


Abstract
The constitutive androstane receptor (CAR) mediates the hepatic induction of various xenobiotic metabolizing enzymes and transporters after specific chemical exposures. Recent reports have established the existence of several human CAR mRNA splice variants, including a prominently expressed form termed CAR3, a receptor that possesses a 5 amino acid insertion within its ligand binding domain. In this study, we demonstrate that, in contrast to the constitutively active reference form of the receptor, CAR3 is ligand-activated, transactivating an optimized DR-4 × 3 reporter in response to the human CAR ligand 6-(4-chlorophenyl)imidazo[2,1-b]thiazole-5-carbaldehyde O-(3, 4-dichlorobenzyl)oxime (CITCO). The transactivation response requires the DNA binding domain and AF-2 motif of CAR3 and is markedly enhanced by retinoid X receptor-α (RXR) cotransfection. The stimulatory effects of RXR involve a unique mechanism, because they were completely dependent on the RXR AF-2 function but independent of both the RXR A/B domain and its C domain/heterodimerization region. Mammalian two-hybrid results demonstrated that RXR enhanced CITCO-dependent interaction of CAR3 with the receptor interaction domain of SRC-1, indicating that RXR augments CAR3 activity by facilitating coactivator recruitment. It is noteworthy that clotrimazole also functions as a ligand activator of CAR3, in contrast to the inverse agonist activity exhibited by this agent on the reference form of the receptor. Furthermore, results of transfection assays reveal that CAR3 is capable of transactivating the natural CYP2B6 and CYP3A4 gene enhancers, exhibiting both ligand- and RXR-dependence. These results demonstrate that CAR3, unlike CAR1, is a ligand-activated receptor and that CAR3 may regulate gene expression in vivo in a manner distinct from the reference form of the receptor.
The constitutive androstane receptor (CAR, MB67, NR1I3) is a member of the nuclear hormone receptor superfamily (Nuclear Receptors Nomenclature Committee, 1999). Its expression is most prevalent in the liver, where it mediates the induction of drug and endobiotic metabolism through a mechanism involving the direct regulation of genes encoding biotransformation enzymes (Baes et al., 1994; Handschin and Meyer, 2003; Wang and LeCluyse, 2003). Studies involving genetically modified mice have demonstrated specific roles for CAR in xenobiotic (Wei et al., 2000; Zhang et al., 2002), heme (Huang et al., 2003; Xie et al., 2003), carcinogen (Xie et al., 2003), bile acid (Guo et al., 2003; Assem et al., 2004; Saini et al., 2004), and thyroid hormone metabolism (Maglich et al., 2004). Furthermore, quantitative trait loci analyses have suggested a role for CAR in HDL homeostasis, an observation that complements other investigations showing that cholesterol precursors (isoprenoids) regulate CAR activity (Kocarek and Mercer-Haines, 2002; Wang et al., 2003b). Together, these findings imply that CAR-mediated activities are potentially important modifiers of human disease processes, such as drug-induced hepatotoxicity, carcinogenesis, cholestasis, atherosclerosis, and obesity.
Identified CAR ligands include 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (mouse CAR-specific agonist; Tzameli et al., 2000), 5β-pregnane-3,20-dione (hCAR-specific agonist; Moore et al., 2000), CITCO (hCAR agonist; Maglich et al., 2003), clotrimazole (hCAR-specific inverse agonist; Moore et al., 2000), and androstanol (mouse CAR inverse agonist) (Forman et al., 1998). Using microarray analyses, a number of CAR-regulated genes have been elucidated (Maglich et al., 2002; Ueda et al., 2002), and many of these genes seem to be directly involved in the metabolism and transport of xenobiotics [e.g., CYP2B6 (Sueyoshi et al., 1999; Wang et al., 2003a), CYP3A4 (Goodwin et al., 2002), CYP2C9 (Ferguson et al., 2002; Gerbal-Chaloin et al., 2002), CYP2C19 (Chen et al., 2003), UGT1A1 (Sugatani et al., 2001), and MDR1 (Geick et al., 2001; Arnold et al., 2004). After inducer treatment in vivo or in primary cultures of hepatocytes, CAR translocates to the nucleus, heterodimerizes with RXR, and interacts with its target genes (Baes et al., 1994; Honkakoski et al., 1998; Kawamoto et al., 1999). An idealized response element for CAR/RXR has been described as a direct repeat-4 element (DR-4) (Frank et al., 2003), although other functional CAR response elements have been characterized, including the DR-2, -3, and -5, everted repeat-6 and -8, and inverted repeat-0 format (Handschin and Meyer, 2003). Furthermore, results from this and other investigations (Frank et al., 2003) suggest that CAR is capable interacting with 5′-extended nuclear receptor response elements as a monomer.
We have identified previously the existence of several CAR splice variants that are expressed concurrently in human liver (Auerbach et al., 2003). These observations have since been substantiated by other laboratories (Savkur et al., 2003; Arnold et al., 2004; Jinno et al., 2004; Lamba et al., 2004). The CAR3 variant in particular is detected in liver tissues at relatively high levels that approach those of CAR1 in certain individuals (Jinno et al., 2004; S. Auerbach and C. Omiecinski, unpublished observations). We have hypothesized that the five-amino acid insertion present in CAR3, occurring in the highly conserved loop 8-9 of the ligand binding domain, may alter the ligand and DNA binding properties of the variant receptor (Auerbach et al., 2003). Although CAR3 exhibits ligand (CITCO)-dependent coactivator interaction (Arnold et al., 2004), significant transactivation by CAR3 of heterologous reporters has not been observed (Auerbach et al., 2003; Arnold et al., 2004; Jinno et al., 2004). In this investigation, we sought to more critically evaluate the functional properties of CAR3. With the use of multiple heterologous reporters, engineered with nuclear receptor half-sites spaced at 1 to 5 base pairs, we now demonstrate that CAR3 mediates transactivation of an optimized DR-4 reporter in a ligand-dependent manner and establish that the CAR1 inverse agonist clotrimazole functions as a bone fide agonist of CAR3. In addition, we report that RXR overexpression markedly enhances CAR3-coactivator interactions as well as CAR3 activation of a DR-4 × 3 reporter. Finally, we demonstrate RXR- and ligand-dependent activation of the endogenous CYP2B6 and CYP3A4 reporters by CAR3, supporting a biological function of the variant receptor, in vivo.
Materials and Methods
Materials. 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) was purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA). Clotrimazole and dimethyl sulfoxide were obtained from Sigma (St Louis, MO) and EM Science (Gibbstown, NJ), respectively. Primers for polymerase chain reaction and electrophoretic mobility shift assays (EMSAs) were purchased from Integrated DNA Technologies (Coralville, IO).
Plasmids. Polymerase chain reaction-based cloning was performed with AccuPOL DNA polymerase (GeneChoice, Frederick, MD). All clones were verified by DNA sequencing. Constructs cloned into the expression vectors contained only the protein coding regions, preceded by a Kozak sequence (Kozak, 1987). Mutagenesis was performed using mutagenesis primers and the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol. Before transfection, plasmids were prepped using the Quantum Prep Plasmid Maxiprep Kit (Bio-Rad, Hercules, CA). All RXR clones referred to in this article are derived from human RXRα. Details of the primers used and design of all of the plasmid constructs used in this study are provided in Tables 1, 2, 3, 4, 5.
pTracer-CMV2 (Invitrogen, Carlsbad, CA) clones The parent plasmid contains a CMV promoter that yields robust expression of cloned sequences when transfected into mammalian cells. This plasmid is referred to in the text as “CMV2”.
pcDNA3.1(+) clones The parent plasmid contains a CMV promoter that yields robust expression of cloned sequences when transfected into mammalian cells. This plasmid is referred to in the text as “3.1”.
p3× FLAG-CMV10 (Sigma,) expression vectors The plasmid was designed with a fusion of three copies of a flu antigen (FLAG) epitope tag to the N terminus of a cloned sequence. Expression of the epitope tagged protein is by way of a CMV promoter. The 3× FLAG-BAP expression vector was supplied by Sigma.
pM and pVP16 (BD Biosciences, Palo Alto, CA) clones In frame cloning of sequences into pM and VP16 generate fusion proteins of the GAL4-DNA binding domain and the viral protein 16 (VP16) activation domain, respectively. These plasmids were used in combinations to access interaction of proteins in mammalian cells. The multiple cloning sites of the plasmids are identical, therefore cloned sequences are interchangeable. The luciferase reporter employed in the mammalian 2-hybrid assays was pFR-Luc (Stratagene, La Jolla, CA).
pGL3-Basic (Promega, Madison, WI) clones The pGL3 basic vector was engineered with the thymidine kinase core promoter as described previously to generate a TK-luc reporter (Auerbach et al., 2003). DR-1X3 through DR-5X3 reporters were made with complimentary primers following their annealing and blunt end ligation into the Sma1 site upstream of TK promoter.
Cell Culture and Transfections. Simian virus 40-transformed African green monkey kidney cells, COS-1 cells, and human embryonic kidney (HEK) 293 cells were maintained and transfected in Dulbecco's modified Eagle medium with 10% FBS, 2 mM l-glutamine, 10 mM HEPES, 0.15% sodium bicarbonate, 50 units/ml penicillin G, and 50 μg/ml streptomycin (all purchased from Invitrogen, Carlsbad, CA).
All transfections for luciferase reporter assays were performed in a 48-well format. On the morning of day 1, cells were plated at a density of 50,000 cells/well. While the cells were attaching, DNA transfection mixtures were assembled using the Fugene6 transfection reagent (Roche Applied Science, Indianapolis, IN). In general, for assays involving standard reporters (not two-hybrid), a combination of 25 ng of CMV2 or CMV2-CAR expression plasmid, 25 ng of pcDNA3.1 or 3.1-RXR expression plasmid, 100 ng of luciferase reporter, and 10 ng of pRL-CMV (for transfection normalization; Promega, Madison, WI) was transfected into each well of a 48-well plate. In one set of experiments, 40 ng of virus protein 16 (VP16) or VP16-CAR3 was transfected in combination with 10 ng of RXR, 100 ng of DR-4 × 3 luciferase reporter, and 10 ng of pRL-CMV. All mammalian two-hybrid assays were performed with 40 ng of pVP16 expression plasmid, 10 ng of pM (GAL4) expression plasmid, 100 ng of pFR-luciferase (luc) reporter, and 10 ng of pRL-CMV. Where indicated, 10 ng of RXR-pcDNA3.1 expression plasmid was used in the two-hybrid assays. The Fugene6 transfection reagent was used at a ratio of 1:3 (micrograms of DNA/microliters of Fugene6 reagent), as recommended in the manufacturer's protocol. Within a given experiment, all transfections contained the same total amount of DNA. At the time of transfection (∼6 h after plating), cells were approximately 80% confluent and had initiated cell division. The following day (16-18 h after transfection), cells were treated with chemical agents as indicated in the figures. If chemical treatment was not performed, cells were lysed and assayed 24 h after transfection. In treated samples, media levels of dimethyl sulfoxide never exceeded 0.1% (v/v). On day 3 (24 h after chemical treatment), cells were washed with phosphate-buffered saline, and luciferase assays were performed using the dual-luciferase reporter assay system (Promega) and analyzed with a Veritas microplate luminometer (Turner Biosystems, Sunnyvale, CA). The luciferase assay and stop and glow reagents were diluted with 1× Tris-buffered saline, pH 8.0, to 0.5× concentration. All other aspects of the assay were performed in accordance with the manufacturer's protocol. Dilution of the luciferase reagent had no effect on normalized luciferase values.
For expression studies and for generating EMSA nuclear extracts, 500 ng of 3×FLAG-tagged CAR1, CAR3, or 3×FLAG-BAP were cotransfected with 500 ng of pcDNA3.1 or 3.1-RXR into a single well of a six-well plate using the Fugene6 transfection reagent. For CITCO treatments, the chemical was added 18 h after transfection, and cells were incubated for an additional 24 h. If chemical treatment was not performed (e.g., in EMSA studies), cells were harvested 24 h after transfection as detailed below.
Nuclear Extraction, Western Blot, and Electrophoretic Mobility Shift Assays. Crude nuclear extracts were prepared from individual wells of a six-well plate that had reached confluence within 24 h of transfection. The protocol has been described in detail elsewhere (Stoner et al., 2002). In brief, cells were trypsinized and collected after the addition of an equal volume of serum-containing media. Pulse centrifugation was used to collect the cells, which were then washed with 100 pellet-volumes of phosphate-buffered saline. Cells were resuspended in 10 pellet-volumes of 1× reporter lysis buffer (Promega, Madison, WI) containing a protease inhibitor cocktail (Calbiochem, La Jolla, CA) and placed on ice for 30 m to allow for passive lysis of the plasma membrane. Every 5 to 10 min, the samples were vortexed gently and then placed back on ice. Nuclei were subsequently collected by centrifugation and resuspended in 10 pellet volumes of 1× reporter lysis buffer supplemented with 500 mM KCl and protease inhibitors. Nuclei were placed on ice and allowed to swell and rupture. Intermittent vortexing was performed to facilitate the nuclear lysis. After 30 m on ice nuclear extract was collected and frozen at -80°C.
Western immunoblotting was performed to determine the level of BAP and CAR protein expression from transfected cells. Crude nuclear protein (40 μg) from each sample was separated on a 10% denaturing polyacrylamide gel. After transfer of protein to a polyvinylidene difluoride membrane, the blot was incubated briefly at room temperature in 5% milk in 1× Tris-buffered saline/0.1% Tween 20 for blocking. Flu Antigen (FLAG) (M2)-horseradish peroxidase-conjugated monoclonal antibody (1:2000 in blocking buffer; Sigma) was incubated with the polyvinylidene difluoride membrane at room temperature for 1 h. The membrane was washed extensively in 1× Tris-buffered saline/0.1% Tween 20, covered with Lumi-Light Western Blotting Substrate (Roche), and exposed to film.
EMSAs were performed as described previously with slight variation (Stoner et al., 2002). Ten picomoles of a double-stranded DNA probe containing a single copy of the DR-4 response element was end-labeled with T4 kinase and [γ-32P]ATP. A 20-μl binding reaction was assembled containing a final concentration of 100 mM KCl (a mixture of low- and high-salt nuclear extract lysis buffers), 6 μg of crude nuclear protein, 1 μg of dI-dC, 5 μM CITCO (or corresponding amount of dimethyl sulfoxide), and 20 fmol of labeled probe. Binding reactions were incubated for 15 m at room temperature. After incubation, where indicated, 1 μg of polyclonal FLAG antibody was added. Antibody/epitope interactions took place at room temperature over 15 m. Protein/DNA complexes were separated on a 5% TBE gel. Gels were dried and exposed to film to visualize mobility shifts.
Statistical Analysis. All quantitative data were examined by analysis of variance. Significance was declared if P < 0.01. Data are expressed as means ± S.E. (n = 4).
Results
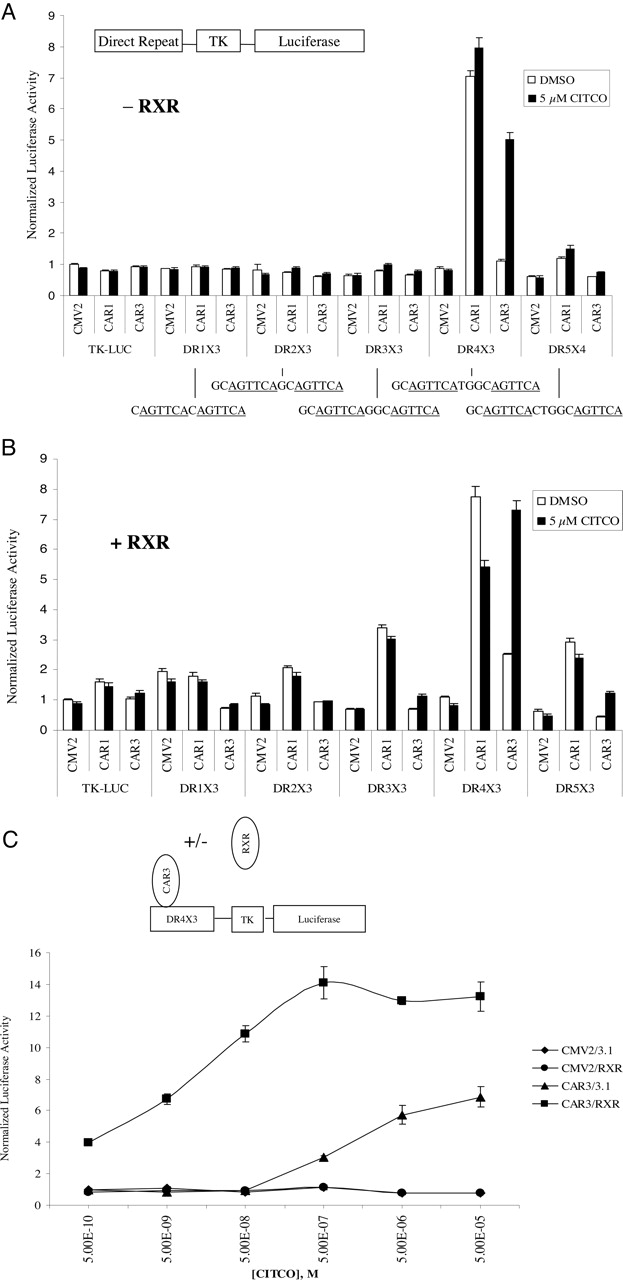
CAR3 Transactivates DR-4 × 3 and DR5 × 3 Luciferase Reporters in a Ligand-Regulated and RXR-Dependent Manner. Other researchers (Arnold et al., 2004) have reported a ligand-regulated recruitment of SRC-1 to CAR3; however, transactivation by this receptor has not been demonstrated previously. We hypothesized that the inserted amino acids in CAR3 produce a unique receptor with a different spectrum of effects on various DNA response elements. To test this hypothesis, heterologous reporters were constructed that contained six copies of an AGTTCA nuclear receptor half-site separated by 1 to 5 base pairs. AGTTCA was chosen over AGGTCA because CAR1 has been demonstrated to prefer binding to this half-site (Frank et al., 2003). Furthermore, the reporters were engineered to allow preferential interaction with CAR/RXR heterodimers (as opposed to CAR monomers) by placing a GC or TC sequence immediately 5′ of each half-site (Frank et al., 2003). In absence of cotransfected RXR, CAR3 activated the DR-4 × 3 reporter only when treated with CITCO (Fig. 1A). However, when CAR3 and RXR were cotransfected, enhanced basal levels of activation together with an overall higher ligand-dependent activation of the DR-4 × 3 reporter were evident, as was a significant ligand-dependent transactivation of the DR-5 × 3 reporter (Fig. 1, A and B). CAR1 did not respond to 5 μM CITCO in our reporter system in the absence of RXR and, surprisingly, was deactivated by CITCO when RXR was cotransfected. As observed with CAR3, RXR cotransfection similarly facilitated a significant transactivation of CAR1 on the DR-2, DR-3, and DR-5 reporters, an effect that was not observed in the absence of cotransfected RXR.
A CITCO dose-response study was performed using the DR-4 × 3 reporter to assess the effect of RXR on ligand-induced CAR3 activity (Fig. 1C). Transfection of RXR alone had no effect on the reporter. CAR3 transfection in the absence of RXR led to a maximum 6-fold activation, with an estimated EC50 of 1 μM. Cotransfection of both receptors resulted in a 4-fold increase in basal activity and a maximal CITCO activation of 14-fold. In addition, the estimated EC50 (based on the adjusted basal activity) was decreased approximately 50-fold, to 20 nM CITCO.
CAR3 Does Not Interact with RXR in EMSA or Two-Hybrid Analyses. EMSA have not been capable of demonstrating an RXR-dependent interaction of CAR3 with a CAR response element (Auerbach et al., 2003; Arnold et al., 2004). These prior assays were performed using purified GST-CAR3 and GST-RXR fusion proteins or CAR3 and RXR expressed from reticulocyte lysates. A potential explanation for these observations may involve the lack of expression of an unknown cofactor protein in COS-1 cells that may otherwise function to facilitate DNA-CAR3/RXR interactions. Furthermore, interaction with such a cofactor might be effected by ligand, as is the case with RXR homodimers and SRC-1 (IJpenberg et al., 2004). To evaluate this scenario, FLAG-tagged CAR1 and CAR3 expression vectors were cotransfected into COS-1 cells in combination with empty pcDNA3.1 or RXR expression plasmid. A FLAG-tagged BAP expression vector was also transfected as a negative control for DNA binding. Crude nuclear extracts were prepared from the cells 24 h after transfection. A Western blot of 40 μg of nuclear extract was performed to evaluate expression of BAP, CAR1, and CAR3 in these extracts (Fig. 2A). RXR expression did not impact the detectable levels of either CAR1 or CAR3. EMSAs were performed with 6 μg of the protein extracts. A CAR1 complex is apparent in the absence and presence of CITCO and is further enhanced by cotransfection of RXR. The CAR1 complex was also supershifted after the addition of FLAG antibody (Fig. 2B). In contrast, a CAR3-containing DNA complex could not be detected (Fig. 2B).
Others researchers (Arnold et al., 2004) have reported that CAR3 was unable to interact with RXR in mammalian 2-hybrid analysis. We performed a similar set of experiments that are presented in Fig. 2C. Although a clear interaction between the ligand binding domains of CAR1 and RXR were evident, a comparable interaction of CAR3 with the RXR was not detected. Neither CITCO treatment nor cotransfection of VP16-SRC-1(RID) was capable of facilitating a CAR3/RXR two-hybrid interaction (data not shown). These results both confirm and extend the results of Arnold et al. (2004).
Activation of DR-4 × 3 by CAR3 Is Dependent on Its DNA Binding Domain and AF-2 Domains. Considering that a CAR3 complex was not observed in the EMSA, we questioned whether CAR3 might interact with the reporter directly. A direct interaction would probably be mediated through the DNA binding domain of the receptor. Deletion of DNA binding domain sequences ablated the CAR3-mediated transactivation of the DR-4 × 3 reporter (Fig. 3), indicating that a CAR3 DNA interaction was important for receptor transactivation. We further reasoned that if the ligand-mediated transactivation occured via the ligand binding domain of CAR3, then deletion of the AF-2 domain of the receptor should block the recruitment of coactivators and hence the ability of CAR3 to transactivate transcription. Deletion of the CAR3 AF-2 led to a complete loss of ligand-mediated transactivation (Fig. 3). These data provide support for a mechanism of ligand-dependent activation by CAR3 involving receptor recruitment to DNA via its DNA binding domain, with the subsequent recruitment of transcriptional coactivators.
Transfection of RXR Has No Effect on CAR3 Cellular Localization. In hepatocytes, CAR1 activity is controlled in part by its cellular localization (Zelko and Negishi, 2000). Furthermore, the CAR interacting protein, GRIP-1, leads to accumulation of CAR in the nucleus independent of ligand treatment (Min et al., 2002). Hence, we considered whether the mechanism of RXR activation of CAR3 might involve an RXR-driven nuclear accumulation of CAR3. Nuclear and cytosolic extracts were generated from transfected COS-1 cells and subjected to Western immunoblot analysis. However, the results demonstrated that the relative nuclear/cytosolic distribution of CAR3 was unchanged either by CITCO treatment or by RXR overexpression (Fig. 4A).
VP16 possesses a strong nuclear localization sequence in addition to a robust transactivation domain. Therefore, a VP16-CAR3 fusion construct should force nuclear accumulation of the receptor. Although the Western blot data of Fig. 4A argue otherwise, if RXR is functioning to enhance the activity of CAR3 by facilitating the nuclear accumulation of the receptor, then fusion of CAR3 receptor with VP16 should serve to eliminate its RXR dependence. In Fig. 4B, we present the results of experiments designed to assess the activity of VP16-CAR3 on the DR-4 × 3 reporter. Transfection of VP16-CAR3 had no effect on DR-4 × 3 reporter activity in the absence of CITCO, whereas its presence stimulated reporter activity 10-fold. Cotransfection of RXR led to a 10-fold activation of the reporter in the absence of CITCO treatment. Although these results were striking in themselves, cotransfection of RXR in combination with the CITCO ligand resulted in a synergistic 170-fold transactivation. VP16-CAR1 activity also was enhanced by cotransfection of RXR, although the effect was not as dramatic as that observed with CAR3.
To demonstrate directly the nuclear localization of transfected VP16-CAR3, COS-1 cells were cotransfected with a CAR3-VP16 fusion construct or a VP16 control vector. As shown in Fig. 4C, CAR3 expression clearly colocalized with 4,6-diamidino-2-phenylindole nuclear fluorescence in the transfected cells. Together, these results support a mechanism involving RXR activation of CAR3 after its nuclear translocation, rather than an RXR-directed facilitation of CAR3 nuclear localization.
The AF-2 of RXR Is Essential for Enhancement of CAR3 Activity. A mutagenized form of RXR (Y397A) that is heterodimerization-deficient has recently been described (Vivat-Hannah et al., 2003). If, as a means of enhancing CAR3 activity, CAR3 forms heterodimeric complexes with RXR on the DR-4 × 3 reporter, then cotransfection of the dimerization mutant Y397A should result in no enhancement of CAR3 activity. When we conducted these assays, we found that the Y397A mutant had the same enhancing effect on CAR3 as the reference form of RXR (Fig. 5), whereas the Y397A RXR mutant was not active in stimulating CAR1 constitutive or ligand-inducible activation (data not shown). These data support a nontraditional, dimerization-independent mechanism of RXR-CAR3 activation, a concept that is also supported by additional results from other transfection studies and mammalian two-hybrid analyses, as described below.

CAR3 transactivation of a DR-4 × 3 reporter is ligand-dependent and facilitated by the overexpression of RXR. In A and B, COS-1 cells were transfected with indicated expression vectors [A, pcDNA3.1 (empty vector), B, pcDNA3.1/RXR] in combination with TK-luciferase reporters containing three copies of a direct repeat spaced by 1 to 5 base pairs. A diagram of the vector scheme and details of the respective direct repeat elements used for the experiments conducted in A (- RXR) and B (+ RXR) are shown at the top. Cells were transfected for 18 h as described under Materials and Methods. On day 2, transfected cells were treated with either DMSO or 5 μM CITCO (A and B) or increasing amounts of CITCO as indicated (C) for 24 h, after which luciferase activity was assayed. CMV2 is an empty expression vector. Data are presented as normalized and adjusted luciferase values in which the activity of the DMSO-treated CMV2/3.1 (A), DMSO-treated CMV2/RXR (B), or 500 pM CITCO-treated CMV2/3.1 (C) data point is set to 1. Each data point represents the mean (± S.E.) of four separate transfections.

Nuclear extracts from CAR3-transfected cells do not bind a DR-4 DNA probe, and CAR3 does not interact with RXR in mammalian 2-hybrid assays. A, Western blot of 40 μg of crude nuclear extract from 24-h transfected COS-1 cells. Nuclear extracts were generated as described under Materials and Methods. Blots were probed with HRP-conjugated M2 FLAG antibody. B, 6 μg of nuclear extract from A was used in an EMSA with a DNA probe representing one copy of the DR-4 element engineered into the DR-4 × 3 reporter. Details of the experiments are found under Materials and Methods. Protein/probe mixtures were incubated with either DMSO or 5 μM CITCO. C, mammalian two-hybrid assays were performed as illustrated in the figure and as described under Materials and Methods. Data are presented as normalized and adjusted values where the luciferase activity of the GAL4/VP16 (empty vector/empty vector) experiment is set to 1. Each data point represents the mean ± S.E. of four separate transfections. *, probe retained in well.
It is well established that the DNA binding domain of RXR is essential for the interaction of heterodimeric receptor complexes with DNA. If RXR is forming a heterodimeric DNA complex with CAR3, then transfection of an expression construct containing only the sequences of the ligand binding domain of RXR would not be expected to enhance CAR3 activity. However, transfection of the RXR ligand binding domain alone resulted in similarly enhanced activities as provided by the full-length receptor (Fig. 5). It is also well accepted that the RXR AF-2 motif is essential for ligand-regulated interaction of coregulators. If the association of RXR with coregulators is essential for enhancing CAR3 activity, then the deletion of this sequence would be predicted to ablate the enhancing effect of RXR. Consistent with this view, transfection of an AF-2-deleted form of RXR was completely ineffective in contributing any stimulation of ligand-independent or -dependent transactivation, indicating that the association of RXR with coregulatory factors is mechanistically essential to its CAR3-enhancing activity (Fig. 5). It is noteworthy that transfection of an AF-1 domain-deleted RXR was still capable of enhancing CAR3 activity, lending additional support to the importance of an AF-2-dependent recruitment mechanism.
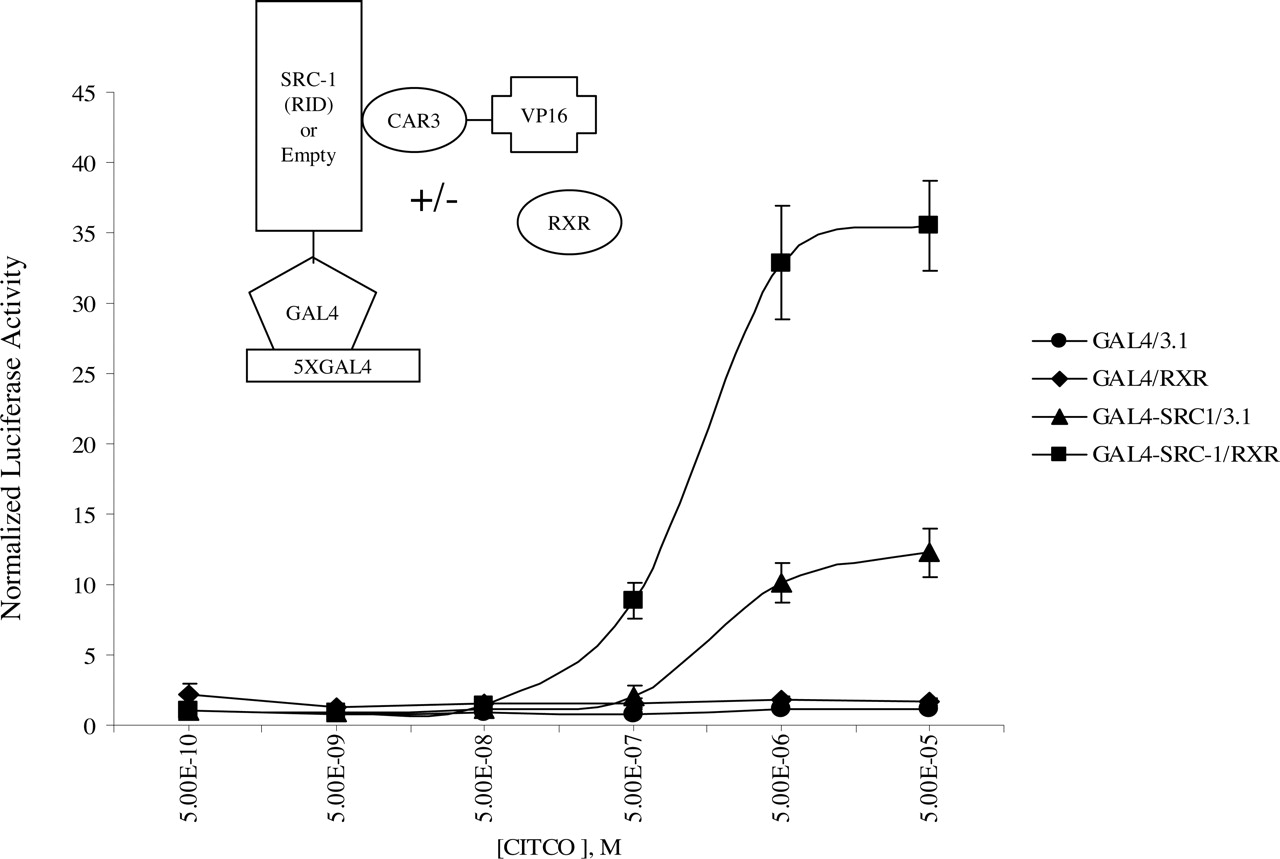
Ligand-Dependent CAR3 Interaction with SRC-1 Is Enhanced by RXR. It has been established previously that CAR3 recruits SRC-1 and other coactivators in response to CITCO (Arnold et al., 2004). To evaluate the effect of RXR on ligand-induced recruitment of coactivators, a mammalian two-hybrid assay was developed in which the receptor interaction domain (RID) of SRC-1 was fused to a GAL4 DNA binding domain and CAR3 was fused to VP16. If the two fusion proteins interact in the transfected cells, then VP16 is recruited to the reporter, leading to increased luciferase expression. As shown in Fig. 6, CITCO additions resulted in a dose-dependent increase in the interaction between CAR3 and SRC-1. Cotransfection of RXR raised the maximum ligand induced interaction between the CAR3 and SRC-1. However, unlike the results obtained in the DR-4 × 3 reporter assays, RXR did not affect ligand sensitivity (EC50 ∼2 μM, ± RXR) and did not enhance basal activity at low levels of ligand. In addition to SRC-1, we evaluated a number of other nuclear receptor coactivators with respect to their interaction with CAR3, including SRC-2, SRC-3, cAMP response element-binding protein binding protein, p300, PGC-1α, and DRIP205. RXR cotransfection resulted in enhanced CAR3 interactions with each of the coactivators tested (results not shown).
Clotrimazole Produces a Dose-Dependent Activation of CAR3 That Is Enhanced by Cotransfection of RXR. By establishing an assay system to allow testing of CAR3 activity, it was possible to evaluate the effect of potential ligands. Initial studies involving known CAR ligands demonstrated an activation of CAR3 by clotrimazole. Clotrimazole is an inverse agonist of CAR1 (Moore et al., 2000). Dose-response experiments using the DR4 × 3 reporter demonstrated EC50 values of approximately 300 nM when RXR was cotransfected with CAR3 and >3 μM in the absence of RXR (Fig. 7A). To further verify that clotrimazole was acting as a ligand activator of CAR3, mammalian two-hybrid studies were performed. In these assays, clotrimazole produced a dose-dependent increase in the interaction of CAR3 with the RID of SRC-1. RXR enhanced the maximal interaction with SRC-1. It was not possible to determine whether the EC50 was affected because of the lack of a clear plateau in the activation curve (Fig. 7B). Two-hybrid assays were also performed in which the CAR3 ligand binding domain was fused to GAL4 and the GAL4-CAR3-ligand binding domain construct was cotransfected with VP16-SRC-1/RID. Treatment of the transfections with CITCO or clotrimazole produced a significant activation of the GAL4-UAS reporter response. The ligand-dependent activation by GAL4-CAR3-ligand binding domain was enhanced by addition of RXR (Fig. 7C). The augmentation of CAR3 activity by RXR in this assay further supports a mechanism of enhancement by RXR that is independent of the interaction of CAR3 with DNA.

Transactivation of the DR4 × 3 reporter by CAR3 is dependent on its DNA binding domain and AF-2 motif. The DR4 × 3 reporter was transfected in the presence of RXR and different forms of CAR3 with deletions illustrated and indicated in figure. Details of the transfection procedure are described under Materials and Methods. DMSO and CITCO treatments were performed 18 h after transfection, and cells were harvested and assayed 24 h after chemical treatment. Data are presented as normalized and adjusted values in which the activity of the DMSO treated 3.1 (empty vector) experiment is set to 1. Each data point represents the mean ± S.E. of four separate transfections

RXR does not stimulate CAR3 activation by enhancing receptor nuclear accumulation. A, nuclear extracts were generated and Western blots were performed as described under Materials and Methods. N, nuclear fraction; C, cytosolic fraction. Molecular mass markers (in kilo-daltons) are shown to the left of the autoradiograph. Treatment with CITCO or cotransfection with RXR had little to no effect on the nuclear/cytosolic distribution of 3×FLAG-CAR3. B, transfections and treatments were performed as described under Materials and Methods and as illustrated in the figure. Data are presented as normalized and adjusted values based on the activity of the DMSO-treated VP16/3.1 experiment as set to 1. C, COS-1 cells were transfected with a CAR3-VP16 fusion construct, or a VP16 control vector. COS-1 nuclei were visualized by 4,6-diamidino-2-phenylindole fluorescence. CAR3 expression and nuclear localization was visualized using a rabbit polyclonal anti-hCAR derived primary antibody (30 μg/ml) and a goat-anti-rabbit-IgG-fluorescein isothiocyanate secondary antibody (1:200; CALTAG Corp.). Magnification, 400×.
Clotrimazole and CITCO Activate the DR-4 × 3 Reporter in HEK293 Cells. To verify that the ligand effects on CAR3 were not cell-type dependent, DR-4 × 3 reporter assays were performed using HEK293 cells. Both CITCO and clotrimazole enhanced CAR3-dependent transactivation of the reporter in these cells (Fig. 8). It is noteworthy that the absolute activation by clotrimazole was even higher than that of CITCO in this cell line.
Ligand and RXR-Dependent CAR3-Mediated Transactivation of CYP2B6 and CYP3A4 Reporters. The DR-4 × 3 reporter employed in the studies described above contains three copies of an optimal CAR response element (Frank et al., 2003), a situation that does not exist in the human genome. Hence, studies were undertaken to determine whether CAR3 was capable of activating two different luciferase reporters derived from sequences present in the endogenous CYP2B6 and CYP3A4 promoters that contain the naturally occurring/degenerate nuclear receptor response elements. The CYP2B6 promoter contains two regions that confer CAR-mediated activation, the phenobarbital-responsive enhancer module (Sueyoshi et al., 1999) and XREM (Wang et al., 2003a). The CYP3A4 promoter also contains two distantly spaced promoter regions that confer xenobiotic inducibility and interact with CAR (Goodwin et al., 2002). These regions are referred to as the proximal ER6 (Barwick et al., 1996) and the XREM, respectively (Goodwin et al., 1999). As described under Materials and Methods, both of the CYP2B6 and CYP3A4 reporters we used possessed single copies of these promoter regions and were arrayed directly upstream of TK-luciferase. The data generated using the CYP2B6 and CYP3A4 reporters are presented in Fig. 9, A and B, respectively. Transfection of CAR3 in the absence of RXR resulted in no basal or ligand-induced (CITCO or clotrimazole) activity on either the CYP2B6 or CYP3A4 promoter. However, cotransfection of CAR3 and RXR yielded a statistically significant CITCO-dependent activation of these reporters. Activation by clotrimazole was attainable only with the CYP2B6 reporter when RXR was cotransfected. It is noteworthy that the constitutive activity of CAR1 on the CYP2B6 and CYP3A4 reporters was enhanced by the cotransfection of RXR. CITCO additions did not significantly activate CAR1 in the absence or presence of RXR on either reporter, although CTICO did down-regulate activity of the receptor on the CYP2B6 reporter when RXR was cotransfected. Perhaps the most remarkable result of these experiments is the opposite effect of clotrimazole on the activity of CAR1 and CAR3. In all cases, clotrimazole treatments decreased CAR1 activity but produced an RXR-dependent induction of CAR3 on the CYP2B6 reporter.
Discussion
CAR3 is a structural variant of the constitutive androstane receptor derived from alternative splicing of the human CAR mRNA. An alternative splice acceptor site in intron 7 inserts 15 nucleotides in the mRNA sequence, which in turn leads to an insertion of five amino acids in loop 8-9 of the ligand binding domain (Auerbach et al., 2003). Homology modeling studies indicate that the ligand binding pockets of CAR3 and CAR1 are identical (Auerbach et al., 2003). Hence, both receptors would be predicted to interact with the same ligands. The CAR3 splice variant is detected in liver tissues at relatively high levels that approach those of CAR1 in certain individuals (Jinno et al., 2004; S. Auerbach and C. Omiecinski, unpublished observations). In contrast to the constitutively active reference form of the receptor, CAR1, the studies presented here demonstrate for the first time that CAR3 possesses unique biological properties, functioning as a ligand-dependent transactivator of both CYP2B6 and CYP3A4 reporters, with the five-amino acid enlargement of loop 8-9 responsible for transforming the reference receptor from one that is normally deactivated by clotrimazole to one that is ligand activated. We also demonstrate that the dependence of the CAR3 transactivation response on RXR cotransfection, an effect that is associated with CAR3 recruitment of transcriptional coactivators such as SRC-1. It is noteworthy that the functional interplay between RXR and CAR3 takes place apparently in the absence of a traditional heterodimerization interaction between the receptors. Together, these results demonstrate that CAR3 has acquired the novel property of being a ligand-activated nuclear receptor and further imply that CAR3 may possess unique capabilities as a regulator of gene expression in vivo.
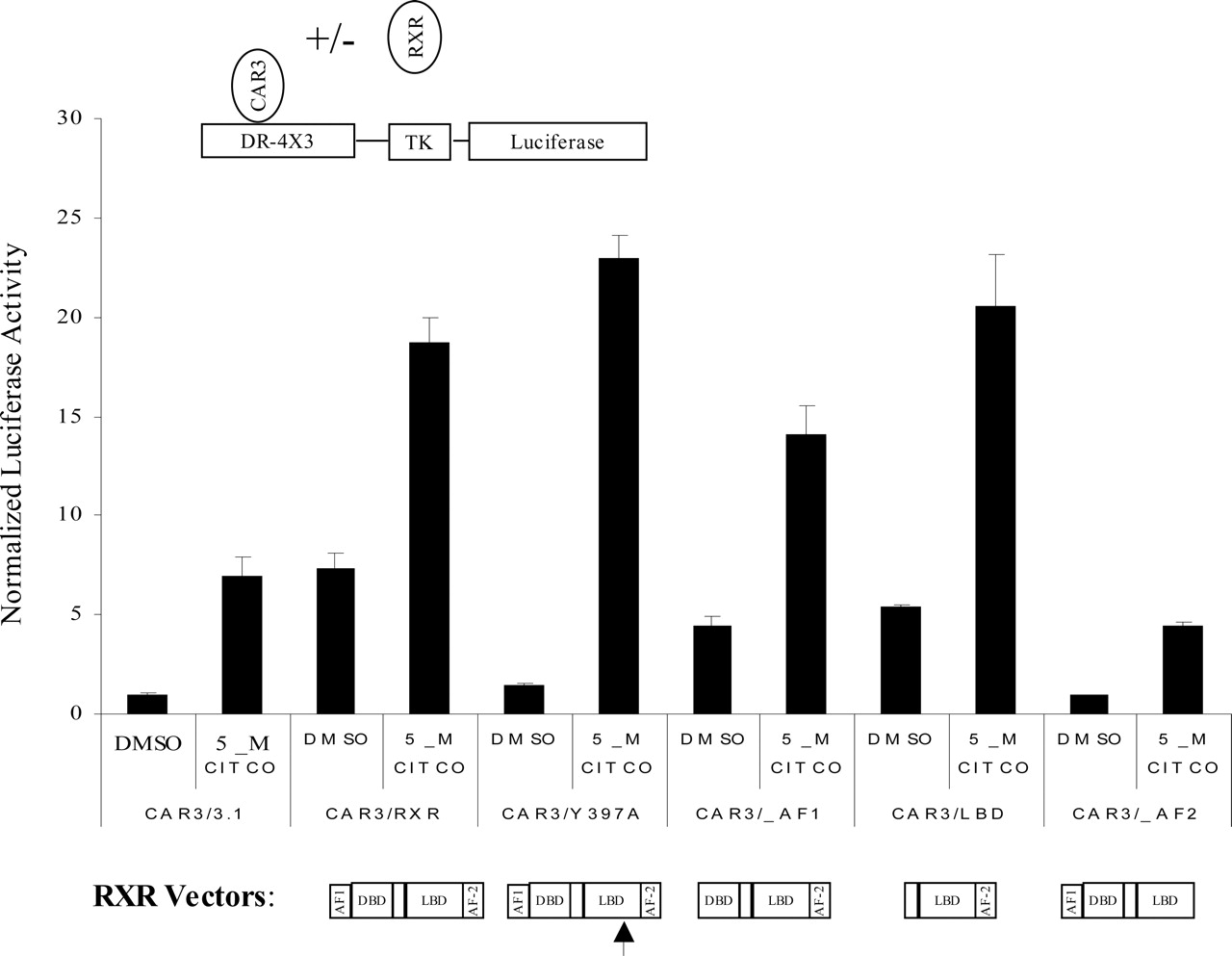
Enhancement of CAR3 activity by RXR is independent of the RXR DNA binding domain but dependent on its AF-2 motif. The DR4 × 3 reporter was cotransfected into COS-1 cells in combination with CAR3 and different forms of RXR as illustrated and indicated in figure. The arrows in the figure refer to the relative positions of the Y397A RXR point mutation in the ligand binding domain region of RXR. Details of the transfection procedure are described under Materials and Methods. Data are presented as normalized and adjusted values in which the activity of the DMSO-treated CAR3/3.1 data point was set to 1. Each data point represents the mean ± S.E. of four separate transfections.
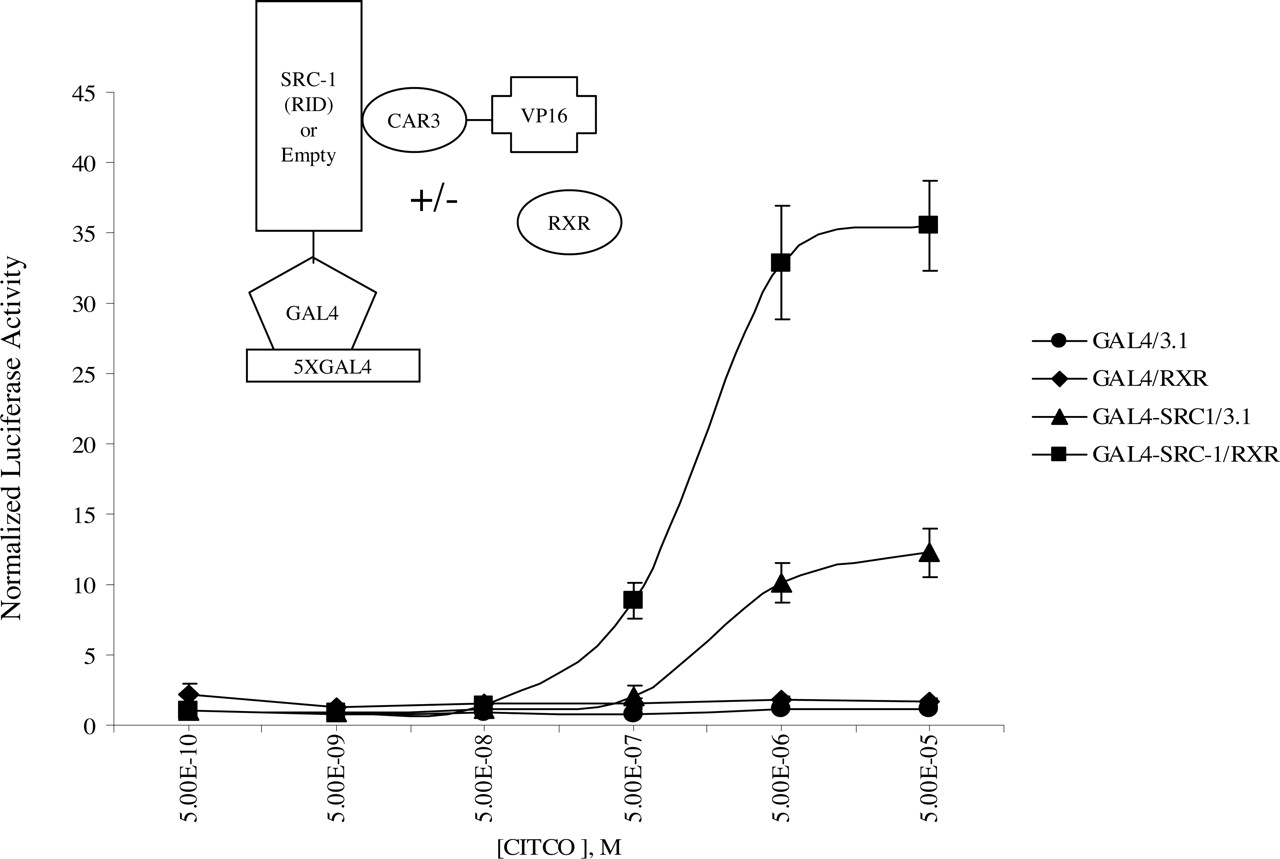
RXR facilitates ligand-dependent interaction between CAR and SRC-1 in mammalian two-hybrid analysis. COS-1 cells were transfected overnight as described under Materials and Methods and as illustrated in the figure. On day 2 (18 h after transfection), cells were treated with increasing amounts of CITCO. Twenty-four hours after treatment, the cells were harvested and assayed for firefly and Renilla reniformis luciferase. Data are presented as normalized and adjusted values in which the activity of the 500 pM CITCO-treated GAL4/3.1 (empty/empty) data point was set to 1. Each data point represents the mean ± S.E. of four separate transfections.

Clotrimazole is a ligand activator of CAR3. Transfections were performed in COS-1 cells as described under Materials and Methods and as illustrated in the figure. Chemical treatments were performed 18 h after transfection, and cells were harvested and assayed 24 h after chemical treatment. 3.1, empty expression vector. Each data point represents the mean ± S.E. of four separate transfections. A, clotrimazole treatment produces a dose- and CAR3-dependent increase in DR-4 × 3 reporter activity. The 1 nM clotrimazole/CMV2/3.1 data point was set to 1, and all data points were adjusted accordingly. B, a mammalian two-hybrid assay demonstrates a clotrimazole-dependent interaction between CAR3 and the SRC-1 RID that is enhanced by the cotransfection of RXR. The 1 nM clotrimazole/GAL4/3.1 data point was set to 1 and all other data points were adjusted accordingly. C, both CITCO and clotrimazole enhance the transactivation by GAL4-CAR3-ligand binding domain. The DMSO/GAL4/3.1 data point was set to 1 and all other data points were adjusted accordingly.
With the exception of a monomeric complex formed between purified GST-CAR3 and the CYP2B6 NR2 element, all efforts to detect a CAR3/DNA complex in vitro have been unsuccessful (Auerbach et al., 2003; Arnold et al., 2004). Therefore, the precise mechanisms that account for the marked CAR3 activation effects in cotransfection assay systems remain to be clarified. From an intuitive standpoint, the evidence presented in this study suggests a direct interaction between the DNA binding domain of CAR3 with selective DNA elements residing in the reporter constructs. Deletion of the CAR3 DNA binding domain disrupts its ability to transactivate a DR-4 × 3 reporter, and CAR3 itself does not transactivate an empty TK-luciferase reporter. These results support the proposal that, when expressed in the cell, CAR3 interacts with nuclear receptor half-sites via its DNA binding domain. EMSA data presented in an earlier manuscript (Auerbach et al., 2003) indicated that a monomeric complex may be the primary means of CAR3 binding to DNA. At first glance, this interpretation seems reasonable, especially in light of another report showing that CAR1 can bind certain DNA sequences in monomeric fashion (Frank et al., 2003). However, the results presented within the current study suggest a more complex scenario. If CAR3 interacts with DNA as a monomer, then spacing of the nuclear receptor half-sites in the direct repeat reporters should not affect the capability of CAR3 to transactivate. In fact, we observe the opposite; CAR3 strikingly transactivates the DR-4 reporter with only limited activation of other DR reporters. We designed the DR-4 × 3 reporter to specifically promote interaction with CAR monomers by including three nuclear receptor half-sites, each separated by 12 base pairs, with the dinucleotide AG placed 5′ in each half-site to facilitate CAR monomer binding (Frank et al., 2003). Although CAR1 produced a weak transactivation of this reporter in the absence of cotransfected RXR, CAR3 did not. Even with the addition of RXR to the transfection mix, CAR3 still only weakly activated the reporter (S. Auerbach and C. Omiecinski, unpublished results). These data strongly suggest that CAR3 is interacting with DNA as a heterodimeric complex, and considering the predicted disruption of the dimerization interface produced by the CAR3 5 amino acid insertion within a conserved region of this domain, formation of a typical stable dimeric complex between CAR3 and RXR in solution seems unlikely. This view is further supported by the results obtained from our mammalian two-hybrid experiments. One may believe that dimerization might be possible via regions of the dimer interface that remain intact, or through the DNA binding domain (Khorasanizadeh and Rastinejad, 2001); however, the latter scenario is not supported by our results showing the expendable nature of the RXR DNA binding domain in enhancing the activity of CAR3. Likewise, if CAR and RXR were interacting through their respective AF-2 motifs, as was proposed for RXR tetramers (Egea et al., 2001), then AF-2-mediated recruitment of coactivators would probably be adversely affected.

Clotrimazole and CITCO produce CAR3-dependent activation of the DR-4 × 3 reporter in HEK293 cells. Transfections were performed as described in Fig. 1, A and B, with plasmids indicated in the figure. On day 1, HEK293 cells were transfected. Eighteen hours after transfection, cells were treated with indicated chemicals (CLOT, clotrimazole). Twenty-four hours after chemical treatment, cells were harvested and luciferase activity was measured as described under Materials and Methods. CMV2 and 3.1 represent empty expression vectors. Data are presented as normalized and adjusted values in which the activity of the DMSO-treated CMV2/RXR data point was set to 1. Each data point represents the mean ± S.E. of four separate transfections.
We further considered whether detection of dimer formation might be dependent on the presence of additional factors that stabilize the complex; for example, in EMSA studies, RXR homodimers do not form stable complexes on DR-1 peroxisome proliferator-responsive elements in the absence of coactivator and ligand (IJpenberg et al., 2004). However, our EMSA experiments were performed with standard nuclear extract preparations, methods that others have used successfully to demonstrate cofactor interactions, such as with SRC-1. We also performed these assays in the presence and absence of the CITCO ligand. We further performed two-hybrid experiments using the combination of GAL4-RXR, VP16-CAR3, and VP16-SRC1/RID, anticipating that SRC-1 would stabilize the CAR/RXR complex; however, no interaction between CAR3 and RXR was observed (S. Auerbach and C. Omiecinski, unpublished results). To better account for our results, we note that standard EMSA assays may not accurately reproduce the chromatin context in which protein/DNA interactions take place in vivo, a context that can be an important determinant of bone fide nuclear receptor-DNA complexation (Urnov and Wolffe, 2001). We suggest that chromatin interactions ultimately determine the nature of CAR3-DNA interactions. A compelling observation that addresses the issue of CAR3-DNA interaction relates to our demonstration of the highly selective and potent transactivation of a DR4 × 3 reporter by CAR3. This result supports a dynamic model of CAR3-DNA interaction, involving transient formation of CAR3/RXR dimers, with the DNA interaction of the complex mediated principally through the DNA binding domain of CAR3. We speculate that DNA binding provides for a limited enhancement of heterodimeric complex stability. This model may account for the necessity of RXR overexpression for CAR3 activities because high levels of RXR would be expected to drive the formation of RXR heterodimers, especially in the presence of high levels of transfected CAR3.

CAR3 activation of CYP2B6 and CYP3A4 reporters is RXR and ligand-dependent. A and B, COS-1 cells were transfected with reporter and expression plasmids as indicated in the figure and as described under Materials and Methods. Eighteen hours after transfection, cells were treated with indicated chemicals (Clot, clotrimazole). Twenty-four hours after chemical treatment, cells were harvested and luciferase activity was measured as described under Materials and Methods. CMV2 and 3.1 represent empty expression vectors. Data are presented as normalized and adjusted values in which the activity of the DMSO treated CMV2/3.1 experiment was set to 1. Each data point represents the mean ± S.E. of four separate transfections. CAR1 transactivation of the CYP2B6 (A) and CYP3A4 (B) reporters was enhanced by RXR.
Our observations are also consistent with a second mechanism whereby RXR might affect CAR3 activity, a mechanism involving the facilitation of ligand-dependent coactivator recruitment to CAR3. In our 2-hybrid experiments, RXR augmented the ligand dependent interaction of CAR3 with SRC-1. Other researchers have demonstrated that the interaction of CAR with SRC-1 was enhanced by RXR (Dussault et al., 2002). In these studies, it was assumed that the enhancement of coactivator recruitment was dependent on CAR-RXR heterodimerization and that the associated allosteric effect of RXR was independent of RXR interaction with coactivator (Dussault et al., 2002). Here, we demonstrate that the AF-2 domain of RXR is necessary for enhancing CAR3 activity, indicating that coactivator interaction with RXR is a critical component of the CAR3 activation complex. We hypothesize that RXR competes for a ligand-oblivious corepressor that mimics a coactivator, such as hairless (Moraitis et al., 2002) or RIP140 (Treuter et al., 1998), thereby preventing competition with coactivators at the coregulator binding cleft of CAR3 (Steinmetz et al., 2001). Such a model would seem to reconcile the noted CAR3/RXR dimerization deficiencies. Furthermore, squelching of a ligand-oblivious corepressor may explain the consistently lower activity of CAR3 in the absence of RXR, even at saturating ligand levels. Although additional studies are necessary to evaluate these potential mechanisms, our results clearly show that RXR overexpression greatly enhances SRC-1 interaction with CAR3.
The activation of the CYP2B6 and CYP3A4 gene reporters by CAR3 lends additional credibility to the proposal that this receptor variant is likely to be a bona fide regulator of these genes in vivo. Our results demonstrated a relatively weak, ∼12-fold activation of the CYP2B6 reporter by CAR1 in the absence of RXR, whereas in its presence, an activation of nearly 60-fold was observed. In the context of a human hepatocyte, phenobarbital induction responses for the CYP2B6 gene range between 30- and 70-fold (Faucette et al., 2004), an induction level that otherwise parallels that obtained in our RXR cotransfection experiments. Furthermore, in human hepatocytes, 100 nM CITCO is sufficient to produce a robust induction of CAR target genes (Maglich et al., 2003). In the presence of cotransfected RXR, 50 to 500 nM CITCO markedly activates our CAR3 reporter, supporting the idea that CAR3 may at least partially mediate the effect of CITCO on gene transcription. Faucette et al. (2004) noted an induction of CYP2B6 expression by clotrimazole between 1 and 10 μM, approximating the dose response data exhibited in our CAR3 studies using a DR4 × 3 element as well as the dose range observed for the CAR3-dependent transactivation of the CYP2B6 reporter by clotrimazole. Additional analyses probing the in vivo relevance of CAR3 expression may require extended studies of humanized CAR3 mice; such studies are under way in our laboratory.
Acknowledgments
We thank Denise Weyant for expert technical assistance and Jack Vanden Heuvel and Eric Tien for gifts of plasmids and suggestions regarding the mammalian two-hybrid assays.
Footnotes
-
Supported by United States Public Health Service grants GM066411 from the National Institute of General Medical Sciences (to C.J.O.) and institutional training grant ES077032 from the National Institute of Environmental Health Sciences (to S.S.A.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.013417.
-
ABBREVIATIONS: CAR, constitutive androstane receptor (CAR1, MB67, NR1I3); hCAR, human constitutive androstane receptor; DR, direct repeat; RXR, retinoid X receptor α; CITCO, 6-(4-chlorophenyl)imidazo[2,1-b]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; EMSA, electrophoretic mobility shift assay; CMV, cytomegalovirus; VP16, virus protein 16; BAP, bacterial alkaline phosphatase; LBD, ligand binding domain; RID, receptor interaction domain; XREM, xenobiotic-responsive enhancer module; TK, thymidine kinase; DMSO, dimethyl sulfoxide; SRC-1, steroid receptor coactivator-1; AF, activation function.
- Received April 11, 2005.
- Accepted August 10, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}