


Abstract
Because our previous studies indicated that squalestatin 1 treatment induces CYP2B expression in primary cultures of rat hepatocytes as a direct consequence of squalene synthase inhibition, we investigated possible underlying mechanisms. Cotransfection of cultured Sprague-Dawley male rat hepatocytes with each of the three sterol regulatory element binding protein (SREBP) transcription factors failed to induce luciferase expression from a squalestatin 1-responsive CYP2B1 reporter plasmid. Squalestatin 1 treatment of primary hepatocyte cultures from male Wistar-Kyoto rats produced a greater induction of CYP2B mRNA than occurred in cultures from female rats, consistent with the previously demonstrated response dimorphism that has been attributed to differences in constitutive androstane receptor (CAR) levels. Cotransfection of female Wistar-Kyoto rat hepatocyte cultures with plasmid expressing either mouse or rat CAR restored squalestatin 1-inducible CYP2B1-reporter expression. Cotransfection of Sprague-Dawley rat hepatocyte cultures with plasmid expressing rat CAR lacking the C-terminal AF-2 subdomain inhibited squalestatin 1-inducible CYP2B1-reporter expression. Squalestatin 1-mediated CYP2B mRNA induction in rat hepatocyte cultures was completely abolished by pretreatment with the 3-hydroxymethyl-3-glutaryl CoA reductase inhibitor pravastatin and was rescued by mevalonate supplementation, whereas phenobarbital-mediated induction was unaffected by these treatments. Finally, direct addition oftrans,trans-farnesol to the culture medium caused the rapid induction of CYP2B mRNA. These results indicate that squalestatin 1 treatment induces CYP2B expression, not by inhibiting sterol synthesis and activating SREBPs, but by evoking the accumulation of an endogenous isoprenoid and activating CAR.
More than 30 years after the phenomenon known as cytochrome P450 induction was first described (Conney, 1967), the mechanisms governing the inducible expression of many members of this superfamily are becoming clarified. A major catalyst for progress in this field has been the discovery that several “orphan” members of the nuclear receptor superfamily are “xenobiotic receptors”. In particular, the CAR, PXR, and PPARα receptors are now known to mediate the effects of myriad xenobiotics on hepatic CYP2B, CYP3A, and CYP4A expression, respectively (Lee et al., 1995; Lehmann et al., 1998; Wei et al., 2000). Besides serving as xenobiotic receptors, each of these nuclear receptors is regulated by at least one class of endogenous molecule. Thus, certain bile acids activate PXR (Staudinger et al., 2001), whereas fatty acids activate the PPARs (Kliewer et al., 1997). By contrast, certain androstanes are “inverse agonists” of CAR, in that they suppress the receptor's constitutive activity (Forman et al., 1998).
It is now recognized that the hepatocyte contains multiple systems for maintaining lipid homeostasis. Prominent among these, the SREBP class of transcription factors is activated after cholesterol deprivation and up-regulates the expression of a large number of genes controlling both sterol and fatty acid metabolism (Edwards et al., 2000). More recently, the oxysterol receptors LXRα and -β have been identified as key regulators of cholesterol metabolism (Lu et al., 2001). Also, as mentioned above, PPARα is a fatty acid receptor whose activation causes peroxisome proliferation, which includes the induction of numerous lipid-metabolizing enzymes. Not surprisingly, there is substantial interplay among these pathways. For example, SREBP1c expression is dependent on the ongoing activation of LXR by oxysterols (DeBose-Boyd et al., 2001).
The fact that the known endogenous regulators of the CAR, PXR, and PPARα receptors are all lipoidal in nature suggests that there may be mechanistic cross-links between the systems responsible for the metabolism of endogenous and exogenous lipophilic compounds. In this regard, we demonstrated previously that several drugs that inhibit cholesterol biosynthesis affect hepatic cytochrome P450 expression. Thus, certain inhibitors of HMG-CoA reductase, including lovastatin, simvastatin, and fluvastatin, induce CYP2B, CYP3A, and CYP4A in primary cultured rat hepatocytes (Kocarek and Reddy, 1996). By contrast, pravastatin treatment has no effect on the expression of any of these P450s (Kocarek and Reddy, 1996), suggesting that HMG-CoA reductase inhibition is not mechanistically responsible for the effects of these agents on P450 expression. However, further studies using another class of agents, exemplified by the endogenous fungal metabolite squalestatin 1 (also known as zaragozic acid A) and the synthetic bisphosphonate SQ-34919, suggested that inhibition of squalene synthase, the first committed step in sterol biosynthesis, is a mechanistic trigger of CYP2B induction in primary cultured rat hepatocytes (Kocarek et al., 1998). Evidence included the findings that 1) squalestatin 1 treatment of primary cultured rat hepatocytes induced CYP2B expression with very high potency (EC50 ∼ 5 nM), 2) squalene synthase inhibitor treatments induced CYP2B and HMG-CoA reductase mRNA levels with comparable concentration-response profiles, and 3) cotreatment with 25-hydroxycholesterol, a model oxysterol that potently suppresses SREBP-activated gene expression, readily reversed squalestatin 1-mediated CYP2B induction (Kocarek et al., 1998). Notably, by contrast to the other “phenobarbital-like” agents that invariably induce both CYP2B and CYP3A gene products, squalene synthase inhibitor treatments were relatively specific inducers of CYP2B in the rat hepatocyte cultures and in rat liver in vivo; CYP4A, not CYP3A, was the only other P450 that was induced, and this only slightly (Kocarek et al., 1998). Using a chicken hepatoma system, Ourlin et al. (2002)recently reported findings that were generally consistent with ours. Thus, squalestatin 1 treatment of LMH cells induced CYP2H1, which was reversed by 25-hydroxycholesterol cotreatment. However, squalestatin 1 treatment also induced CYP3A37, suggesting that there may be some mechanistic differences in regulation of the rodent and avian P450s.
In this study, we have further investigated the mechanism of CYP2B induction by squalestatin 1 in primary cultured rat hepatocytes. In particular, we first considered the possibility that squalestatin 1 treatment induces CYP2B expression through the activation of an SREBP and found this not to be substantiated. We next established a central role for CAR in squalestatin 1-inducible CYP2B expression; furthermore, we provided evidence that squalestatin 1 does not directly activate CAR but induces CYP2B through a mechanism that requires the ongoing biosynthesis of an endogenous isoprenoid.
Materials and Methods
Materials.
Squalestatin 1, fluvastatin, and ciprofibrate were gifts from GlaxoSmithKline (Research Triangle Park, NC), Novartis (Summit, NJ), and Sterling Winthrop (Rensselaer, NY), respectively. Pravastatin and SQ-34919 were gifts from Bristol-Myers Squibb (Wallingford, CT). Phenobarbital, dexamethasone, pregnenolone 16α-carbonitrile, mevalonate, trans,trans-farnesol and 22(R)-hydroxycholesterol were purchased from Sigma Chemical Co. (St. Louis, MO). Chenodeoxycholic acid was purchased from Steraloids (Newport, RI). Collagenase (type IV) was purchased from Worthington Biochemical Corp. (Freehold, NJ). Matrigel was purchased from Collaborative Biomedical Products (Bedford, MA). Vitrogen was purchased from The Collagen Corporation (Palo Alto, CA). Recombinant human insulin (Novolin R) was purchased from Novo Nordisk Pharmaceuticals, Inc. (Princeton, NJ). Other cell culture supplies and Lipofectin and TRIzol reagent were purchased from Invitrogen (Carlsbad, CA). Oligonucleotides were purchased from Sigma-Genosys (The Woodlands, TX).
Primary Culture of Rat Hepatocytes.
Hepatocytes were isolated from the livers of adult male Sprague-Dawley rats (250–350g; Harlan Sprague-Dawley, Indianapolis, IN), or from adult male or female Wistar-Kyoto rats (200–300g; Harlan Sprague-Dawley), as described previously (Kocarek and Reddy, 1996). After isolation, 3 million viable hepatocytes were plated onto 60 mm Matrigel-coated dishes (for Northern blot experiments) or 300,000 viable hepatocytes were plated onto Vitrogen-coated wells in 12-well plates (for transient transfection experiments) and maintained in Williams' Medium E supplemented with 0.25 U/ml insulin, 0.1 μM triamcinolone acetonide, 100 U/ml penicillin, and 100 μg/ml streptomycin. Culture medium was renewed every 24 h. For treatments, drugs were added to the culture medium as concentrated stock solutions in water (squalestatin 1, SQ-34919, phenobarbital, fluvastatin, pravastatin, or mevalonate), DMSO (dexamethasone, pregnenolone 16α-carbonitrile, or ciprofibrate), or ethanol [chenodeoxycholic acid, 22(R)-hydroxycholesterol ortrans,trans-farnesol]. When added, the final concentration of organic solvent in the culture medium was 0.1%.
Northern Blot Analysis.
Beginning 48 h after plating, cultures were treated with drugs (three dishes per treatment group), as described in the individual figure legends. After treatment, the three dishes of hepatocytes representing each treatment group were pooled for the preparation of total RNA, as described previously (Kocarek and Reddy, 1996). Samples of the pooled RNAs (10 μg) were resolved on denaturing agarose gels and analyzed by Northern blot hybridization, using the cDNA probes to CYP2B1, CYP3A23, CYP4A1, HMG-CoA reductase, and 7S that have been described previously (Kocarek and Reddy, 1996). After hybridization with the P450 or HMG-CoA reductase cDNA probes, hybridizable bands were identified by autoradiography. Radiolabeled probes were then removed from the filters by incubation in 1% SDS at 90°C, and blots were re-hybridized with 7S cDNA to control for RNA loading and transfer. For quantification, band intensities were estimated using a scanning laser densitometer (Molecular Dynamics, Sunnyvale, CA) equipped with ImageQuant software.
Transient Transfection of Primary Cultured Rat Hepatocytes.
Primary cultures of rat hepatocytes were transiently transfected with reporter and expression constructs, essentially as described previously (Kocarek et al., 1998). The following luciferase reporter plasmids were used in these studies. Plasmid pGL3-Basic containing 2413 nucleotides of the CYP2B1 5′-flanking region was described previously (Kocarek et al., 1998). A corresponding plasmid containing only 2151 nucleotides of the CYP2B1 5′-flanking region was prepared by PCR. Plasmid pGL2-Basic containing nucleotides −324 to −225 of the HMG-CoA synthase promoter fused to the HMG-CoA synthase TATA box (pSYNSRE-Luc) (Jackson et al., 1995) was a gift from Dr. Peter Edwards (UCLA, Los Angeles, CA). Plasmid pCYP3A4-XREM-Luc, containing proximal and distal enhancer elements of the CYP3A4 5′-flanking region (Goodwin et al., 1999), was a gift from Dr. Bryan Goodwin (GlaxoSmithKline Research and Development, Research Triangle Park, NC). Plasmids containing three concatamerized copies of the CYP3A23-DR3 motif [top strand sequence, 5′-GTAGATGAACTTCATGAACTGTCTA-3′; bases corresponding to the hexameric-repeat nuclear receptor motif are underscored (Quattrochi et al., 1995)], six copies of a consensus FXR-responsive IR1 motif [top strand sequence, 5′-ACAAGAGGTCATTGACCTTGTCC -3′ (Laffitte et al., 2000)], or four copies of an LXR-responsive DR4 motif [top strand sequence, 5′-TGCTTAGTTCACTCAAGTTCAAGTTA-3′ (Lehmann et al., 1997)] were prepared by ligating the double-stranded oligonucleotides upstream of a minimal herpes simplex virus thymidine kinase promoter, which had been preligated into pGL3-Basic (Promega Corporation, Madison, WI). A fragment containing the peroxisome proliferator responsive region of CYP4A1 (Aldridge et al., 1995) was prepared by PCR, using rat genomic DNA as template, Pfupolymerase (Stratagene, LaJolla, CA) and primers corresponding to nucleotides 154 to 170 (5′-GGAGATCTTGGGTAAAGAGGGAAAAT-3′) and 290 to 274 (5′-GGAGATCTTAGAGCAAAGGGGAATG-3′) of a published sequence (GI 758269); the underscored nucleotides of the primers indicate BglII sites. After amplification for 35 cycles, the product was digested with BglII and ligated into pGL3-Promoter (Promega, Madison, WI), which drives luciferase expression through the simian virus 40 promoter. The following expression plasmids were used in these studies. Plasmids containing full-length human SREBP1a and SREBP2 were purchased from the American Type Culture Collection (Manassas, VA). cDNAs encoding the first 480 amino acids of SREBP1a (Shimano et al., 1996) and the first 481 amino acids of SREBP2 (Magana and Osborne, 1996) were prepared essentially as described previously (Shimano et al., 1996), and ligated into pcDNA3.1 (Invitrogen). A plasmid expressing the first 436 amino acids of human SREBP1c was purchased from American Type Culture Collection. Plasmid pCR3 expressing mouse CAR (Sueyoshi et al., 1999) was a gift from Dr. Masahiko Negishi (National Institute of Environmental Health Sciences, Research Triangle Park, NC). A cDNA for rat CAR was prepared by PCR, using reverse-transcribed rat liver RNA as template, TaqPlusPrecision Polymerase (Stratagene, LA Jolla, CA) and primers corresponding to nucleotides 1 to 29 (GI 12621107) and 1077 to 1058. The sequence of the forward primer was 5′-GGGGATCC GAGACCATGACAGCTACTCTAACACTAGAGACCAT-3′ and the sequence of the reverse primer was 5′-GGCTCGAGTCAGCTGCAAATCTCCCCAA-3′. The underscored nucleotides of the primers indicate BamHI andXhoI restriction sites; the bold nucleotides of the forward primer represent a short section of the mouse CAR 5′-untranslated region, to ensure efficient translation of the expressed mRNA. After amplification for 35 cycles, the product was digested withBamHI and XhoI, and ligated into pcDNA3.1. To prepare a cDNA encoding a putative dominant negative rat CAR receptor, PCR was used to prepare a CAR fragment containing all but the eight C-terminal amino acids of the receptor, corresponding to the deletion described previously by Choi et al. (1997) for mouse CAR. PCR was performed for 20 cycles, using 100 ng of the rat CAR plasmid as template and Pfu polymerase. The forward primer was that described above; the reverse primer, spanning nucleotides 1051 to 1031, was 5′-GGCTCGAG TCATCACATCATAGCAGACAGTCCCT-3′ (the underscored and bold nucleotides represent a XhoI site and tandem translation stop codons, respectively). The PCR fragment was again digested with BamHI and XhoI and ligated into pcDNA3.1. Identities of all cloned fragments were verified by the Center for Molecular Medicine and Genetics DNA Sequencing Facility at Wayne State University.
For each transfection, on the day after plating, hepatocytes were incubated with 0.6 ml of OptiMEM (Invitrogen) containing a premixed complex of 6.25 μg of Lipofectin reagent (Invitrogen) and plasmid mixture consisting of 800 ng of one of the reporters, the appropriate amount of one of the expression plasmids (i.e., 0.5 or 1.0 ng of SREBPs, 100 ng of wild type rat or mouse CAR or AF-2–deleted rat CAR, or pcDNA3.1 as empty vector control), 0.5 ng pRL-CMV (to normalize for transfection efficiency among samples), and sufficient pBlueScript II KS+ to balance total amounts of DNA to 1 μg. After transfection and overlay with 160 μg of Matrigel (Kocarek et al., 1998), transfectants (three wells per treatment group) were incubated as described in the individual figure legends and then harvested for the measurement of luciferase activities (firefly andRenilla reniformis) using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer's instructions and a Dynex model MLX luminometer. Transient transfection data were analyzed by one-way ANOVA followed by the Newman-Keuls multiple comparison test (Prism; GraphPad Software, San Diego, CA).
Results
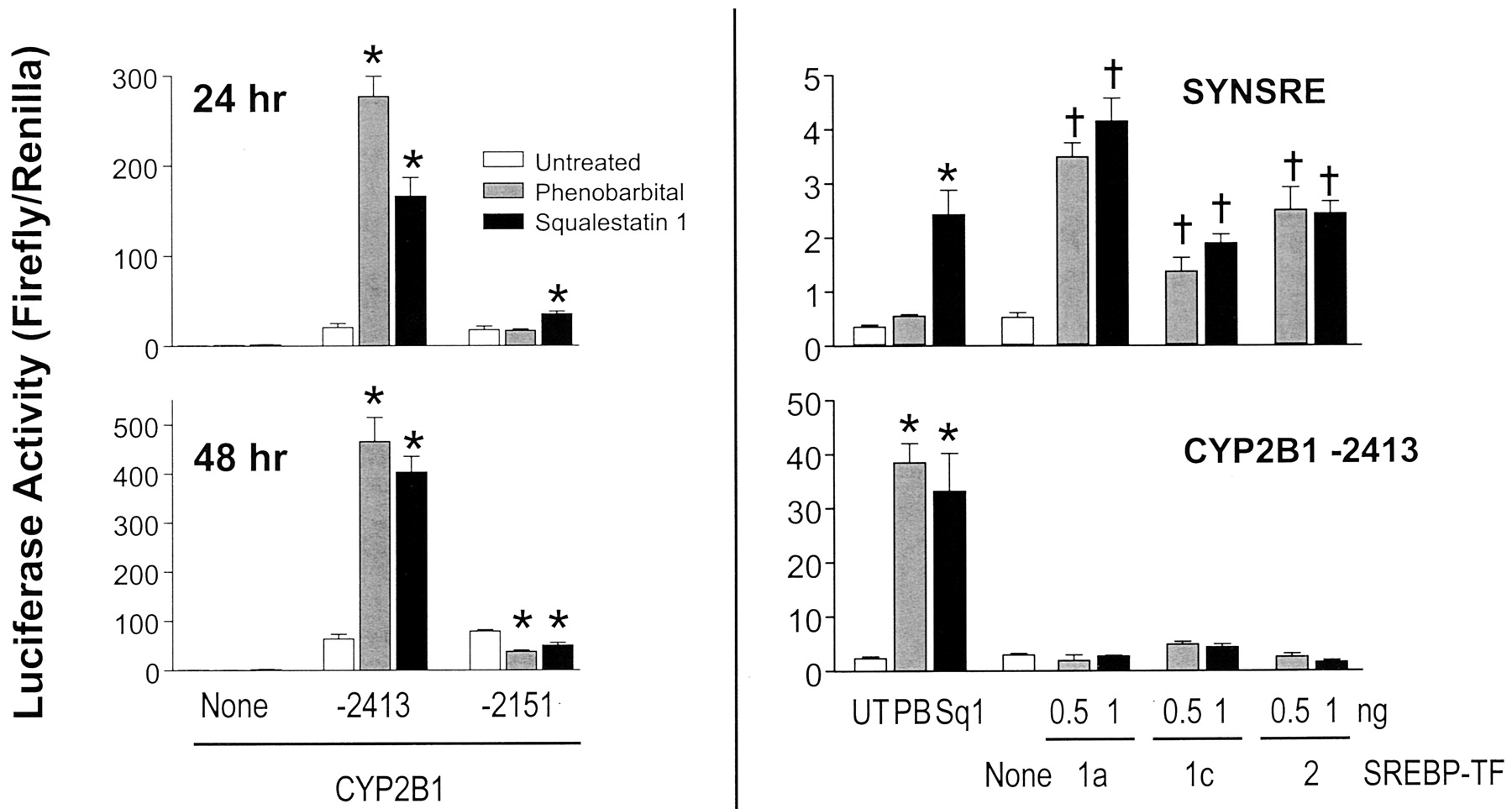
We reported previously that squalestatin 1 treatment potently induces CYP2B mRNA and immunoreactive protein in primary cultured rat hepatocytes and in rat liver and provided several lines of evidence that this effect was likely to be a consequence of squalene synthase inhibition (Kocarek et al., 1998). We also demonstrated that squalestatin 1 treatment is able to activate CYP2B1 transcription, as demonstrated by its ability to increase reporter gene expression from a plasmid containing 2413 nucleotides of the CYP2B1 5′-flanking region, with the previously identified PBRU region (Trottier et al., 1995). Squalestatin 1 treatment also induced transcription from a reporter plasmid containing the CYP2B1 PBRU region alone, and driving transcription through a heterologous (simian virus 40) promoter (Kocarek et al., 1998). To determine whether the squalestatin 1-sensitive cis-elements were confined solely to the PBRU region, we prepared a reporter plasmid containing 2151 nucleotides of CYP2B1 5′-flanking region, beginning immediately downstream of the PBRU region, and tested its responsiveness to squalestatin 1 treatment in transient transfection experiments (Fig.1, left). Like PB, squalestatin 1 treatment significantly induced reporter gene activity in hepatocytes that were transfected with the −2415 construct containing the PBRU but not with the −2151 construct lacking the PBRU. The results were qualitatively the same whether the hepatocytes were treated with drug for 24 or 48 h; quantitatively, however, the effect of squalestatin 1 relative to that of phenobarbital seemed to be greater after 48 h of treatment (Fig. 1, left). Thus, these results indicate that the squalestatin 1-sensitive region of CYP2B1 is confined to the PBRU, as it is for phenobarbital.

Effects of squalestatin 1 treatments or SREBP transcription factor cotransfections on reporter gene activity in primary cultured rat hepatocytes transiently transfected with a CYP2B1 or HMG-CoA synthase promoter construct. Freshly isolated rat hepatocytes were placed into primary culture and transiently transfected with a promoter-luciferase construct containing either 2151 (lacking the PBRU) or 2413 nucleotides (containing the PBRU) of CYP2B1 5′-flanking sequence or nucleotides −324 to −225 of the HMG-CoA synthase promoter (SYNSRE), either in the absence or presence of 0.5 or 1.0 ng of plasmid expressing the active transcription factor (TF) of SREBP-1a, SREBP-1c or SREBP-2, as described under Materials and Methods (Bottom right, None, control group transfected with 1.0 ng of pcDNA3.1 plasmid lacking insert). Forty-eight hours after plating, indicated cultures were incubated in medium alone (UT) or containing either 10−4 M phenobarbital (PB) or 10−7 M squalestatin 1 (Squal 1). Ninety-six hours after plating (72 h after plating for the top left), cultures were harvested for the measurement of luciferase activities, as described underMaterials and Methods. Each bar represents the mean ± S.D. of normalized (firefly/Renilla) luciferase measurements (three wells per treatment group). *,p < 0.05, significantly different from untreated control group; †, p < 0.05, significantly different from group transfected with pcDNA3.1 plasmid.
Most known effects of drugs that inhibit cholesterol biosynthesis on gene expression are mediated through activation of the SREBP class of transcription factors, which is composed of three gene products, SREBP1a, SREBP1c, and SREBP2, that are derived from two genes (Edwards et al., 2000). These factors exist in sterol-replete cells as inactive precursors associated with the endoplasmic reticulum. By this mechanism, inhibition of sterol biosynthesis results in the reduction of membrane cholesterol levels. Sensing this, the inactive SREBPs are trafficked to the Golgi apparatus, where first site 1 protease and then site 2 protease cleave the SREBPs, liberating the amino-terminal ∼500 amino acids of the factors from the membrane. The mature factors then translocate to the nucleus, where they activate the transcription of responsive genes through sterol response elements. Thus, a convenient way to test whether the expression of a particular gene is under the regulatory control of an SREBP is to transfect the mature factor into a cell. Although a computer-based search [MatInspector V2.2 (Quandt et al., 1995)] indicated that the PBRU-containing region of CYP2B1 does not contain a consensus sterol response element, several sequences with core and matrix similarities of at least 0.75 and 0.6, respectively, to SREBP-binding elements (TRANSFAC 4.0 matrices V$SREBP1_01 and V$SREBP1_02) were identified. We therefore wished to test directly the possibility that the PBRU may nevertheless be activated by an SREBP, because it contains either a nonconsensus, but functional, sterol response element or an element that is indirectly activated through an SREBP. As an example of the latter type of mechanism, Kim et al. (1998) previously reported that expression of SREBP1c in 3T3-L1 preadipocytes resulted in the production of an endogenous ligand for PPARγ. We therefore examined the effects of cotransfecting the mature form of SREBP1a, SREBP1c, or SREBP2 on reporter gene activity in hepatocyte cultures transfected with the squalestatin 1-responsive CYP2B1 reporter (i.e., containing 2413 nucleotides of 5′-flanking region) or, as a positive control, the SREBP-responsive region of the HMG-CoA synthase gene (Fig. 1, right). We tested each of the known SREBPs, because substantial evidence indicates that there are differences among them in terms of expression and biological activities. Thus, although SREBP1a is a stronger transcriptional activator than SREBP1c, SREBP1c and SREBP2 are the predominant SREBPs expressed in normal liver. Also, whereas the SREBP1 forms have been most closely associated with the regulation of fatty acid biosynthesis and insulin-mediated actions, SREBP2 seems to be primarily responsible for regulation of the genes involved in maintaining cholesterol homeostasis (Edwards et al., 2000). In these experiments, we wished to transfect amounts of the mature SREBP factors that would be comparable with the amounts existing after endogenous activation. Thus, in preliminary experiments, we identified 0.5 and 1.0 ng as amounts of SREBPs that would produce approximately the same activation of the HMG-CoA synthase reporter plasmid as produced by squalestatin 1 treatment. When these amounts of the SREBPs were transiently transfected into the hepatocyte cultures, each significantly increased reporter gene expression from the HMG-CoA synthase plasmid, relative to the levels that were observed after transfection with empty plasmid. However, none of the SREBPs produced any increase in reporter gene activity in hepatocytes transfected with the CYP2B1 reporter plasmid, despite the fact that reporter expression was strongly increased by squalestatin 1 treatment in this experiment. These findings indicate that SREBP activation is not the mechanism by which squalestatin 1 treatment induces CYP2B1 transcription in primary cultured rat hepatocytes.
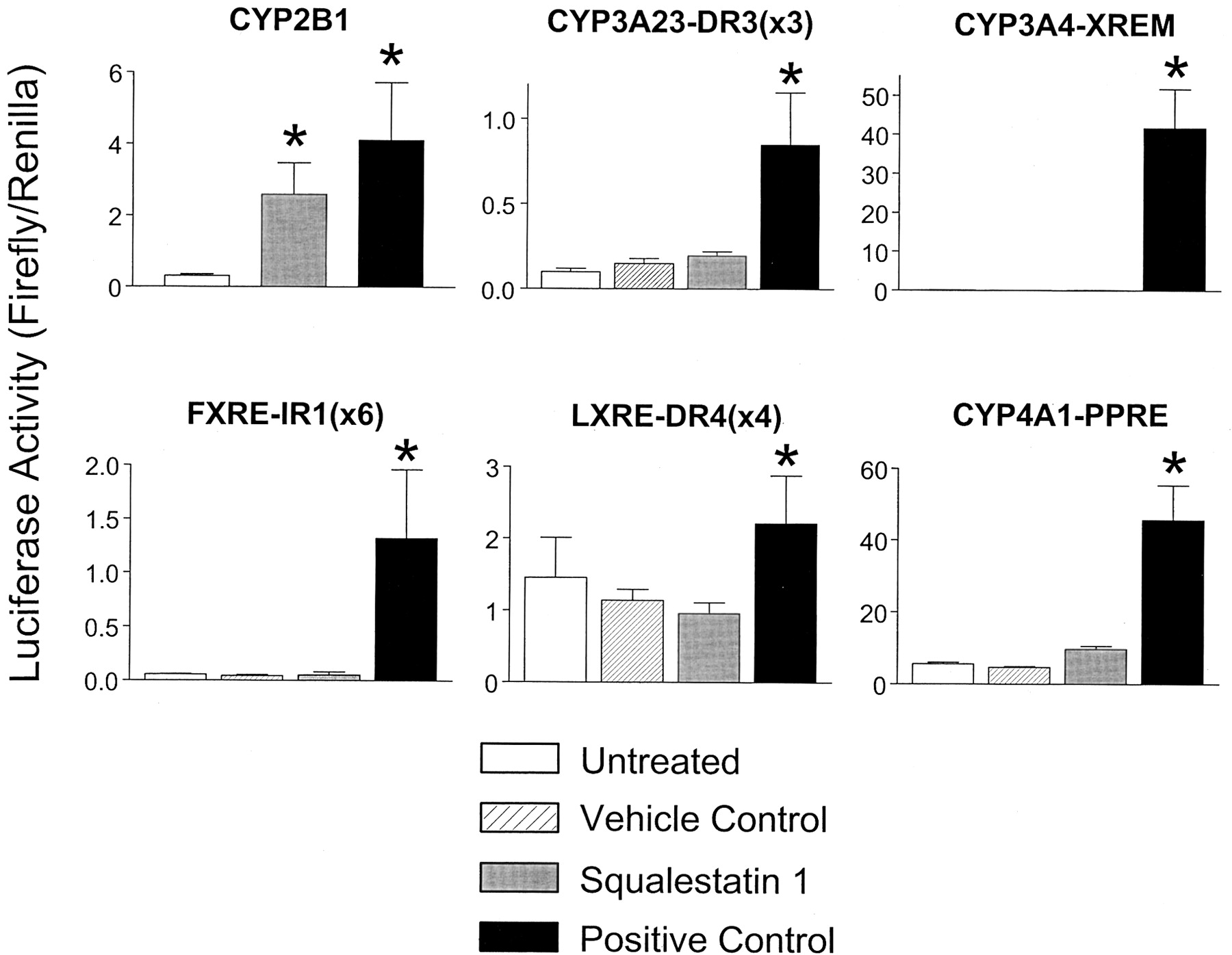
It is now well accepted that “orphan” members of the nuclear receptor superfamily mediate the effects of many endogenous and exogenous agents on gene transcription. In particular, CAR has been definitively shown to mediate the effects of phenobarbital and other phenobarbital-like inducers on CYP2B expression in mice (Wei et al., 2000). Our findings that squalestatin 1-inducible CYP2B1 transcription was restricted to the PBRU region and that the effect was not mediated through SREBP activation suggested that the effect of squalestatin 1 was also likely to be mediated through CAR. Indeed, when we examined the ability of squalestatin 1 treatment to regulate luciferase expression from a series of reporter plasmids containing motifs known to be responsive to nuclear receptors found in the hepatocyte (i.e., CAR, PXR, FXR, LXR, and PPARα), only the CAR-responsive CYP2B1 reporter construct was significantly activated (Fig.2). Consistent with our previous finding that squalestatin 1 treatment did not induce CYP3A expression in primary cultured rat hepatocytes (Kocarek et al., 1998), this drug also did not induce luciferase activity in hepatocytes treated with either of two PXR-responsive reporter plasmids. Indeed, squalestatin 1 was without effect on the CYP3A4-XREM reporter, containing proximal and distal enhancer elements of CYP3A4, and which we have routinely found to be induced more than 200-fold by pregnenolone 16α-carbonitrile treatment of the cultured rat hepatocytes.
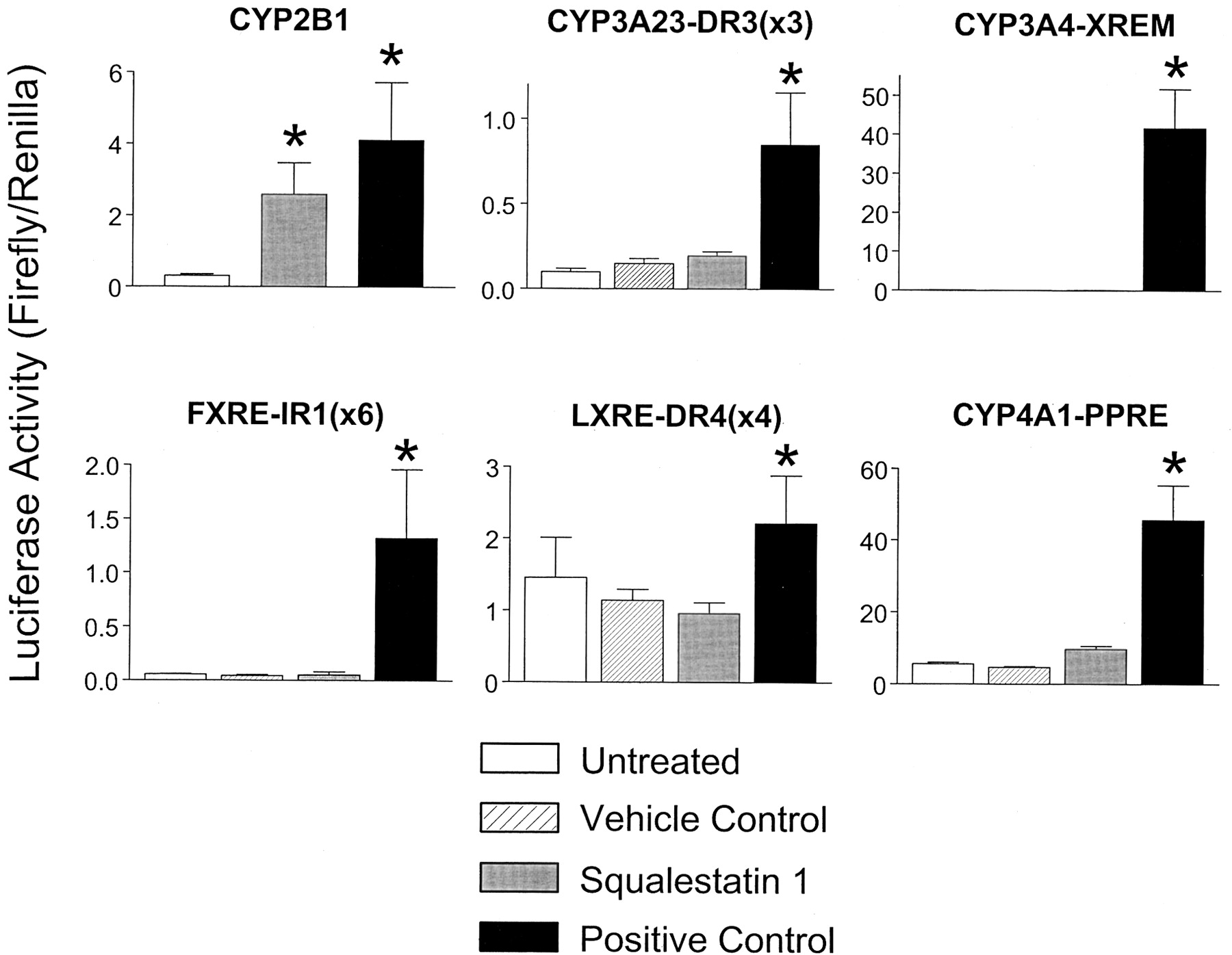
Effects of squalestatin 1 treatments on luciferase expression from nuclear receptor-responsive reporter plasmids. Primary rat hepatocyte cultures were transiently transfected, as described under Materials and Methods, with each of a series of a reporter plasmids activated by the following nuclear receptors: CAR (CYP2B1, containing 2413 nucleotides of the CYP2B1 5′-flanking region; 10−4 M phenobarbital positive control), PXR [CYP3A23-DR3 (×3), 10−5 M pregnenolone 16α-carbonitrile positive control, DMSO vehicle control], PXR (CYP3A4-XREM, 10−5 M pregnenolone 16α-carbonitrile positive control, DMSO vehicle control), FXR [FXRE-IR1 (×6), 3 × 10−5 M (2 doses) chenodeoxycholic acid positive control, ethanol vehicle control], LXR [LXRE-DR4 (×4), 3 × 10−5 M (two doses) 22(R)-hydroxycholesterol positive control, ethanol vehicle control] or PPARα (CYP4A1-PPRE, 10−4 M ciprofibrate positive control, DMSO vehicle control). After transfection, cultures were incubated for 24 h in medium alone (untreated) or containing 10−7 M squalestatin 1, the indicated positive control agent or vehicle control. Twenty-four hours after treatment (i.e., 72 h after plating), hepatocytes were harvested for the measurement of luciferase activities. Each bar represents the mean ± S.D. of normalized (firefly/Renilla) luciferase measurements (three wells per treatment group). *, p < 0.05, significantly different from the appropriate negative control group (i.e., untreated or vehicle-treated).
We next wished to determine specifically whether the effects of squalestatin 1 were mediated through CAR. To do this, we ideally would have tested the ability of this agent to induce CYP2B expression in the CAR-null mouse model (Wei et al., 2000). However, preliminary experiments to establish whether squalestatin 1-mediated CYP2B expression was conserved in primary cultured mouse hepatocytes indicated that treatments with squalestatin 1 produced little, if any, CYP2B induction. It was therefore necessary to employ a rat-based model in these studies. The Wistar-Kyoto strain of rat has been reported to exhibit a sexual dimorphism in hepatic CYP2B induction, whereby female rats show a much attenuated response, relative to male rats, to treatment with phenobarbital and other phenobarbital-like inducers (Larsen et al., 1994). Recently, this dimorphism has been attributed to differences in the hepatic levels of CAR protein, with male rats containing much greater amounts than female rats (Yoshinari et al., 2001). We therefore examined the ability of squalestatin 1 to induce CYP2B1 mRNA in primary cultured hepatocytes prepared from male and female Wistar-Kyoto rats (Fig. 3). As controls, we included phenobarbital as well as fluvastatin, an HMG-CoA reductase inhibitor that is also a highly efficacious CYP2B inducer in primary cultured rat hepatocytes (Kocarek and Reddy, 1996,1998). As occurred for phenobarbital, the CYP2B mRNA induction response was much greater (∼3-fold) in squalestatin 1-treated male hepatocytes than it was in female hepatocytes (Fig. 3, A and B), consistent with a CAR-mediated response. To demonstrate the specificity of this response, we examined dexamethasone-mediated CYP3A and ciprofibrate-mediated CYP4A mRNA induction in the hepatocyte cultures, which are mediated through PXR and PPARα, respectively. In each case, inducible expression was approximately the same in the male and female rat hepatocyte cultures (Fig. 3, A and B). To examine the role of CAR further, we next examined the ability of squalestatin 1 to activate luciferase expression in female Wistar-Kyoto hepatocytes that were transfected with the CYP2B1 reporter plasmid, in either the absence or the presence of cotransfected plasmid expressing mouse or rat CAR. In cultures not cotransfected with expression plasmid or cotransfected with pcDNA3.1 without insert, basal reporter gene expression was very low, and treatment with phenobarbital, fluvastatin, or squalestatin 1 produced modest, nonsignificant increases (Fig. 3C). In cultures cotransfected with either of the CAR expression plasmids, basal reporter activity was increased (>10-fold relative to cultures transfected with empty plasmid), and each of the drug treatments produced statistically significant enhancements (Fig. 3C), again consistent with the conclusion that squalestatin1-induced CYP2B expression is mediated through CAR.

Squalestatin 1-mediated CYP2B induction in primary cultures of male and female Wistar-Kyoto (WKY) hepatocytes. A, hepatocytes from the livers of adult male or female WKY rats were maintained in primary culture as described under Materials and Methods. Forty-eight hours after plating, cultures (three replicate dishes per treatment group) were incubated for 24 h with medium alone (UT) or containing 0.1% DMSO (vehicle control), 10−4 M phenobarbital (PB), 10−5 M dexamethasone (Dex, in DMSO), 10−4 M ciprofibrate (Cipro, in DMSO), 10−7 M squalestatin 1 (Squal1), or 3 × 10−5 M fluvastatin (Fluva). After treatment, hepatocytes from each treatment group were harvested for the preparation of total RNA, and pooled samples were analyzed for levels of CYP2B, CYP3A, and CYP4A mRNA by Northern blot hybridization. Autoradiographs are shown for one set of male and female hepatocyte cultures; comparable results were obtained in a second set of cultures. Also shown is a representative autoradiograph of a blot that was rehybridized with 7S cDNA, to demonstrate consistency of RNA loading and transfer among samples. B, graphical representations of the densitometrically quantified data from all hepatocyte culture experiments. To normalize these data across Northern blots, the CYP2B, CYP3A, and CYP4A band intensities that were measured for the PB-treated, Dex-treated, and Cipro-treated male hepatocytes were defined as 100%, respectively. Other treatment groups are shown as the mean ± range relative to those values. C, effects of cotransfected CAR plasmids on inducible reporter gene activity. Primary cultures of female WKY rat hepatocytes were transiently transfected with a reporter plasmid containing 2413 nucleotides of the CYP2B1 5′-flanking region, either alone (None) or in combination with 100 ng of pcDNA3.1 plasmid expressing either no insert (pcDNA), rat CAR (rCAR), or mouse CAR (mCAR). After transfection, cultures were incubated for 24 h in medium alone (UT) or containing 10−4 M PB, 3 × 10−5 M Fluva, or 10−7 M squalestatin 1. After treatment, hepatocytes were harvested for the measurement of luciferase activities. Each bar represents the mean ± S.D. of normalized luciferase (firefly/Renilla) measurements (three wells per treatment group). *, p < 0.05, significantly different from the corresponding UT group.

Effects of cotransfected rat CAR plasmids on inducible reporter gene activity in primary cultured rat hepatocytes. Primary cultured rat hepatocytes were transiently transfected with a reporter plasmid containing 2413 nucleotides of the CYP2B1 5′-flanking region (top) or proximal and distal enhancer elements of the CYP3A4 5′-flanking region (CYP3A4-XREM), either alone (UT or 0) or in combination with the pcDNA3.1 plasmid without insert (EV, empty vector control) or expressing rCAR (WT, wild-type) or dominant negative rCAR (DN). After transfection, cultures were incubated for 24 h in medium alone (UT) or containing 10−4 M phenobarbital (PB), 10−7 M squalestatin 1 (Squal 1), or 3 × 10−5 M fluvastatin (Fluva) (top) or 10−5 M pregnenolone 16α-carbonitrile (PCN) (bottom). After treatment, hepatocytes were harvested for the measurement of luciferase activities. Each bar represents the mean ± S.D. of normalized luciferase (firefly/Renilla) measurements (three wells per treatment group). *, p < 0.05, significantly different from the corresponding UT group; †, significantly different from the corresponding noncotransfected “0” group.
In a previous study by Choi et al. (1997), deletion of the eight C-terminal amino acids, containing the AF-2 region of the ligand-binding domain, from mouse CAR yielded a receptor that was unable to trans-activate a CAR-responsive reporter but was still able to bind to a DR5 nuclear receptor motif as a heterodimer with RXR. Other studies have demonstrated that nuclear receptors lacking the AF-2 subdomain function in a dominant-negative fashion (Venkateswaran et al., 2000). We therefore prepared a corresponding AF-2–deleted rat CAR and tested its ability to inhibit inducible CYP2B1 transcription in cotransfection assays, in primary cultured rat (Sprague-Dawley) hepatocytes. As controls, cultures were cotransfected with pcDNA3.1 plasmid without insert or with plasmid expressing wild-type rat CAR. Whereas cotransfection with wild-type CAR significantly enhanced the CYP2B1-driven luciferase induction produced by phenobarbital or squalestatin 1 treatment, cotransfection with the AF-2–deleted CAR completely inhibited inducible expression. The CAR transfections had no effect on pregnenolone 16α-carbonitrile–inducible expression from the PXR-responsive CYP3A4-XREM reporter plasmid, demonstrating specificity of the effect for CYP2B.
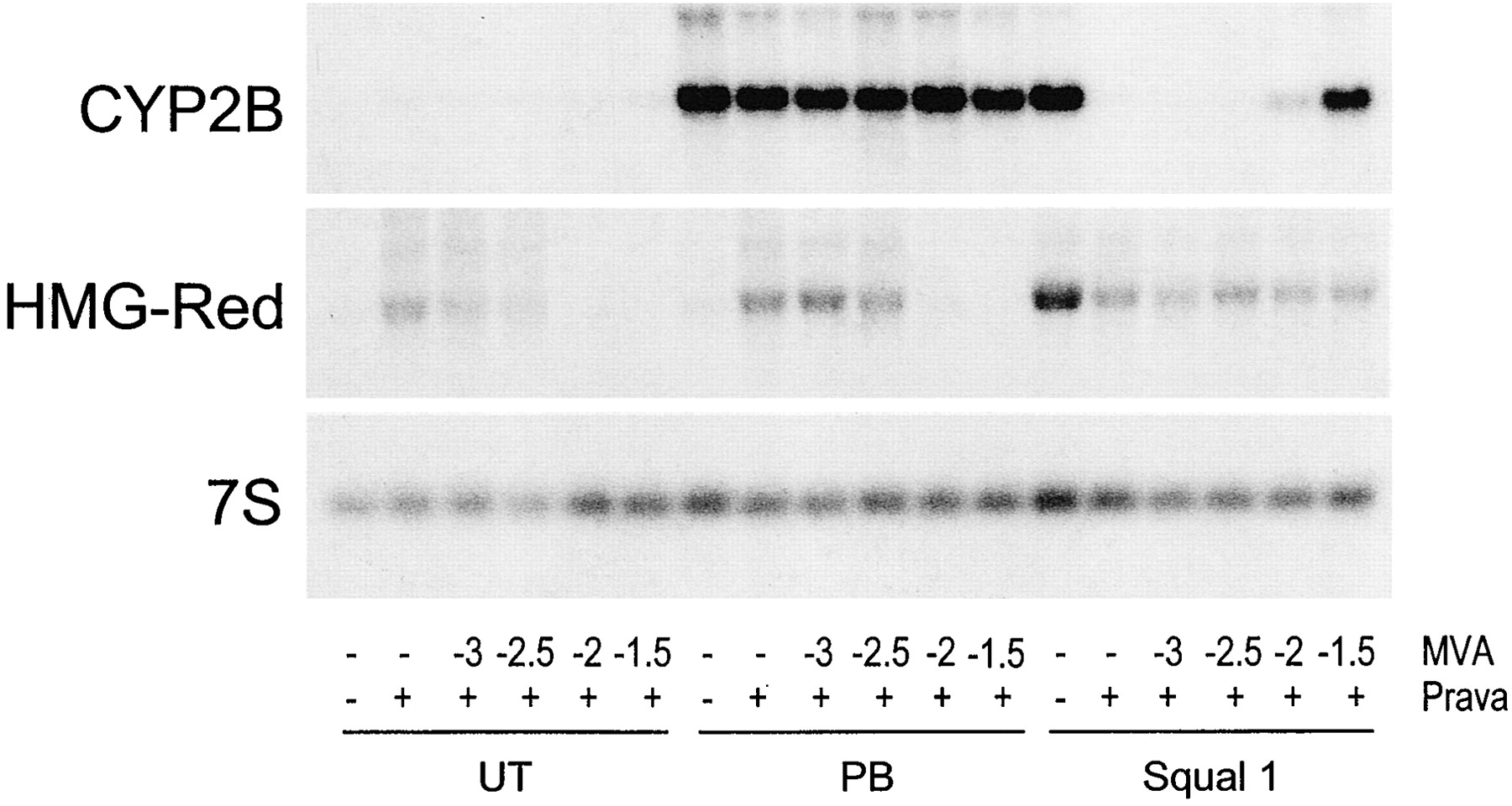
Thus, our data indicate that CAR activation, not SREBP activation, underlies squalestatin 1-mediated CYP2B induction. This prompts the question of whether squalestatin 1 is a direct activator of CAR or the effect is mediated indirectly, as a result of squalene synthase inhibition, as our previous results suggested. We reported previously that squalestatin 1 treatment induced CYP2B mRNA and protein with a slower time course than did phenobarbital (Kocarek et al., 1998). Here, we compared the time courses of treatment with phenobarbital, fluvastatin, squalestatin 1, or the bisphosphonate squalene synthase inhibitor SQ-34919 on the induction of CYP2B and HMG-CoA reductase mRNAs (Fig. 5). Elevated levels of CYP2B mRNA were clearly evident 3 h after treatment with either phenobarbital or fluvastatin (Fig. 5). By contrast, no increase in the amount of CYP2B mRNA was seen after treatment with either of the squalene synthase inhibitors until 12 h after treatment (Fig. 5). The delayed effects of the squalene synthase inhibitors, relative to phenobarbital or fluvastatin, on CYP2B mRNA expression were not the result of slower cellular uptake, because squalestatin 1 treatment up-regulated HMG-CoA reductase mRNA levels with the same time course as fluvastatin (Fig. 5). Fluvastatin and squalestatin 1 induce HMG-CoA reductase mRNA through the same mechanism as a consequence of sterol synthesis inhibition and SREBP activation. Thus, these results suggest that although phenobarbital and fluvastatin activate CAR through a rapidly occurring mechanism, the squalene synthase inhibitors more probably act through an indirect mechanism, whereby squalene synthase inhibition causes some perturbation in cellular metabolism, which takes time to occur. If this is so, then squalene synthase inhibition could evoke its effect by causing either depletion of a downstream metabolite (e.g., a sterol) or accumulation of an upstream metabolite. As evidence against the former, we have reported that whereas some inhibitors of HMG-CoA, such as CYP2B, CYP3A, and CYP4A, induce P450s, this is not always the case (Kocarek and Reddy, 1996). In particular, pravastatin has no effect on the expression of CYP2B, CYP3A, or CYP4A in primary cultured rat hepatocytes (Kocarek and Reddy, 1996). Regarding the latter possibility, squalestatin 1 treatment of rats, mice, and dogs has been reported to cause a massive excretion of farnesol-derived dicarboxylic acids, which are derived from the progressive metabolism of farnesyl pyrophosphate, the substrate of squalene synthase (Bostedor et al., 1997; Vaidya et al., 1998). Thus, it seemed possible that squalestatin 1 treatment might lead to the increased production and accumulation of a cellular isoprenoid, which might then activate CYP2B expression through CAR. Our finding that pravastatin, when used alone, has no effect on CYP2B expression enabled us to use this drug as a tool to shut down the sterol biosynthetic pathway upstream of squalene synthase. We therefore examined the effect of pravastatin pretreatment on the ability of squalestatin 1 to induce CYP2B mRNA. Pravastatin pretreatment of primary cultured rat hepatocytes completely abolished squalestatin 1-mediated CYP2B mRNA induction (Fig.6). Furthermore, addition of mevalonate, the product of the HMG-CoA catalyzed reaction, to the pravastatin-treated cultures, restored squalestatin 1-inducible CYP2B expression to the hepatocyte cultures (Fig. 6). As controls, pravastatin treatment, with or without mevalonate supplementation, had no effect on either basal or phenobarbital-induced CYP2B expression (Fig. 6). Also, whereas addition of mevalonate produced the expected suppression of pravastatin-mediated up-regulation of HMG-CoA reductase mRNA, reflecting efficient conversion to sterols, it had no effect in the presence of squalestatin 1, demonstrating that squalene synthase blockade was complete and that only isoprenoids, not sterols, were being produced (Fig. 6). These results clearly demonstrate that squalestatin 1-mediated CYP2B induction requires the ongoing biosynthesis of an endogenous isoprenoid.

Time courses of CYP2B and HMG-CoA reductase mRNA induction in primary cultured rat hepatocytes treated with phenobarbital, fluvastatin, squalestatin 1or SQ-34919. Forty-eight–hour–old primary cultures of rat hepatocytes were either harvested (0 h) or incubated with medium containing 10−4 M phenobarbital (PB), 3 × 10−5 M fluvastatin (Fluva), 10−7 M squalestatin 1 (Squal 1), or 10−6 M SQ-34919. At treatment times ranging from 3 to 24 h, three dishes of hepatocytes per treatment group were harvested for the preparation of total RNA, and pooled samples were analyzed for levels of CYP2B and HMG-CoA reductase (HMG-Red) mRNAs by Northern blot hybridization. Autoradiographs are shown for a representative experiment; comparable results were obtained in an additional hepatocyte culture experiment. Also shown are the autoradiographs of the blots after their rehybridization with 7S cDNA, to demonstrate consistency of RNA loading and transfer among samples.

Effect of coincubations with pravastatin and/or mevalonate on squalestatin 1-mediated CYP2B mRNA induction. At 48 h after plating, hepatocytes were incubated in medium alone or containing 3 × 10−5 M pravastatin (Prava +). One hour later, indicated cultures were treated with either 10−4 M phenobarbital (PB), 10−7 M squalestatin 1 (Squal 1), or no inducer (UT) in either the absence (−) or presence of mevalonate (MVA), at concentrations ranging from 10−3 M to 3 × 10−2 M (indicated as log molar concentrations). After 24 h of treatment, the three dishes of hepatocytes representing each treatment group were harvested for preparation of total RNA, and pooled samples were analyzed for levels of CYP2B and HMG-CoA reductase (HMG-Red) mRNA by Northern blot hybridization. Also shown is a representative autoradiograph of a blot that was rehybridized with 7S cDNA. Data are representative of two independent hepatocyte culture experiments.
The first intermediate in the conversion of farnesyl pyrophosphate to “farnesoids” is trans,trans-farnesol. Thus, to obtain initial evidence as to whether farnesoid accumulation might be involved in squalestatin 1-mediated CYP2B induction, we examined the time-dependent effects of direct addition oftrans,trans-farnesol to the hepatocyte cultures. To demonstrate the degree of consistency among multiple experiments, the results of three independent hepatocyte culture experiments are shown (Fig. 7). The effects of phenobarbital and squalestatin 1 were highly reproducible among the three experiments and recapitulated the results shown in Fig. 5. Although the effects of farnesol were somewhat more variable among experiments, principally in terms of response magnitudes, in all cases, farnesol treatment evoked the induction of CYP2B mRNA with the same rapid time course as occurred for phenobarbital (Fig. 7). In these experiments, the highly lipophilic farnesol was added to the cultures at a relatively high concentration (3 mM) in ethanol, and was not freely soluble in the culture medium, probably contributing to the variability among experiments. Thus, for comparison, we added farnesol to the cultures in association with bovine serum albumin. We found that when farnesol was added in this manner, CYP2B induction was observed at a lower farnesol concentration (0.3 mM), although the magnitude of response was no greater than those shown in Fig. 7 (data not shown). In any case, the direct addition of a metabolite that would be expected to be produced endogenously by the hepatocyte after squalene synthase blockade rapidly induced CYP2B mRNA.

Time course of CYP2B mRNA induction in primary cultured rat hepatocytes treated with phenobarbital, squalestatin 1 or farnesol. Forty-eight–hour–old primary cultures of rat hepatocytes were either harvested (0 h) or incubated with 10−4 M phenobarbital (PB), 10−7 M squalestatin 1 (Squal 1), or 3 × 10−3 M trans,trans-farnesol. At treatment times ranging from 3 to 24 h, three dishes of hepatocytes per treatment group were harvested for the preparation of total RNA, and pooled samples were analyzed for levels of CYP2B mRNA by Northern blot hybridization. Autoradiographs are shown for three different hepatocyte culture experiments.
Discussion
In this study, we provide evidence that inhibition of squalene synthase induces CYP2B expression in primary cultured rat hepatocytes, not by inhibiting sterol biosynthesis and activating an SREBP transcription factor, but by evoking the accumulation of an endogenous isoprenoid that is an activator of the CAR nuclear receptor. Supporting evidence includes:
- 1.
- The absolute dependence of squalestatin 1-mediated induction on the PBRU-containing portion of the CYP2B1 5′-flanking region;
- 2.
- The failure of any cotransfected SREBP to activate a squalestatin 1-responsive CYP2B1 reporter;
- 3.
- The sex difference in response to squalestatin 1 treatment of primary cultured Wistar-Kyoto rat hepatocytes and the attainment of response in female Wistar-Kyoto hepatocytes transfected with either rat or mouse CAR;
- 4.
- The inhibition of squalestatin 1-mediated CYP2B1-luciferase induction by cotransfection with a dominant negative CAR;
- 5.
- The substantially delayed time course of CYP2B mRNA induction by treatment with squalestatin 1 or SQ-34919, relative to that observed for phenobarbital;
- 6.
- The blockade of squalestatin 1-mediated CYP2B mRNA induction by pravastatin pretreatment, and its rescue by mevalonate supplementation; and
- 7.
- The rapid induction of CYP2B mRNA after direct addition of farnesol to rat hepatocyte cultures.
Our findings are bolstered by reports that treatment of animals with squalestatin 1 causes the excretion of massive amounts of farnesol-derived dicarboxylic acids, indicating that squalene synthase blockade substantially increases flux through the farnesol pathway (Bostedor et al., 1997; Vaidya et al., 1998).
The concept that endogenous isoprenoids are regulators of nuclear receptor activity is not new. FXR bears its name because farnesol and related molecules were the first described activators of this receptor (Forman et al., 1995). PPARα and PPARγ are also activated by certain isoprenoids (Takahashi et al., 2002), whereas LXRα activity is suppressed by geranylgeraniol (Forman et al., 1997). When we examined the effects of squalestatin 1 treatment on expression from series of nuclear receptor-responsive reporters, only the CAR-responsive CYP2B1 plasmid was activated, suggesting that CAR may be the major isoprenoid-sensitive nuclear receptor in the hepatocyte. Relatively high concentrations of mevalonate were necessary to overcome the pravastatin-mediated blockade of squalestatin 1-inducible CYP2B expression, suggesting that substantial ongoing isoprenoid biosynthesis is necessary to achieve accumulation of the biologically active molecule. Such a finding is consistent with our failure to elicit a high level of CYP2B expression when farnesol was added to the cultures exogenously. Thus, the limited solubility of farnesol in culture medium and the mode of treatment (i.e., addition of a single “dose” to the cultures) may have precluded the accumulation of large amounts of active intermediate.
Our results clarify some of our previous findings (Kocarek and Reddy, 1996; Kocarek et al., 1998). Thus, if sterol biosynthesis inhibition was a cause of CYP2B induction, then all inhibitors of sterol biosynthesis should have induced CYP2B. However, pravastatin had no effect (Kocarek and Reddy, 1996). Also, the time course of CYP2B induction after treatment with fluvastatin, which was as rapid as that for phenobarbital, suggests that fluvastatin induces CYP2B by activating CAR directly, rather than through an indirect mechanism secondary to HMG-CoA reductase inhibition. Our proposed mechanism for squalestatin 1-mediated CYP2B induction also suggests an explanation for its failure to coinduce CYP3A. Phenobarbital and phenobarbital-like agents induce CYP2B expression at lower concentrations than are necessary to achieve CYP3A induction in cultured rat hepatocytes (Kocarek et al., 1990). If squalestatin 1 induces CYP2B as a consequence of squalene synthase inhibition, then the maximum induction will occur at the drug concentration that produces complete enzyme inhibition. If the concentration of active intermediate that accumulates at this concentration is sufficient to achieve CAR, but not PXR, activation, then only CYP2B induction will occur.
One aspect of our previous work (Kocarek et al., 1998), verified byOurlin et al. (2002), that requires explanation is the observation that squalestatin 1-mediated CYP2B induction was reversed by 25-hydroxysterol cotreatment. Indeed, this classic response was a major finding that led us to hypothesize that the effects of squalestatin 1 on CYP2B expression were secondary to sterol synthesis inhibition and SREBP activation. Because we have eliminated SREBP activation as the mechanism of squalestatin 1-mediated CYP2B induction, an alternative explanation for the sterol-suppressible component must be sought. One possibility might be that an endogenous sterol serves as an inverse agonist of CAR (Forman et al., 1998), and that squalestatin 1 treatment would inhibit biosynthesis of the inverse agonist. However, this mechanism seems unlikely, as pravastatin treatment should also inhibit the synthesis of such a regulator.
Because our results indicate that squalestatin 1-mediated CYP2B induction is triggered not through inhibition of sterol biosynthesis but through accumulation of an endogenous isoprenoid, the sterol-suppressive component cannot rely on reversal of inhibited sterol biosynthesis. In their study, Ourlin et al. (2002) hypothesized that endogenous sterols may negatively regulate P450 expression by enabling LXR to out-compete CXR for interaction with the DR4 motifs within the PBRU. In support of their hypothesis, these investigators (Handschin et al., 2002) have recently provided evidence that LXR can bind to the CYP2H1 and CYP2B6 PBRU in vitro, at the same as DR4 sites bound by CXR, CAR, or PXR, and can antagonize the actions of these “xenosensor” receptors. By this mechanism, inhibition of sterol biosynthesis would deprive LXR of ligand, thereby permitting CXR (or CAR) to bind to the PBRU and activate transcription. By itself, such a mechanism is again incompatible with our findings, because, if true, pravastatin treatment should also induce CYP2B. However, adding to this mechanism's plausibility are data indicating that geranylgeranyl pyrophosphate, which may also accumulate after squalene synthase blockade, can inhibit the binding of LXRα-RXR to a DR4 (Forman et al., 1997). If LXR is bound to the PBRU in vivo, the question then arises as to whether the evoked dissociation of LXR from the PBRU after squalestatin 1 treatment might be sufficient to permit CAR binding and CYP2B transcription, without the additional need to produce CAR activation. However, arguing against this possibility, CAR is primarily cytosolic under basal conditions, necessitating some stimulus to evoke nuclear translocation (Kawamoto et al., 1999). Also, we have found that primary cultures of mouse hepatocytes lacking LXRα and -β do not display basal expression of CYP2B mRNA (but they remain fully responsive to phenobarbital or dexamethasone treatment), as would be expected if the mere absence of LXR from the PBRU was sufficient to permit CYP2B expression (S. Shenog, T. Spencer, D. Mangelsdorf, T. Kocarek, manuscript in preparation). These findings thus suggest that CAR activation per se is a necessary step in the induction mechanism.
As an additional mechanism for sterol-mediated suppression, we suggest the now-classic “squelching” effect, in which the simultaneous activation of two pathways, each requiring the use of common cofactors, results in mutual inhibition. As support, Kakizaki et al. (2002)recently reported that retinoic acid treatment suppressed TCPOBOP-inducible Cyp2b10 expression in primary cultured mouse hepatocytes. Coexpression of retinoic acid receptor increased repression, whereas coexpression of RXR decreased repression, suggesting that competition between CAR and retinoic acid receptor for limiting amounts of RXR was the responsible mechanism. Along these lines, we have found that pretreatment of primary cultured hepatocytes with dexamethasone or pregnenolone 16α-carbonitrile, both of which strongly activate PXR, suppresses phenobarbital-mediated CYP2B induction (Kocarek and Reddy, 1998; T. Kocarek and S. Shenoy, unpublished observations). In addition, we have found that 25-hydroxycholesterol and certain other sterols are inducers of CYP3A in cultured rat and mouse hepatocytes (S. Shenog, T. Spencer, D. Mangelsdorf, T. Kocarek, manuscript in preparation). Thus, we suggest that sterols may activate one or more competing pathways (e.g., PXR, LXR), thus suppressing CYP2B induction, at least in part, through a squelching mechanism.
We have not, in this study, identified the isoprenoid species that produces CYP2B induction. Certainly, our results with farnesol and the high flux through the farnesoid pathway after squalestatin 1 treatment (Bostedor et al., 1997; Vaidya et al., 1998) implicate farnesol itself or a downstream metabolite, such as farnesal, farnesoid acid, or a dicarboxylic acid. However, the finding of Forman et al. (1997) that geranylgeranyl pyrophosphate can interact with LXR in vitro raises the possibility that farnesyl pyrophosphate itself (which is membrane-impermeable, necessitating specialized methods for efficient delivery to intact cells) may interact with CAR.
Finally, the observed rat-mouse species difference offers an opportunity to understand additional intricacies of CYP2B regulation by endogenous metabolites. As noted above, the difference does not seem to reside at the level of CAR, because mouse CAR supported squalestatin 1-mediated CYP2B1 activation in transfected female Wistar-Kyoto rat hepatocytes. We have recently found that we can achieve squalestatin 1-inducible CYP2B expression in primary cultured mouse hepatocytes if the medium is supplemented with additional mevalonate (T. Kocarek and N. Mercer-Haines, unpublished observations). We speculate that mouse hepatocytes more rapidly metabolize the active isoprenoid, and therefore do not sufficiently accumulate the molecule in the absence of mevalonate supplementation. In this regard, recent evidence indicates that farnesol is a substrate for P450 metabolism (Raner et al., 2002). Thus, our findings have implications not only for the identification of endogenous regulators of CAR but also for the further elucidation of farnesoid metabolism.
Acknowledgments
We thank Drs. Peter Edwards, Bryan Goodwin, and Masahiko Negishi for providing plasmid constructs used in these studies.
Footnotes
- Received June 28, 2002.
- Accepted August 15, 2002.
-
↵1 Because the CYP2B1, CYP3A23, and CYP4A cDNA probes used in this study hybridize to multiple P450 subfamily memebers, we refer to mRNAs detected on the Northern blots generically as CYP2B, CYP3A, and CYP4A, respectively.
-
This work was supported by National Institutes of Health Sciences grant HL50710, and by services provided by the Cell Culture and Imaging and Cytometry Facility Cores of National Institute of Environmental Health Sciences Center Grant P30-ES06639.
Abbreviations
- P450
- cytochrome P450
- CAR
- constitutive androstane receptor
- PXR
- pregnane X receptor
- PPAR
- peroxisome proliferator-activated receptor
- SREBP
- sterol regulatory element binding protein
- LXR
- liver X receptor
- HMG
- hydroxymethylglutaryl
- DMSO
- dimethyl sulfoxide
- PCR
- polymerase chain reaction
- AF-2
- activator function 2
- PBRU
- phenobarbital-responsive unit
- RXR
- retinoid X receptor
- FXR
- farnesoid X receptor
- CXR
- chicken X receptor
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}