derived from insect cells over expressing human MDR1/Pgp (SOLVO Biotechnology, Budaörs, Hungary) were used to measure the inhibition of Pgp substrate-stimulated ATPase activity by artemisinin. Pgp membranes were incubated with 40 μM verapamil, as Pgp substrate, in the absence or presence of increasing concentrations of artemisinin. The amount of inorganic phosphate, generated of ATP by the ATPase activity of Pgp, was determined according to the protocol of the manufacturer. The IC50 value of inhibition by artemisinin was calculated with the mean percentage of ATPase activities of five measurements derived from two independent experiments (using Prism 4.01).
Results
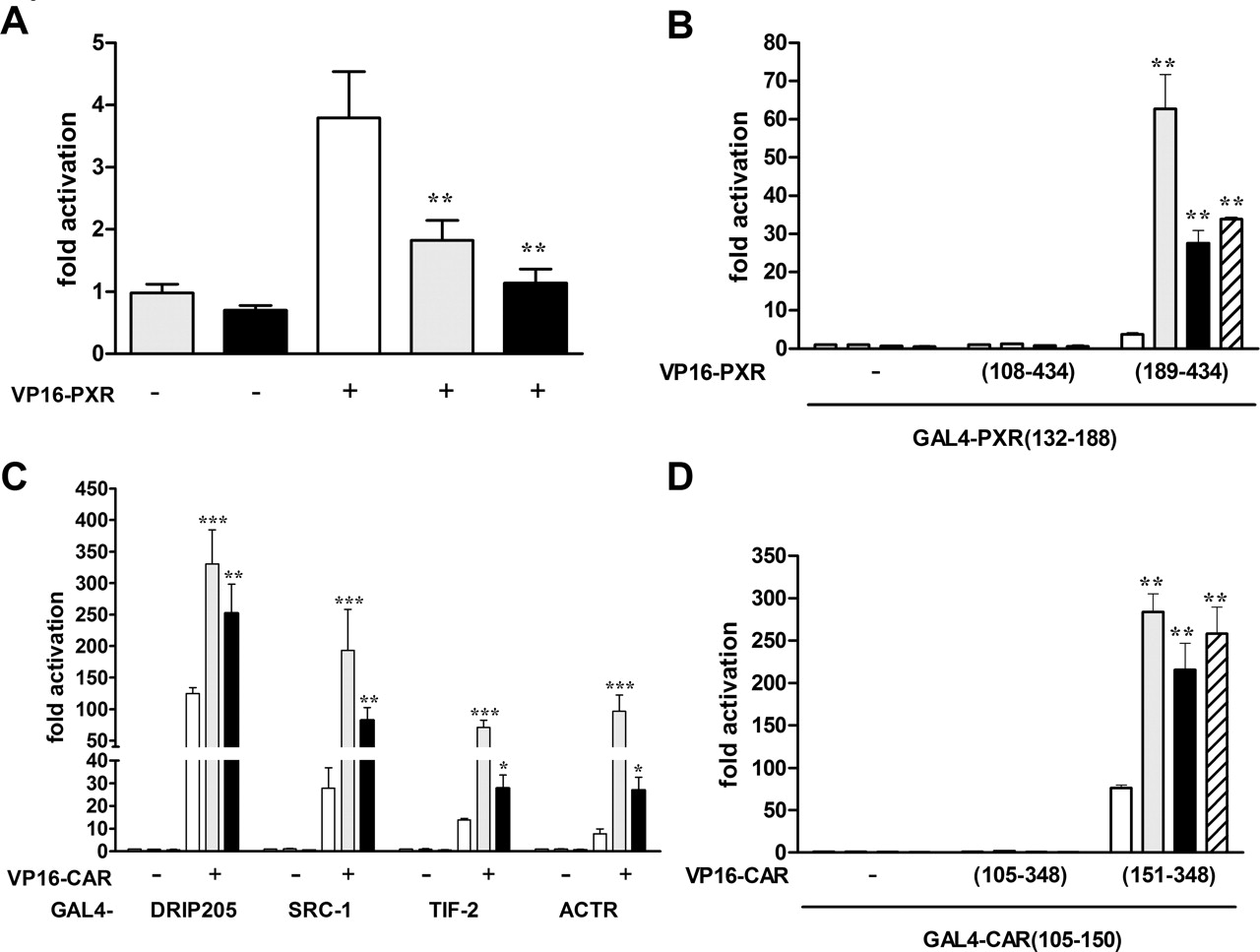
Artemisinin Drugs Specifically Modulate the Activity of PXR and CAR. Expression plasmids encoding different nuclear receptors were cotransfected together with appropriate promoter reporter gene plasmids into COS-1 cells, which were subsequently treated with artemisinin. Figure 1 shows that artemisinin specifically activated human (h) PXR, hCAR, and mouse (m) CAR. The activity of the other human nuclear receptors tested (TRα,TRβ, VDR, HNF4α, and GRα) was not altered significantly, whereas mouse PXR was clearly inhibited by artemisinin. Wherever applicable, we confirmed in control experiments that appropriate prototypical ligands activated the investigated nuclear receptors (data not shown).
Furthermore, we compared the activation potential of artemisinin to that of rifampin, a prototypical ligand of hPXR. Using two different artificial promoter reporter gene constructs, which contained oligomerized DR3 motifs of CYP3A23 or oligomerized DR4(I) motifs of the –7.8-kb enhancer of human MDR1, respectively, we observed that 100 μM artemisinin activated cotransfected hPXR as strong as 10 μM rifampin (Fig. 2A). Dose-response analysis revealed that artemisinin activated hPXR with an EC50 value of 34 μM (95% CI 27–45 μM) (Fig. 2B).
It has been suggested that some clinically used derivatives of artemisinin also induce drug metabolism (for review, see Giao and de Vries, 2001). Therefore, we investigated the potential of the artemisinin derivatives artemether, arteether, and dihydroartemisinin to activate hPXR. Figure 2C shows that 50 μM derivatives artemether and arteether activated hPXR as strong as 100 μM artemisinin. In contrast, 25 μM dihydroartemisinin did not significantly activate hPXR. Concentrations above 25 μM might activate hPXR; however, this possibility could not be tested because of the cytotoxic effects of the compound.
Because CAR demonstrates a strong constitutive activity in cotransfection experiments (Fig. 2D), treatment with artemisinin resulted in only a weak induction of both mouse and human CAR activity (Fig. 1). To confirm activation of CAR by artemisinin, we examined whether the compound can induce CAR-dependent transactivation in the presence of the inverse agonist androstenol that was shown to inhibit CAR activity. Figure 2D shows that androstenol completely repressed mCAR. hCAR activity was inhibited to approximately 50%, thus confirming previous results (Moore et al., 2000). Cotreatment with artemisinin then significantly reinduced CAR-dependent transactivation as efficiently as TCPOBOP and CITCO, prototypical agonists of mCAR and hCAR, respectively. We also analyzed induction of androstenol-repressed hCAR by artemether and arteether. At 50 μM, both derivatives of artemisinin reinduced hCAR as efficiently as 100 μM artemisinin (data not shown).

Artemisinin specifically induces human PXR as well as human and mouse CAR activity. COS1 cells were cotransfected with expression plasmids encoding the indicated human or mouse (m) nuclear receptors and appropriate promoter reporter gene plasmids. The following reporter gene plasmids have been used: CYP3A4 (PXR and mouse PXR), CYP2B6 (CAR and mCAR), DR3 (VDR), GR-dependent reporter (GRα), DR4 (TRα1 and TRβ1), and HNF4α-dependent reporter (HNF4α1). Columns show the mean induction factors (±S.D.) by treatment with 100 μM artemisinin. The activity of each nuclear receptor in the presence of Me2SO only was designated as 1. Statistically significant differences from this value were analyzed by one-sample t test and indicated by asterisks (**, p < 0.01; ***, p < 0.001).
Artemisinin Acts as a Ligand of hPXR and hCAR. Our previous experiments suggest that artemisinin may act as a ligand of hPXR and CAR. If so, it should promote the release of corepressors and induce the interaction with coactivators. To address this issue, we performed mammalian two-hybrid assays that report the interaction of the LBD of hPXR and the RID of corepressors or coactivators, respectively. In the absence of inducers, hPXR constitutively interacted with corepressor SMRT. Artemisinin reversed this interaction as efficiently as rifampin (Fig. 3A). In contrast to rifampin, artemisinin (up to 300 μM) did not induce the interaction of hPXR with coactivators steroid receptor coactivator-1, transcriptional intermediary factor 2, and activator of thyroid hormone and retinoic acid receptors of the p160 family of coactivators. A weak but significant induction of interaction by artemisinin (1.9-fold) was observed with coactivator DRIP205 (data not shown). To prove that artemisinin is bound by hPXR, we applied a recently developed assay based on the ligand-dependent assembly of fragments of the LBD of nuclear receptors. In the absence of ligand, a fragment containing LBD helix 1 of a particular nuclear receptor cannot stably associate with the rest of its LBD. Ligand binding promotes the interaction of helix 1 with the remainder of the LBD (Pissios et al., 2000). Figure 3B shows that the known hPXR ligand rifampin efficiently induced interaction of the hPXR helix 1 fragment with the remainder of the LBD. Artemisinin also induced this interaction, thereby reaching up to 54% of the activity of rifampin.
CAR interacts with coactivators even in the absence of ligands. However, this constitutive interaction can be enhanced by ligand binding. Therefore, we analyzed interaction of hCAR and coactivators by mammalian two-hybrid assays. Figure 3C shows that hCAR-specific agonist CITCO significantly enhanced the interaction of hCAR with the coactivators of the p160 family and with DRIP205. Likewise, artemisinin significantly enhanced the interaction of hCAR with these coactivators. The effects of 1 μM CITCO and 100 μM artemisinin were similar regarding induction of the interaction of hCAR with DRIP205; however, artemisinin seemed to enhance interaction with the coactivators of the p160 family somewhat weaker than CITCO. Increasing the artemisinin concentration up to 300 μM did not significantly enhance induction of these interactions (data not shown). In contrast to PXR, helix 1 of hCAR constitutively interacted with the remainder of the LBD in the assembly assay. CITCO and artemisinin further enhanced this assembly significantly and to a similar extent (Fig. 3D).
Artemisinin Induces CYP2B6, CYP3A4, and MDR1 in LS174T Cells.CYP3A4 is a major target gene of hPXR-mediated induction. However, it has been reported that artemisinin did not induce CYP3A4 enzymatic activity in vivo (Svensson et al., 1998). In an attempt to investigate this inconsistency, we analyzed the induction by artemisinin of different reporter gene constructs containing the authentic enhancer and proximal promoter regions of CYP2B6, CYP3A4, or MDR1, respectively, which have been shown to be required for hPXR-mediated induction. We furthermore made use of the fact that LS174T cells strongly express endogenous hPXR (40% of the expression level in liver; Fig. 4B, inset), so that we could compare the activities of endogenous and cotransfected hPXR. On top of that, hCAR expression is barely detectable in LS174T cells. CAR expression came up to only 0.01% of the expression in liver (Fig. 4B, inset). Figure 4A shows that artemisinin activated cotransfected hPXR to induce the activity of all the reporter genes tested, as did the prototypical hPXR-ligand rifampin. In contrast, induction in the absence of cotransfected hPXR, thus relying on endogenous hPXR only, seemed to be reporter gene-specific: The MDR1 reporter was induced by artemisinin and rifampin to a similar extent, whereas the CYP3A4 reporter was differentially activated by the two compounds. Artemisinin induced the activity of the CYP3A4 reporter only weakly. In the absence of cotransfected hPXR, the CYP2B6 reporter was not significantly induced by artemisinin and rifampin at all. Increasing the artemisinin concentration up to 300 μM did not change the differential induction of reporter activity via activation of endogenous PXR (data not shown).

Artemisinin drugs activate PXR and CAR as efficiently as prototypical inducers. A, LS174T cells were cotransfected with the indicated promoter reporter genes and the hPXR expression plasmid. Gray and black columns denote treatment with 10 μM rifampin or 100 μM artemisinin, respectively. B, dose-response analysis of PXR-dependent induction by artemisinin. LS174T cells were cotransfected with hPXR expression plasmid and the DR3 reporter gene and treated with increasing doses of artemisinin. C, LS174T cells were cotransfected as described in B and treated subsequently as indicated: RIF, 10 μM rifampin; ART, 100 μM artemisinin; AM, 50 μM artemether; AE, 50 μM arteether; DHA, 25 μM dihydroartemisinin. The results of cotransfection experiments are presented as mean induction factors (±S.D.). Reporter gene activity in the presence of Me2SO only was designated as 1 (A–C). Statistically significant differences from this value were analyzed by one-sample t test and indicated by asterisk (*, p < 0.05). D, COS1 cells were cotransfected with the indicated CAR expression plasmids and the CYP2B6 promoter/enhancer reporter gene. The columns show the mean activation factors (±S.D.) by CAR in the presence of the indicated compounds, alone or in combination: AND, 5 μM androstenol; ART, 100 μM artemisinin; CITCO, 1 μM CITCO; TCPOBOP, 0.25 μM TCPOBOP. The activity in the presence of empty expression vector pcDNA3 and treatment with Me2SO only, was designated as 1. Statistically significant differences, as analyzed by one-way analysis of variance with the Student-Newman-Keuls post test, are indicated by asterisks (*, p < 0.05; ***, p < 0.001).
Because these results might point to a gene-specific role for artemisinin in PXR activation, at least regarding endogenous hPXR of LS174T cells, we analyzed the induction of endogenous CYP3A4, MDR1, and CYP2B6 mRNA in these cells. With the exception of MDR1, basal expression of these genes proved to be very low in LS174T cells. Expression of CYP3A4 and CYP2B6 came up to only 0.003 and 0.1% of the respective expression levels in liver, whereas MDR1 expression reached 60% of the hepatic level (Fig. 4B, inset). Therefore, we analyzed induction of these genes by quantitative real-time PCR. The mRNA expression of all three genes was induced in a concentration-dependent manner by artemisinin (Fig. 4B), which is not reflected in the gene-specific induction of reporter activity via activation of endogenous PXR by artemisinin. Treatment with the prototypical PXR activator rifampin induced CYP3A4, MDR1 and CYP2B6 mRNA expression 15.2 ± 4.9-, 4.1 ± 1.1-, and 1.6 ± 0.5-fold, respectively (data not shown). Thus, artemisinin and rifampin induced CYP3A4 and MDR1 mRNA in the intestinal LS174T cells equally well, whereas a significant induction of CYP2B6 was only achieved by artemisinin. In conclusion, these results do not support the hypothesis of a gene-specific role for artemisinin in induction via PXR.

Artemisinin modulates the interaction of coregulators with PXR and CAR and induces the assembly of their LBDs. A, HepG2 cells were transfected with a plasmid encoding GAL4-DBD/SMRT-RID fusion protein, together with a plasmid encoding VP16-AD/hPXR-LBD(108–434) fusion protein (+) or empty expression vector pVP16-AD (–). Columns show the mean activation factors (±S.D.) of the activity of cotransfected reporter plasmid pGL3-G5 by treatment with 10 μM rifampin (gray), 100 μM artemisinin (black), or Me2SO (white). The activity of the combination of the GAL4-SMRT expression plasmid and pVP16, treated with Me2SO only, was designated as 1. B, assembly assay of a GAL4-DBD fusion containing helix 1 of hPXR with two different VP16-AD fusions of hPXR LBD including or lacking helix 1. HepG2 cells were cotransfected with a plasmid encoding GAL4-DBD/hPXR-LBD(132–188) fusion protein and plasmids encoding the indicated VP16-AD/PXR-LBD fusion proteins or empty expression vector pVP16-AD (–). Results are presented as described for A, treatment with 300 μM artemisinin (hatched). The activity of the combination of the GAL4-helix 1 expression plasmid and pVP16, treated with Me2SO only, was designated as 1. C, COS1 cells were cotransfected with combinations of plasmids encoding GAL4-DBD/coactivator-RID fusion proteins, as indicated, and the VP16-AD/hCAR-LBD(105-348) fusion protein (+) or empty vector pVP16-AD (–). The columns show the mean activation factors (± S.D.) of pGL3-G5 reporter activity by treatment with Me2SO (white), (gray), 1 μM CITCO or 100 μM artemisinin (black). The activity of the combination of the respective GAL4-coactivator expression plasmid and pVP16, treated with Me2SO only, was designated as 1. D, assembly assay of a GAL4-DBD fusion containing helix 1 of hCAR with two different VP16-AD fusions of hCAR LBD including or lacking helix 1. COS1 cells were cotransfected with a plasmid encoding GAL4-DBD/hCAR-LBD(105–150) fusion protein and plasmids encoding the indicated VP16-AD/CAR-LBD fusion proteins or empty expression vector pVP16-AD (–). Results are presented as described for C; treatment with 300 μM artemisinin (hatched). The activity of the combination of the GAL4-helix 1 expression plasmid and pVP16, treated with Me2SO only, was designated as 1. Statistical significant differences of treatments versus Me2SO control, as analyzed by one-way analysis of variance with Dunnett's post test (A, B, and D) or the Student-Newman-Keuls post test (C), are indicated by asterisks (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Artemisinin Specifically Induces CAR-Dependent Activation of CYP2B6. Next, we were interested whether CAR may be differentially activated by artemisinin. Figure 5 shows that hCAR, cotransfected into COS-1 cells, activated CYP2B6, CYP3A4, and MDR1 enhancer promoter reporter genes 13-, 7-, and 11-fold, respectively. Treatment with the known hCAR agonist CITCO further enhanced hCAR activity at the CYP3A4 reporter. It did not significantly induce activation of CYP2B6 and MDR1 reporter genes. In contrast, artemisinin exclusively induced the activity of hCAR at the CYP2B6 reporter gene. Thus, induction of hCAR activity by artemisinin may specifically affect CYP2B6 expression.
Artemisinin Induces CYP2B6, CYP2C19, CYP3A4, and MDR1 in Primary Human Hepatocytes. Having shown that artemisinin activates PXR and CAR in vitro and induces the expression of genes regulated by PXR in the intestinal cell line LS174T, we then were interested whether artemisinin is able to induce PXR and CAR target genes in primary human cells. Therefore, cultures of primary human hepatocytes were treated with artemisinin at different concentrations. Of the four genes investigated, CYP3A4, CYP2B6, and MDR1, demonstrated a concentration-dependent induction by artemisinin (Fig. 6). Concentration-dependent induction was not clearly noticeable for CYP2C19. Table 1 shows that artemisinin induced mRNA expression of CYP3A4 in hepatocytes derived from all four donors, although consistently weaker than that induced by rifampin. In contrast, induction of CYP2B6 by artemisinin was slightly stronger than by rifampin. Induction of MDR1 was only detectable in three donors, and in summary both compounds induced MDR1 mRNA expression equally well. CYP2C19 expression was induced only in two donors, with rifampin inducing stronger than artemisinin. As has been described for other inducers, induction by artemisinin was highly variable between individual hepatocyte cultures. CYP3A4 and CYP2B6 were induced 5- to 37-fold and 7- to 17-fold, respectively, whereas MDR1 and CYP2C19 were induced up to 5-fold.
Artemisinin induces P450 and MDR1 expression in human hepatocytes
Induction of mRNA expression by treatment with 30 μM rifampin (RIF) or 300 μM artemisinin (ART) in primary human hepatocytes derived from four donors. Data represent fold induction relative to treatment with vehicle Me2SO alone.
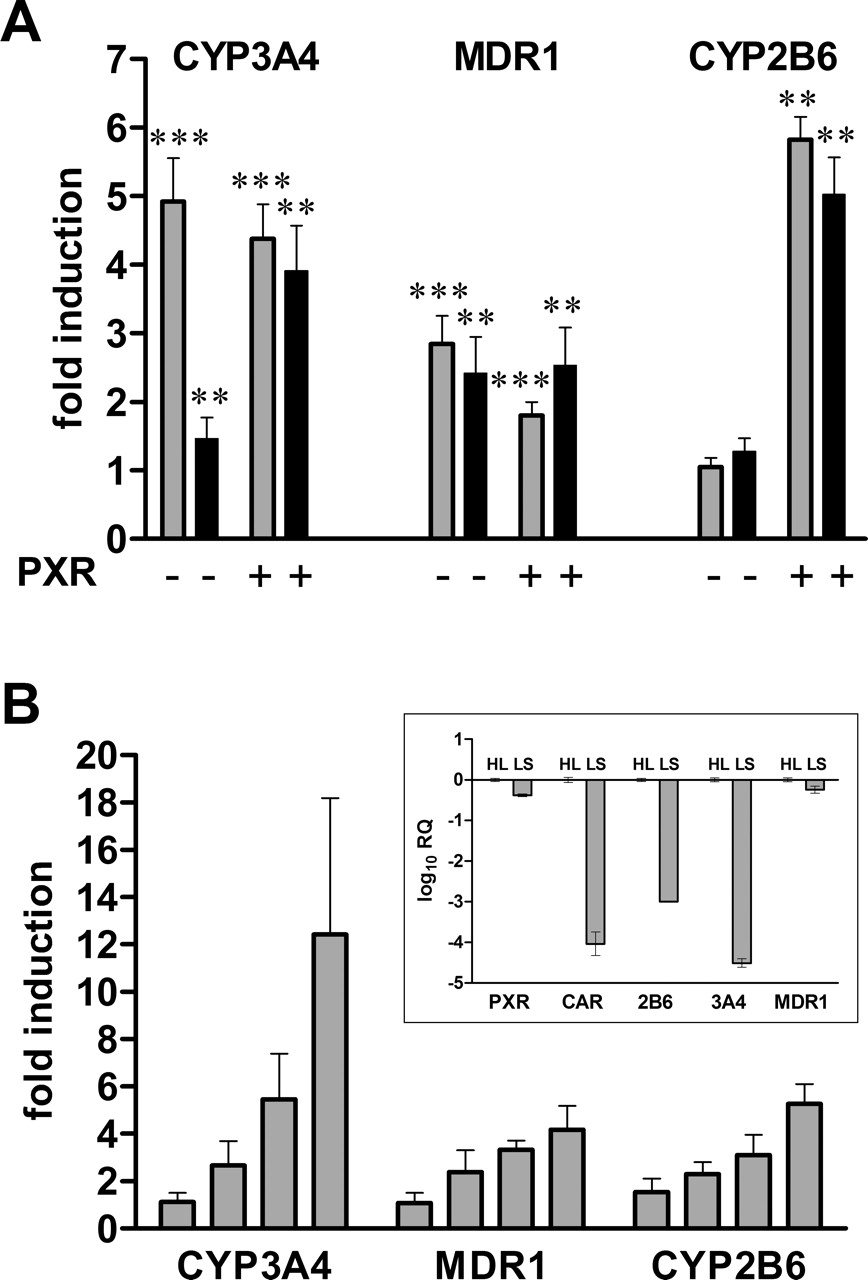
Artemisinin induces P450 and MDR1 reporter genes and endogenous mRNA in LS174T cells. A, LS174T cells were cotransfected with the indicated promoter reporter genes and the human PXR expression plasmid (+) or empty expression vector pcDNA3 (–). The columns show the mean induction factors (± S.D.) of the respective reporter activity by treatment with 10 μM rifampin (gray) or 100 μM artemisinin (black). The corresponding reporter gene activity in the presence of Me2SO only was designated as 1. Statistically significant differences from this value were analyzed by onesample t test and indicated by asterisks (**, p < 0.01; ***, p < 0.001). B, LS174T cells were treated for 48 h with increasing concentrations of artemisinin (10, 30, 100, and 300 μM). The expression of the indicated genes was analyzed by TaqMan real-time reverse transcription-PCR and normalized with respect to the expression of 18S rRNA. The columns show mean fold induction (±S.D.) of four independent experiments, relative to treatment with Me2SO only, which was designated as 1. The inset shows the relative quantification (RQ) of expression of the indicated genes in Me2SO-treated LS174T cells (LS), compared with a human liver pool (HL), as analyzed by TaqMan real-time RT-PCR (see Materials and Methods for details). RNA preparations of liver tissue of six individuals were pooled. The expression level of each gene in human liver was designated as 1.
Artemisinin Is Not a Substrate but an Inhibitor of MDR1/Pgp. We here have demonstrated the induction of MDR1 by artemisinin in the intestinal LS174T cells and in primary human hepatocytes. Therefore, it was of interest to see whether MDR1 also may play a role in the autoinduction of artemisinin elimination. If artemisinin is a substrate of MDR1/Pgp, induction of its expression especially in the intestine may contribute to the autoinduction of artemisinin elimination. Therefore, we investigated vectorial transport of artemisinin by Pgp. As has been shown previously, Caco-2 cells demonstrated a vectorial basal to apical transport of the Pgp substrate digoxin, which was nearly abolished after addition of the Pgp inhibitor PSC-833 (digoxin net transport: digoxin alone versus digoxin + PSC-833, 19.4 ± 4.8 versus 4.7 ± 1.2; p < 0.0001; Fig. 7A; data not shown). In contrast, artemisinin (0.52–10.25 μM) did not show any vectorial transport dependent on Pgp activity in Caco-2 cells (e.g., at 10.25 μM, artemisinin alone versus artemisinin + PSC-833, –2.4 ± 4.8 versus –1.5 ± 4.7.; N.S.; Fig. 7A). Thus, artemisinin did not seem to be a substrate of Pgp. However, artemisinin partially inhibited the Pgp-mediated digoxin transport in Caco-2 cells (Fig. 7A). The compound demonstrated a concentration-dependent inhibition of digoxin net transport with an IC50 of approximately 33 μM. The highest concentration tested (200 μM) reduced digoxin net transport to 48% (Fig. 7B). To confirm these results by means of an independent in vitro assay, we analyzed the transport activity of Pgp membrane vesicles, measured as ATPase activity, in the absence or presence of artemisinin. Figure 7C shows that artemisinin inhibited the ATPase activity of Pgp, which was stimulated by transport of the Pgp-substrate verapamil, to 66% of control level with an IC50 of 32 μM (95% CI 14–75 μM). In conclusion, these results demonstrate that artemisinin is a partial inhibitor of MDR1/Pgp transport activity.

Artemisinin differentially activates hCAR. COS1 cells were cotransfected with the indicated reporter genes and the human CAR expression plasmid (+) or empty expression vector pcDNA3 (–). The columns show the mean activation factors (±S.D.) of the respective reporter activity by treatment with Me2SO (white), 1 μM CITCO (gray), or 100 μM artemisinin (black). The activity in the presence of empty expression vector pcDNA3 and treatment with Me2SO only, was designated as 1. Statistically significant differences, as analyzed by one-way analysis of variance with the Student-Newman-Keuls post test, are indicated by asterisks (***, p < 0.001).
Discussion
In this study, we have demonstrated that artemisinin activates human PXR and CAR, as well as mouse CAR. Thus, we here provide for the first time a molecular mechanism that explains the previously observed induction of P450 enzymatic activity by artemisinin in vivo (Svensson et al., 1998; Simonsson et al., 2003). Artemisinin activates human PXR with an EC50 of approximately 30 μM in vitro. This concentration is 10 to 15 times higher than artemisinin peak plasma concentrations in healthy volunteers and patients (Svensson et al., 1998). Activation of human CAR requires the same range of concentration as PXR; however, EC50 could not be calculated accurately because of the small effect on CAR activity in vitro. Because animal and human studies have indicated that artemisinin is eliminated mainly by extensive hepatic extraction (Dien et al., 1997; Ashton et al., 1998; Ashton et al., 1999), it can be assumed that the concentration of artemisinin is considerably higher in the intestinal epithelium and in the liver, which also are the relevant sites of PXR and CAR expression, than in the systemic circulation. In conclusion, activation of PXR and CAR most probably represents the molecular mechanism of induction by artemisinin. This notion is further supported by the concentration-dependent induction of PXR/CAR target gene expression in the intestinal LS174T cells and primary human hepatocytes by artemisinin, which demonstrates that similar concentrations are required for induction of gene expression as for nuclear receptor activation.
Artemisinin released the interaction of corepressor SMRT with human PXR as efficiently as the prototypical ligand rifampin and also promoted the ligand-dependent interaction of helix 1 with the remainder of the LBD. In contrast to rifampin, artemisinin failed to induce the interaction of the coactivators of the p160 family with human PXR. However, this is not necessarily arguing against artemisinin being a ligand of PXR, because the chemical may promote the interaction of other coactivators with PXR. Alternatively, the release of corepressor interaction by artemisinin may by itself be sufficient to explain activation of PXR. The importance of corepressor release is exemplified by docetaxel, which was shown to promote the interaction of coactivators with PXR however it did not activate the receptor. The failure of docetaxel to activate PXR was assigned to its inability to displace corepressors (Synold et al., 2001). Regarding human CAR, artemisinin significantly enhanced coactivator interaction, as the prototypical agonist CITCO does. Furthermore, artemisinin strengthened the assembly of the LBD, similar to CITCO. In conclusion, artemisinin most probably acts as a ligand of PXR and CAR.
In contrast to the induction of endogenous mRNA, the CYP2B6 and CYP3A4 enhancer/promoter reporter genes are not or only weakly induced by treatment of LS174T cells with artemisinin in the absence of cotransfected PXR. Treatment with artemisinin should induce almost exclusively by activation of endogenous PXR, because LS174T cells barely express CAR. However, exogenous cotransfected PXR did not demonstrate the specificity that was observed for endogenous PXR activated by artemisinin. First, these results indicate that endogenous and exogenous PXR may be functionally different, possibly because of the action of a PXR-modifying system, which is rapidly saturated by cotransfection. Second, further regulatory regions, missing in the reporter constructs, may be required for induction of CYP2B6 and CYP3A4 by specific inducers. These regions may harbor additional PXR binding sites or, alternatively, binding sites for transcription factors interacting with PXR. The reporter gene-specific effects of endogenous PXR activated by artemisinin may also be explained by the variable arrangement and composition of enhancer/promoter modules, which mediate induction of the different genes. Solely far distal PXR binding sites have been identified in the MDR1 gene. Three DR4 motifs and one everted repeat (ER)6 motif, which partially overlap, are clustered in the –7.8-kb enhancer. However, a single motif, the DR4(I) site, almost exclusively mediates PXR-dependent induction (Geick et al., 2001). In contrast, both distal and proximal PXR and CAR binding sites are known in the CYP2B6 and CYP3A4 genes. CYP3A4 harbors a distal XREM, located around –7.7 kb, which consist of single DR3 and ER6 elements (Goodwin et al., 1999). An additional proximal ER6 motif is present around –170 bp. In the CYP2B6 gene, two closely spaced proximal DR4 sites reside at –1.7 kb, whereas the distal XREM around –8.5 kb harbors a cluster of potential binding sites, among which two DR4 motifs proved to be functional binding sites for PXR and/or CAR (Wang et al., 2003). To achieve full induction, the distal XREM modules of CYP3A4 and CYP2B6 have to operate synergistically with their respective proximal regions (Goodwin et al., 1999; Wang et al., 2003), thus indicating a more complex regulation of induction of these genes than in case of the MDR1 gene.
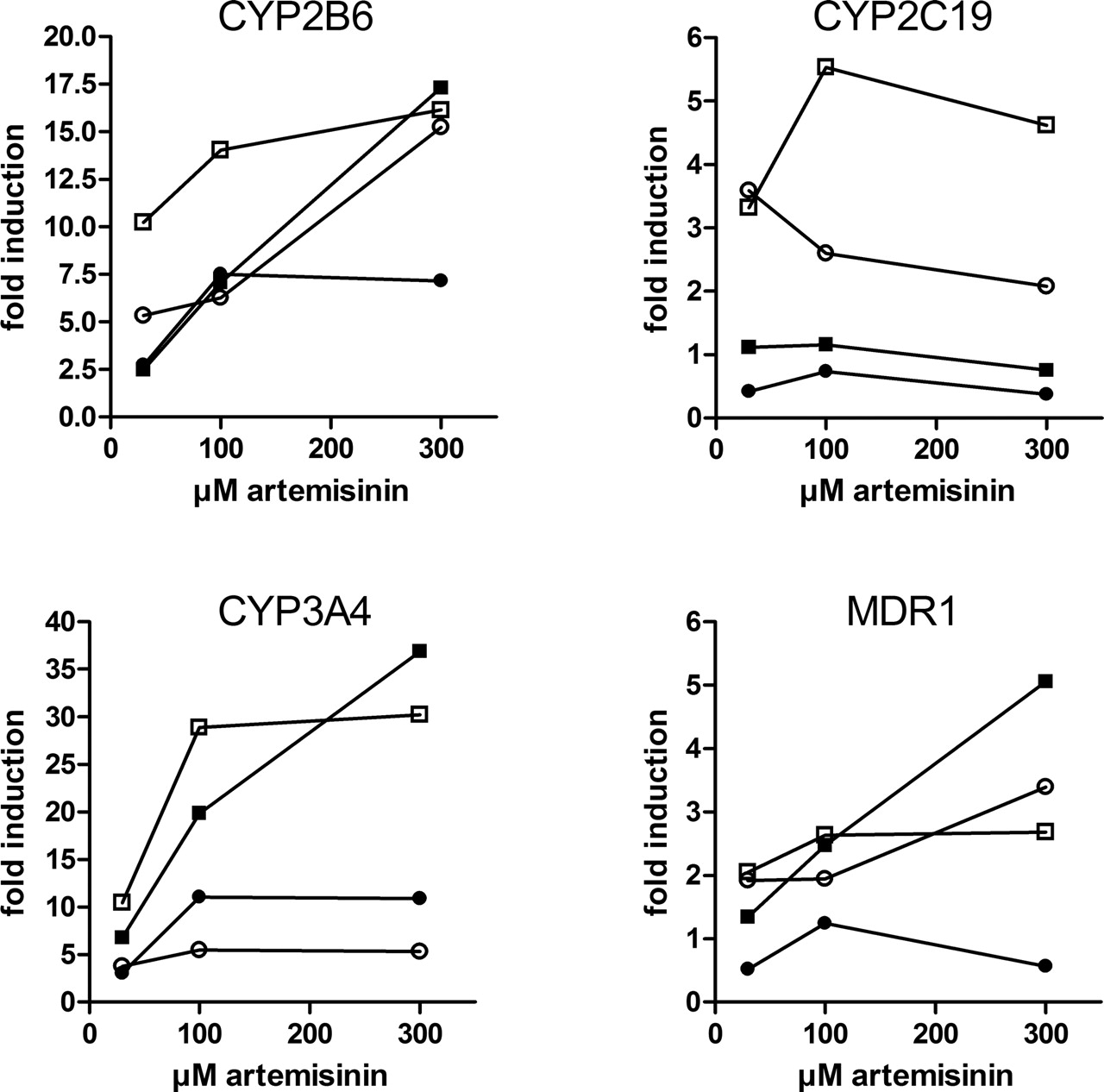
Artemisinin induces P450 and MDR1 mRNA in primary human hepatocytes. Primary human hepatocyte cultures derived from four donors were treated for 48 h with 0.1% Me2SO or increasing concentrations of artemisinin (30, 100, and 300 μM). The expression of the indicated genes was analyzed by TaqMan real-time RT-PCR and normalized with respect to the expression of 18S rRNA. Expression is presented as fold induction relative to treatment with Me2SO only, which was designated as 1. Symbols denote the different cultures: ▪, BH-01; •, BH-02; □, BH-03; and ○, BH-04.
Induction of CYP2B6 and CYP2C19 enzymatic activity by artemisinin was demonstrated previously in vivo (Svensson et al., 1998; Simonsson et al., 2003). We here confirm induction of both genes at the mRNA level. The induction of CYP3A4 mRNA expression is somewhat surprising, because a previous study showed that CYP3A4 enzymatic activity was not induced. The mean values of urinary 6β-hydroxycortisol/cortisol and blood omeprazole sulfone/omeprazole metabolic ratios, which were used as in vivo markers of CYP3A4 activity, remained unchanged during treatment with artemisinin (Svensson et al., 1998). Several possible explanations may account for this discrepancy between induction of expression and activity: First, the assays that have been used to determine CYP3A activity may not always reflect the actual in vivo activity, as it has only recently been shown for the urinary ratio of 6β-hydroxycortisol/cortisol, which is hampered by pronounced variabilities in cortisol renal clearance (Furuta et al., 2003). Second, expression and activity of CYP3A4 may not be correlated in the presence of artemisinin. It has previously been suggested that treatment with artemisinin results in an initial inhibition of CYP3A4 activity (Mihara et al., 1999). Thus, it may be speculated that artemisinin plays a dual role in the regulation of CYP3A4: expression is induced and activity inhibited. A similar behavior has been reported for ritonavir, which induces CYP3A4 expression via activation of PXR (Dussault et al., 2001) and inhibits the activity of the enzyme (Eagling et al., 1998). Inhibition of CYP3A4 activity may therefore mask the effect of an induced expression. As induction of CYP3A4 expression seemed to be highly variable between individuals, at least in vitro (Table 1), the net effect of artemisinin on CYP3A4 activity might also be highly variable. This implication is supported by Svensson et al. (1998), who showed that although the mean value of omeprazole sulfone/omeprazole metabolic ratio remained unchanged, the ratios determined in four of nine subjects clearly increased (Svensson et al., 1998).

Artemisinin partially inhibits MDR1/P-glycoprotein activity. A, vectorial transport was studied using 7-day postconfluent cultures of Caco-2 cells grown as polarized monolayers on semiporous filters. Top, time-dependent, basal-to-apical and apical-to-basal translocation (mean ± S.D.) of artemisinin without and with addition of the Pgp inhibitor PSC-833. Bottom, results of translocation of the Pgp substrate digoxin through the Caco-2 monolayers in the absence and presence of artemisinin. B, dose-response analysis of digoxin net transport in the absence or presence of the indicated doses of artemisinin. Vectorial transport of digoxin was studied in Caco-2 cells as described in A, and net transport rate was calculated by subtracting the apical-to-basal from the basal-to-apical transport rate at 4 h. The columns denote mean ± S.D. of at least four independent experiments performed in triplicates. Statistical significant differences of treatments with artemisinin versus Me2SO control, as analyzed by one-way analysis of variance with Dunnett's post test, are indicated by asterisks (*, p < 0.05; **, p < 0.01). C, dose-response analysis of the inhibition of Pgp ATPase activity by artemisinin. Transport activity in Pgp membrane vesicles was studied by means of ATPase activity stimulated by the Pgp substrate verapamil, in the absence or presence of increasing doses of artemisinin. Results are presented as mean ± S.D. Pgp-dependent ATPase activity in the absence of artemisinin was designated as 100%.
We unequivocally demonstrated induction of MDR1 expression by artemisinin in primary human hepatocytes and intestinal LS174T cells. However, results obtained with a rat in situ perfusion model have shown that pretreatment with artemisinin did not change transport activity of intestinal Pgp (Svensson et al., 1999). This discrepancy may be explained by a lack of correlation between expression and activity of MDR1/Pgp in the presence of artemisinin. We could not show that artemisinin is transported by Pgp, thus confirming previous results (Augustijns et al., 1996). However, we further demonstrated that artemisinin partially inhibited the activity of Pgp. Inhibition of Pgp activity by artemisinin may therefore mask induction of MDR1 expression, as mentioned above for CYP3A4. Again, artemisinin would match ritonavir, which was shown to induce expression of MDR1 by PXR (Dussault et al., 2001) and to inhibit Pgp activity (Penzak et al., 2004). Alternatively, species differences in the activation of PXR may account for the discrepancy. We showed that artemisinin specifically activated human PXR against what mouse PXR was even inhibited. The identity between mouse and rat PXR LBD is 97% and only subtle differences have been identified up to now between the two rodent receptors, regarding ligand specificity (Jones et al., 2000). Thus, we may assume that rat PXR is also not activated by artemisinin. Therefore, the observed auto-induction of artemisinin metabolism in the rat (Gupta et al., 2001) has to depend exclusively on the activation of CAR.
Showing that artemisinin drugs activate PXR and CAR clearly expands the theoretical drug interaction potential of these medicines. It has to be analyzed in future experiments whether the entire battery of detoxifying enzymes and transporters, known to be activated by PXR and CAR (for review, see Handschin and Meyer, 2003), is also induced by artemisinin. In particular, the induction of CYP3A4 and MDR1 expression expands the drug interaction potential of artemisinin, because most drugs, which are coadministered in the combination therapy of malaria, are metabolized by CYP3A4 and/or transported by Pgp (Pham et al., 2000; Giao and de Vries, 2001; Li et al., 2003). A second area of concern may be the treatment of patients being coinfected with human immunodeficiency virus, because many antiretroviral drugs used in the therapy of AIDS are substrates of CYP3A and Pgp (de Maat et al., 2003). Thus, coadministration of artemisinin may result in subtherapeutic levels of these drugs. In conclusion, we suggest that artemisinin drugs may have a much higher risk of drug interactions as previously recognized.
Acknowledgments
We are indebted to K. Abuazi de Paulus and S. Rekersbrink for expert technical assistance. S. Kliewer, T. Lang, E. L. LeCluyse, M. Negishi, and I. Talianidis kindly provided plasmids.
Footnotes
-
This work was supported by Deutsche Forschungsgemeinschaft Grant Bu 1249/1-2, 3, and the Robert Bosch Foundation (Germany).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.009019.
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; CAR, constitutive androstane receptor; MDR, multidrug resistance; CITCO, 6-(4-chlorophenyl)imidazo[2,1-b]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; TCPOBOP, 1,4-bis-[2-(3,5-dichloropyridyloxy)]-benzene; PSC-833, cyclosporin D, 6-[(2S,4R,6E)-4-methyl-2-(methylamino)-3-oxo-6-octenoic acid]; VDR, vitamin D receptor; TR, thyroid hormone receptor; PCR, polymerase chain reaction; HNF, hepatic nuclear factor; GRα, glucocorticoid receptor α; kb, kilobase(s); XREM, xenobiotic-responsive enhancer module; SMRT, silencing mediator for retinoid and thyroid hormone receptors; ER, everted repeat; DBD, DNA binding domain; LBD, ligand binding domain; AD, activation domain; RID, receptor interaction domain; DRIP205, vitamin D receptor interacting protein 205; FAM, 6-carboxy-fluorescein; MGB-NFQ, minor groove binder and nonfluorescent quencher; P-gp, P-glycoprotein; hPXR, human pregnane X receptor; hCAR, human constitutive androstane receptor; mCAR, mouse constitutive androstane receptor; CI, confidence interval.
- Received November 9, 2004.
- Accepted March 10, 2005.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}