


Abstract
After the recent description of β-arrestin2 recruitment to the human histamine H4 receptor (hH4R) in response to the well known H4R antagonist 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methyl-piperazine (JNJ 7777120), we evaluated in this study the efficacy of 31 known hH4R ligands to induce Gαi protein signaling and β-arrestin2 recruitment by the hH4R. The selected hH4R ligands belong to nine different structural classes that partly cover (pre)clinical trial candidates. We have identified hH4R ligands with a significant bias for the Gαi protein or β-arrestin2 pathway on the basis of efficacy differences. In addition, hH4R antagonists that did not show positive efficacy in either functional readouts were found. A common trend in pathway preference for the nine different ligand classes could not be observed. In particular, the isothiourea class shows very diverse results, varying from Gαi protein-biased or β-arrestin2-biased to nonbiased antagonists upon minor structural changes. The identified biased hH4R ligands are important pharmacological tools to unravel the significance of biased hH4R signaling in H4R pharmacology.
Introduction
Since its discovery in 2000, the human histamine H4 receptor (hH4R) is the most recent addition to the histamine G protein-coupled receptor (GPCR) subfamily (Nakamura et al., 2000; Oda et al., 2000; Liu et al., 2001; Morse et al., 2001; Nguyen et al., 2001; Zhu et al., 2001). The exact function of hH4R is still to be elucidated, but its predominant expression in cells of the hematopoietic lineage links this receptor to allergic and inflammatory responses (Thurmond et al., 2008; Leurs et al., 2011). During the last decade, hH4R signaling has been the topic of many studies, in which Gαi proteins appear to be the key signal transducer in a variety of cell types (Oda et al., 2000; Liu et al., 2001; Morse et al., 2001; Zhu et al., 2001). Activation of the hH4R leads to inhibition of adenylyl cyclase and a subsequent reduction in cAMP production (Liu et al., 2001; Lim et al., 2005), as well as increases in intracellular calcium levels, actin polymerization, and chemotaxis (O'Reilly et al., 2002; Hofstra et al., 2003; Ling et al., 2004; Bäumer et al., 2008). Moreover, in mouse mast cells, the H4R has been shown to signal to kinases such as ERK1/2 and AKT as well as phosphatidylinositol 3-kinase γ, leading to the production of interleukin 6 (Desai and Thurmond, 2011). Besides ligand-induced signaling, hH4R exhibits Gαi-mediated signaling in the absence of histamine, i.e., the hH4R shows constitutive activity (Lim et al., 2005; Schneider et al., 2009). These reported hH4R activities are all pertussis toxin (PTx)-sensitive, indicating Gαi-mediated signaling routes. In addition, as for many GPCR agonists, histamine stimulation of hH4R leads to recruitment of β-arrestin2 and downstream ERK phosphorylation (Rosethorne and Charlton, 2011).
The well explored reference H4R antagonist JNJ 7777120 (Thurmond et al., 2004) was described to be the first biased H4R ligand that selectively recruits β-arrestin2 in a Gαi protein-independent manner, as revealed by an enzyme fragment complementation (EFC) assay (Rosethorne and Charlton, 2011). Moreover, JNJ 7777120 induces ERK phosphorylation in a time-dependent manner typical for β-arrestin2-mediated signaling. In contrast, the inverse agonist thioperamide is not able to recruit β-arrestin2 but antagonizes the β-arrestin2 recruitment by both histamine and JNJ 7777120 (Rosethorne and Charlton, 2011). The ability of GPCR ligands to exhibit biased signaling is an emerging concept that has the potential to improve the efficacy and specificity and reduce the side effects of newly developed drugs (Galandrin et al., 2007; Kenakin, 2007; Rajagopal et al., 2010). Biased ligands that target the angiotensin II type 1 receptor have been shown to antagonize G protein signaling but to induce β-arrestin2 recruitment as well as downstream phosphorylation events (Violin et al., 2010). In vivo this resulted in reduced blood pressure and favorable cardiac functioning, in contrast to nonbiased ligands that decreased cardiac performance (Violin et al., 2010; DeWire and Violin, 2011).
The biased agonism of JNJ 7777120, together with the incomplete characterization of previous H4R lead compounds, prompted us to fully explore the extent of ligand-biased signaling at the H4R. Moreover, the identification of biased tool compounds will help to evaluate the impact of individual signaling pathways in H4R-mediated (patho)physiological responses. In recent years, both industry and academia have discovered and characterized many new hH4R ligands, mainly on the basis of hH4R binding affinity and specificity (Jablonowski et al., 2003; Terzioglu et al., 2004; Thurmond et al., 2004; Lim et al., 2005, 2006; Liu et al., 2008; Smits et al., 2008; Igel et al., 2009; Strakhova et al., 2009; Schneider et al., 2010; Smits et al., 2010). The therapeutic value of such hH4R ligands is potentially very interesting, not only for pathologies such as airway inflammation and allergic rhinitis but also for other inflammatory conditions (inflammatory bowel disease and atopic dermatitis) (Leurs et al., 2011). Therefore, H4R antagonists and inverse agonists are rapidly proceeding in preclinical tests, and Palau Pharma (Barcelona, Spain) has tested the first H4R antagonist (UR-63325), which is already in clinical phase II trials (NCT01260753).
In the present study, we determined the efficacy of 31 known hH4R ligands to induce Gαi protein-dependent signaling and β-arrestin2 recruitment. This broad selection of ligands partly covers the lead compounds that are currently being tested in preclinical studies.
Materials and Methods
Materials.
U2OS cell culture media (Gibco) were obtained from Invitrogen (Carlsbad, CA). HEK293T cell culture media were purchased from PAA (Pasching, Austria). Forskolin was purchased from Sigma-Aldrich (St. Louis, MO). [3H]Histamine (10.6–13.4 Ci/mmol) was obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA). The H4R compounds tested were synthesized in the medicinal chemistry department of VU University Amsterdam (Amsterdam, The Netherlands), except for UR-PI376, which was a kind gift of Prof. Dr. A. Buschauer, and histamine and clozapine, which were obtained from Sigma-Aldrich.
Cell Culture and Transfection.
PathHunter U2OS β-arrestin2:EA cells stably expressing the human histamine H4 receptor (U2OS-H4R) (Rosethorne and Charlton, 2011) were cultured in minimal essential medium containing l-glutamine supplemented with fetal bovine serum (10% v/v), penicillin (100 IU/ml), streptomycin (100 μg/ml), G418 (Geneticin; 500 μg/ml), and hygromycin (250 μg/ml) at 37°C and 5% CO2. One day before the β-arrestin2 recruitment assay, 10,000 cells/well were seeded in white, clear-bottomed 384-well ViewPlates (PerkinElmer Life and Analytical Sciences) in 20 μl of minimal essential medium supplemented as described above and incubated at 37°C and 5% CO2.
HEK293T cells were cultured in Dulbecco's modified Eagle's medium containing l-glutamine and sodium pyruvate supplemented with fetal bovine serum (10% v/v), 50 IU/ml penicillin, and 50 μg/ml streptomycin.
HEK293T cells were transiently cotransfected with 500 ng of hH4R/pcDEF3 and 2.5 μg of pTNLC1–21 cAMP-response element (CRE) luciferase (containing 21 cAMP-responsive elements upstream of the luciferase gene) by means of the linear polyethyleneimine (25 kDa) method. The next day, 50,000 cells/well were transferred to white-bottomed 96-well plates (Greiner Bio-One, Longwood, FL).
β-Arrestin2 Recruitment Assay.
U2OS-H4R cells were stimulated with hH4R ligands or dimethyl sulfoxide (1%) for 2 h at 37°C and 5% CO2 diluted in Hanks' balanced salt solution supplemented with 20 mM HEPES and 0.1% bovine serum albumin. Then 25 μl of Flash detection reagent (DiscoveRX, Fremont, CA) was added, and cells were further incubated for 15 min at room temperature on a table shaker. Luminescence was subsequently measured on the LeadSeeker imaging system (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK).
CRE Luciferase Reporter Gene Assay.
Transiently transfected HEK293T cells were stimulated for 6 h with H4R ligands or dimethyl sulfoxide (1%) in serum-free Dulbecco's modified Eagle's medium supplemented with 1 μM forskolin at 37°C and 5% CO2. After 6 h, the stimulation medium was aspirated and 25 μl of luciferase assay reagent (0.83 mM ATP, 0.83 mM d-luciferin, 18.7 mM MgCl2, 0.78 μM Na2HPO4, 38.9 mM Tris-HCl (pH 7.8), 0.39% glycerol, 0.03% Triton X-100, and 2.6 μM dithiothreitol) was added to each well. Luminescence (1 s/well) was measured after 30 min of incubation at 37°C and 5% CO2 in a Victor3 1420 multilabel reader (PerkinElmer Life and Analytical Sciences).
[3H]Histamine Saturation and Displacement Binding Experiments.
Two days after transfection, cells were washed once with phosphate-buffered saline and subsequently scraped from their culture dish in 1 ml of phosphate-buffered saline. Cell pellets were collected by centrifugation at ∼2000g for 10 min at 4°C and dissolved in 50 mM Tris-HCl binding buffer (pH 7.4 at room temperature). The cell suspension was subsequently sonicated for 10 s to obtain a homogeneous solution of crude membrane extracts. Both the displacement and saturation binding assays were performed on crude membrane extracts from H4R transfected cells. Displacement binding was done on crude membrane extracts coincubated with increasing amounts (10 pM–100 μM) of H4R ligand and ∼10 nM [3H]histamine in a total volume of 100 μl/well. Saturation binding was performed on crude membrane extracts with increasing amounts of [3H]histamine (0–40 nM) in the presence and absence of 10 μM thioperamide. The reactions were incubated for 1.5 h at room temperature on a shaking table (750 rpm). Bound radioligand was separated from free radioligand via rapid filtration over a 0.5% polyethyleneimine-presoaked glass fiber C plate (GF/C, PerkinElmer Life and Analytical Sciences). GF/C plates were subsequently washed three times with ice-cold 50 mM Tris-HCl wash buffer (pH 7.4 at 4°C). The retained radioactivity on the GF/C plates was counted by liquid scintillation counting in a Wallac MicroBeta counter (PerkinElmer Life and Analytical Sciences).
Ligand Selection.
To evaluate the efficacy of structurally diverse hH4R ligands, we selected 31 ligands (Ki = <3 μM) from nine scaffold classes: I) histamine analogs (e.g., 4-methylhistamine), II) triazoles, III) guanidines [e.g., 2-(2-guanidinoethyl)isothiourea (VUF8430)], IV) isothioureas (e.g., clobenpropit), V) dibenzodiazepines (e.g., clozapine), VI) aminopyrimidines, VII) indole-carboxamides (e.g., JNJ 7777120), VIII) quinoxalines; and IX) quinazoline sulfonamides (Fig. 1; Supplemental Tables 1 and 2).

Selection of hH4R ligands that belong to nine different chemical classes: I, histamine analogs; I, triazoles; III, guanidines; IV, isothioureas; V, dibenzodiazepines; VI, aminopyrimidines; VII, indole-carboxamides; VIII, quinoxalines; IX, quinazoline sulfonamides.
Operational Model Calculations.
Concentration-response curves of both functional assays were fitted to the operational model (Black and Leff, 1983) (variable slope) in GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA). To analyze the data using global fitting, all concentration-response curves for each assay were plotted in one graph. Coupling efficiency (τ) was calculated via the formula:
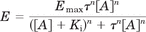 The pKi values that were used to calculate the Ki input values can be found in Supplemental Tables 1 and 2. Subsequently, effective signaling (σ) was calculated with the formula as described previously (Rajagopal et al., 2011):
The pKi values that were used to calculate the Ki input values can be found in Supplemental Tables 1 and 2. Subsequently, effective signaling (σ) was calculated with the formula as described previously (Rajagopal et al., 2011):
 The σ values from both assays are graphically plotted against each other to visualize the ligand bias (Fig. 7A). The ligand bias factor was calculated via the formula
The σ values from both assays are graphically plotted against each other to visualize the ligand bias (Fig. 7A). The ligand bias factor was calculated via the formula

Data Analysis.
Curve-fitting and statistical analysis were performed using GraphPad Prism 5.0. Results shown are from pooled data (mean ± S.E.M.) from at least three independent experiments performed in duplicate. Radioligand binding data were fitted to a one-site binding model (competition and saturation binding).
Results
Gαi Signaling and β-Arrestin2 Recruitment by the hH4R.
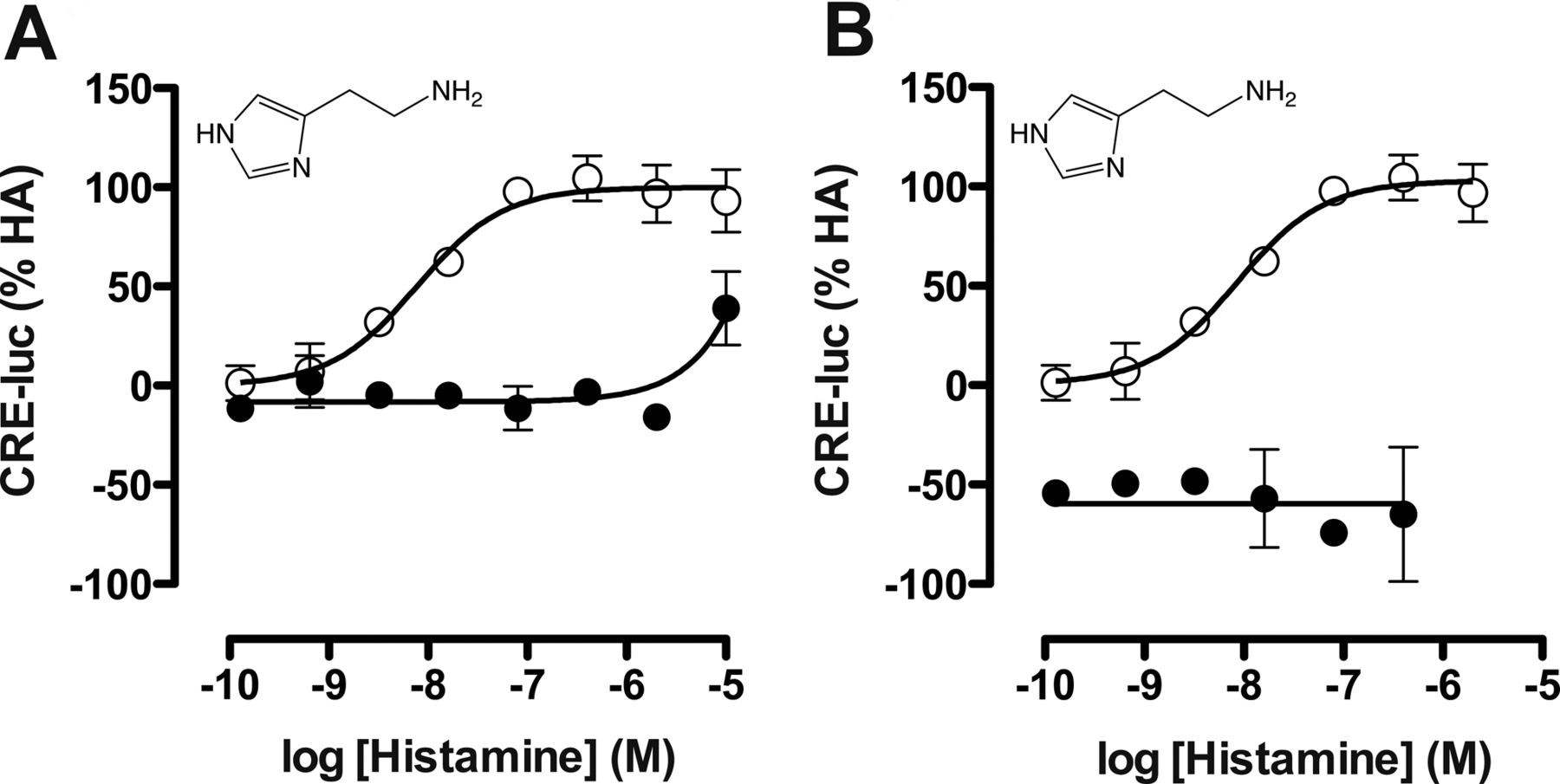
Gαi protein signaling by the hH4R in response to ligand stimulation (1 nM–100 μM) was measured as CRE reporter gene activity, whereas β-arrestin2 recruitment was detected by β-galactosidase enzyme fragment complementation (DiscoveRX PathHunter assay). To confirm that observed responses were mediated through the hH4R, the CRE assay was first performed with histamine in the absence or presence of a 10 μM concentration of the selective hH4R antagonist JNJ 7777120. Histamine inhibited 1 μM forskolin-stimulated CRE activity with a pEC50 value of 8.2 ± 0.03 (n = 3), which was slightly left-shifted compared with its pKi value of 7.8 (Supplemental Table 2). JNJ 7777120 antagonized this histamine-induced hH4R modulation of CRE activity (Fig. 2A). Moreover, pretreatment with the Gαi inhibitor PTx also completely abolished the histamine responsiveness (Fig. 2B). Using the PathHunter assay we could confirm that histamine was also able to recruit β-arrestin2 with a pEC50 value of 7.3 ± 0.08 (n = 3). The signal specificity of the hH4R in recruiting β-arrestin2 as well as the ineffectiveness of PTx in this assay were reported previously (Rosethorne and Charlton, 2011).
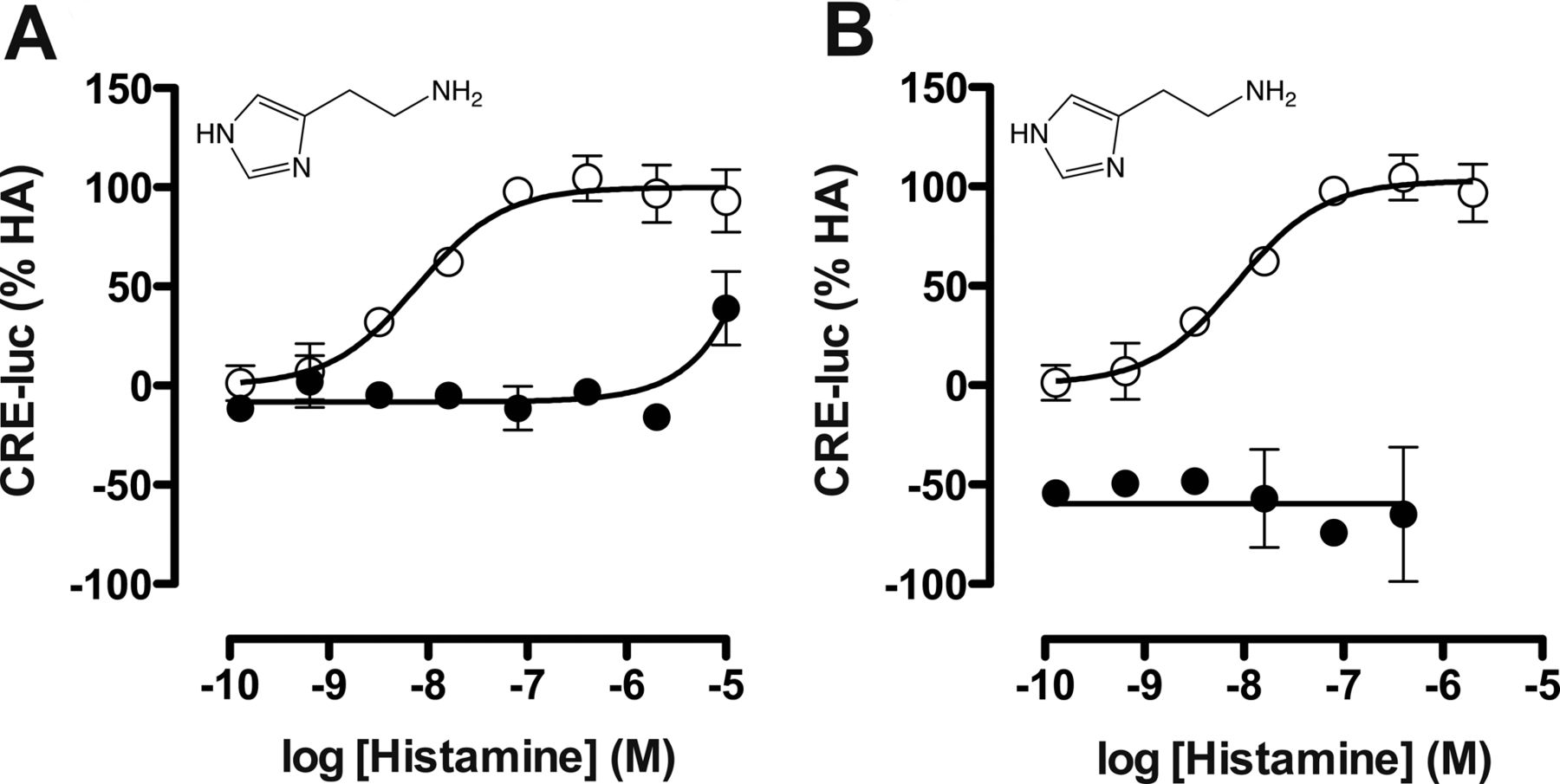
Inhibition of ligand-induced hH4R signaling with JNJ 7777120 and PTx. hH4R and CRE-luciferase (CRE-luc) transfected cells were stimulated with histamine and (A) coincubated with buffer (○) or 10 μM hH4R antagonist JNJ 7777120 (●) or (B) coincubated with buffer (○) or 100 ng/ml PTx (●). Graphs shown are pooled data from at least two experiments performed in duplicate. Data are normalized to histamine (HA; 100% efficacy). Error bars indicate S.D. values.
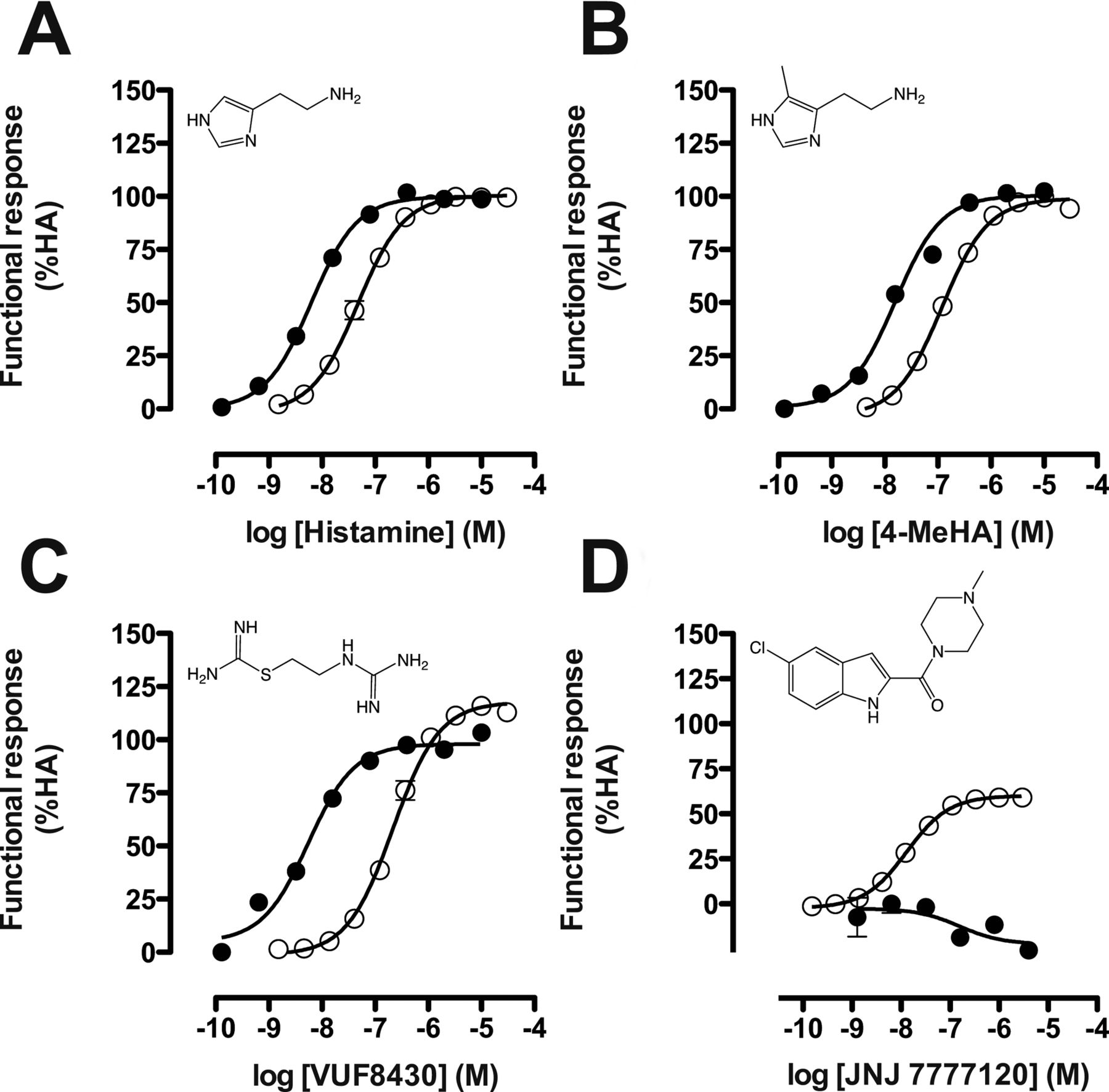
Ligand-induced responses were normalized to the observed activity of histamine (agonist in both assays; efficacy = 100%) or thioperamide (inverse agonist in CRE assay; efficacy = −100%). Therefore, histamine is defined as a nonbiased, full agonist in both Gαi protein-dependent signaling and β-arrestin2 recruitment (Fig. 3A). Concentration-response curves for the well characterized hH4R ligands, histamine, 4-methylhistamine (4-MeHA), VUF8430, and JNJ 7777120, were assessed to compare their potency and efficacy in both assays. Similar to histamine, 4-MeHA was a full agonist in both assays with a comparable 10-fold potency difference between the Gαi protein signaling and β-arrestin2 recruitment (Fig. 3B). The potencies of histamine (pEC50 = 7.3 ± 0.08) and 4-methylhistamine (I) (pEC50 = 6.9 ± 0.06) for β-arrestin2 recruitment were comparable to the potencies (pEC50 = 6.9 and 7.0, respectively) described for the Tango assay from Invitrogen (2008). In contrast, VUF8430 (III) tended to show a slightly higher efficacy (117 ± 3.8%) toward β-arrestin2 recruitment compared with the Gαi-mediated pathways (98 ± 5.5%). Moreover, the potency difference between the two functional responses was larger (∼30-fold) than that for histamine or 4-MeHA (Fig. 3C). The indole-carboxamide JNJ 7777120 (VII) was a weak inverse agonist in the CRE assay used but acted as a partial agonist (pEC50 = 7.8 ± 0.04) in the β-arrestin2 recruitment assay as reported previously (Rosethorne and Charlton, 2011) (Fig. 3D).

Functional responses of selected well known hH4R ligands in both CRE-luciferase (●) and β-arrestin2 recruitment (○) assays. Graphs shown are pooled data from at least three experiments performed in duplicate. Data are normalized to histamine (HA; 100% efficacy) or thioperamide (−100% efficacy) responses, and error bars indicate S.E.M. values. A, histamine. B, 4-methylhistamine. C, VUF8430. D, JNJ 7777120.
Upon assessment of the other selected ligands, we were able to identify hH4R ligands that showed a bias toward one of the pathways tested. In particular, clobenpropit (Fig. 4A), VUF5222 (Fig. 4B), VUF10778 (Fig. 4C), and VUF10185 (Fig. 4D) showed a bias for Gαi protein activation. VUF10185 showed only minor recruitment of β-arrestin2 (17 ± 2.0%); therefore, increasing concentrations of VUF10185 were added to antagonize the stimulatory effects of histamine (Fig. 4E). Indeed, VUF10185 concentration dependently right-shifted the histamine concentration-response curves. The Schild plot that was constructed from the concentration ratios showed that VUF10185 is a competitive antagonist (pA2 = 6.8) in the β-arrestin2 recruitment assay (Fig. 4F).

Functional responses of Gαi protein-biased hH4R ligands in both CRE-luciferase (●) and β-arrestin2 recruitment (○) assays. Pooled data are shown from at least three experiments performed in duplicate. Data are normalized to histamine responses (HA; 100% efficacy) and error bars indicate S.E.M. values. A, clobenpropit. B, VUF5222. C, VUF10778. D, VUF10185. Increasing amounts (0, ○; 316 nM, ♢; 1 μM, ▵; 3.16 μM, □; and 10 μM, ▿) of VUF10185 were added to a concentration-response curve of histamine (E) in a β-arrestin2 recruitment assay to finally construct a Schild plot from the calculated concentration ratios (F). DR, dose ratio.
Furthermore, in addition to JNJ 7777120, 6 of the 31 ligands tested (VUF10214, VUF10056, VUF6002, VUF11273, VUF11012, and VUF5223) showed efficacy toward β-arrestin2 recruitment but possessed neutral or negative efficacy in Gαi-dependent signaling (Fig. 5; Supplemental Table 1). Inverse agonism was percentualized to the hH4R-mediated signaling in response to the inverse agonist thioperamide; hence, compounds that were able to better inhibit the hH4R constitutive activity showed efficacy >−100%. Of interest, VUF10214 displayed a higher efficacy (86 ± 4.2%) toward the β-arrestin2 pathway than the previously identified partial agonist JNJ 7777120 (60 ± 5.4%).

Functional responses of β-arrestin2-biased hH4R ligands in both CRE-luciferase (●) and β-arrestin2 recruitment (○) assays. Graphs shown are pooled data from at least three experiments performed in duplicate. Data are normalized to histamine (HA; 100% efficacy) or thioperamide (Thio; −100% efficacy) responses, and error bars indicate S.E.M. values. A, VUF10214. B, VUF10056. C, VUF6002. D, VUF5223.
A graphical overview of the relationship between affinity, potency, and efficacy values in both functional screens of all hH4R ligands tested is plotted in Fig. 6. All ligands that have positive efficacy in both pathways were more potent (pEC50) for activating Gαi protein-mediated signaling than for recruiting β-arrestin2. The correlation between the agonist potencies for both assays was, however, linear (slope 1.2 ± 0.1) (Fig. 6A). The slope for the correlation between affinity versus potency in the β-arrestin2 recruitment or the CRE assay was, respectively, 1.0 ± 0.1 and 1.2 ± 0.1 (Fig. 6, B and C). However, no correlation was observed between the efficacies in Gαi protein-dependent signaling and β-arrestin2 recruitment for most ligands (Fig. 6D). Of interest, the majority of compounds had a higher efficacy toward the Gαi signaling pathway than toward the β-arrestin2 recruitment and are situated in the upper left-hand corner of the β-arrestin2 versus Gαi protein efficacy plot (Fig. 6D). These observed efficacies were not caused by differences in hH4R expression levels, which were found to be comparable in HEK293T cells (1–3 pmol/mg protein) and U2OS-H4R cells (2.2 ± 0.2 pmol/mg protein) (Rosethorne and Charlton, 2011). Compounds located near the x-axis of the graph show efficacy toward the β-arrestin2 recruitment assay but do not or negatively affect Gαi protein activation. Hence these compounds are classified as β-arrestin2-biased (Fig. 6D).
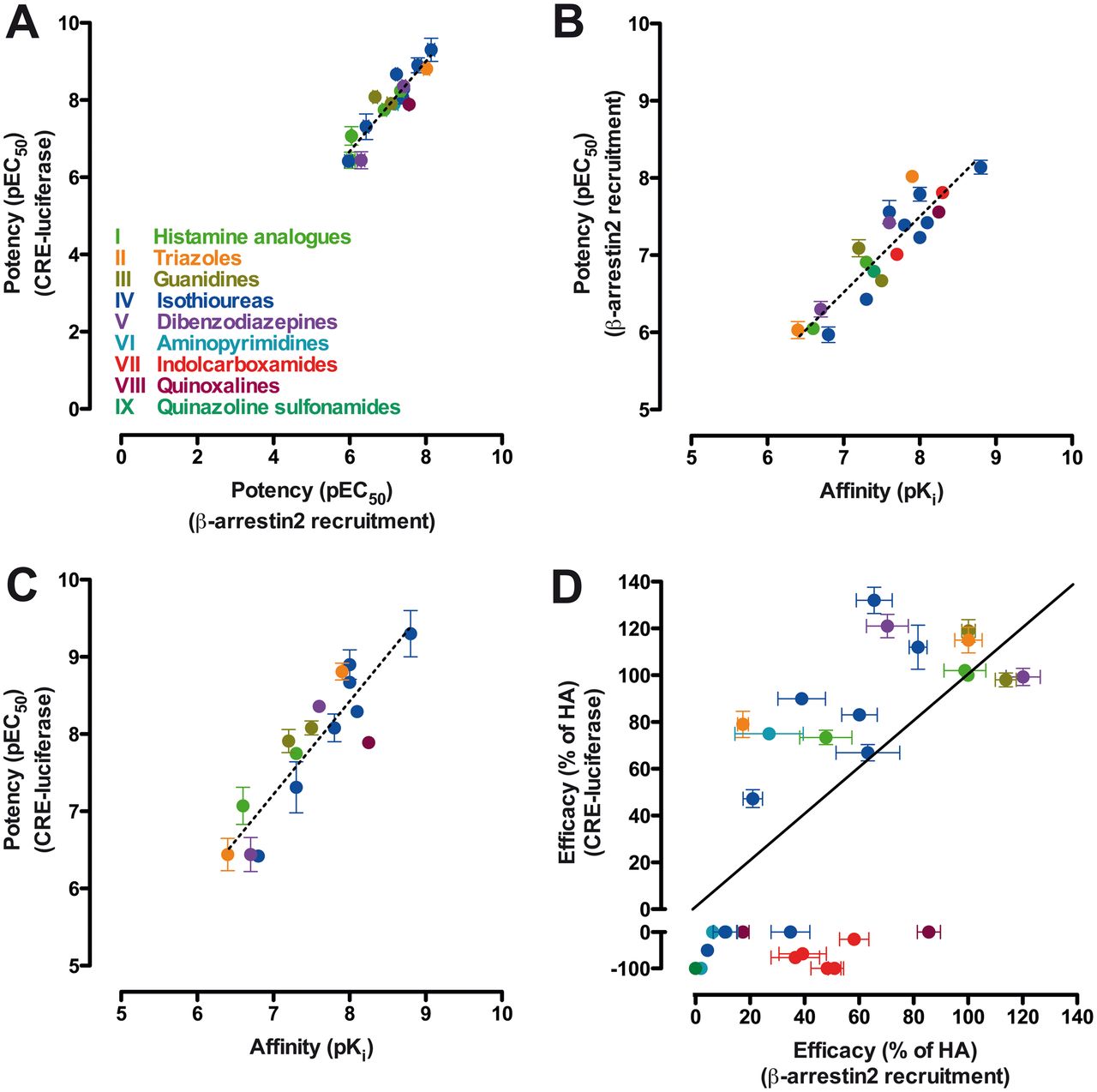
Correlation plots between affinity, potency, and efficacy values for all tested hH4R compounds. Efficacies in each assay were normalized to histamine (100% efficacy) or thioperamide (−100% in CRE assay) responses. A, potency values between different assays show a linear relation. B, linear correlation between potency in β-arrestin2 recruitment assay and affinity values. C, linear correlation between potency in CRE reporter gene assay and affinity values. D, comparison of efficacy values between different assays shows that few ligands display equal efficacy toward the two pathways. Dotted lines in A, B, and C represent regression lines; the continuous line in D is the unity line. Data are means ± S.E.M. values of at least three experiments performed in duplicate. pKi values were previously published or determined via a heterologous competition binding experiment.
Seven compounds induced neither Gαi protein activation nor β-arrestin2 recruitment (thioperamide, VUF9107, VUF11422, VUF10460, VUF10777, VUF10558, and VUF10519), although they have been shown to bind to hH4R (Supplemental Table 2). Consequently, these ligands are located around the origin of the efficacy plot (Fig. 6D).
Determination of Ligand Bias Using the Operational Model for Pharmacological Agonism.
The hH4R ligands that show efficacy in both assays were further examined for biased signaling using the operational model for pharmacological agonism (Black and Leff, 1983). The coupling efficiency (τ) between each ligand-hH4R complex and its downstream signaling components as well as the effective signaling (σ) was calculated (see formulas under Materials and Methods). The σ values are graphically plotted against each other to visualize the ligand bias (Fig. 7A). Ligands that are located at the right side of the unity line show a possible β-arrestin bias, whereas ligands located at the left side of this line show a possible Gαi bias. Finally, the ligand bias factor was calculated (Fig. 7B). A positive bias factor indicated that a ligand is Gαi-biased, whereas a negative value suggested β-arrestin2 bias (Fig. 7B). Using this method, we could identify additional Gαi-biased ligands (i.e., VUF10192, UR-PI376, VUF4656, and clozapine) as well as a β-arrestin2-biased ligand (i.e., VUF5228), which were not readily apparent from their concentration-response curves in both assays (Fig. 7, C–K). Surprisingly, earlier identified Gαi-biased ligands clobenpropit and VUF10185 did not show up as biased ligands on the basis of these calculations.

Evaluation of hH4R ligand bias: operational model versus efficacy comparisons. A, σ was calculated using τ values obtained from an operational model fit of pooled data. Error bars represent S.E.M. values. Dotted line represents unity line. Ligands shown are the following: *, histamine; ⊗, VUF4656; ○, 4-MeHA; □, VUF8430; ♦, VUF10192; ♢, VUF10306; ▵, VUF6884; ⊠, clozapine; ●, UR-PI376; ▿, VUF4704; ★, VUF8328; and  , VUF5228; ■, clobenpropit; ●, VUF10185; ▴, VUF5222; ▾, VUF10778. B, bar graph of bias factor. Positive values indicate a possible Gαi protein bias; negative values indicate a possible β-arrestin2 bias. Statistical differences were determined using an unpaired two-tailed t test (*, p < 0.05; **, p < 0.01). C–H, CRE-luciferase (●) and β-arrestin2 recruitment (○) assay data for possible Gαi-biased compounds based on operational model fit. I–K, CRE-luciferase (●) and β-arrestin2 recruitment (○) assay data for possible β-arrestin2-biased compounds based on operational model fit. HA, histamine.
, VUF5228; ■, clobenpropit; ●, VUF10185; ▴, VUF5222; ▾, VUF10778. B, bar graph of bias factor. Positive values indicate a possible Gαi protein bias; negative values indicate a possible β-arrestin2 bias. Statistical differences were determined using an unpaired two-tailed t test (*, p < 0.05; **, p < 0.01). C–H, CRE-luciferase (●) and β-arrestin2 recruitment (○) assay data for possible Gαi-biased compounds based on operational model fit. I–K, CRE-luciferase (●) and β-arrestin2 recruitment (○) assay data for possible β-arrestin2-biased compounds based on operational model fit. HA, histamine.
Discussion
Biased GPCR signaling is an emerging concept that has the potential to improve efficacy and specificity and reduce side effects of newly developed GPCR targeting drugs (Violin et al., 2010). Many hH4R ligands have been developed in recent years, with the first promising candidates now progressing in preclinical and clinical studies (Leurs et al., 2011). Classically, hH4R ligands were assessed for their ability to modulate Gαi protein-dependent signaling pathways. However, recently the widely used hH4R neutral antagonist/inverse agonist JNJ 7777120 was identified as a partial agonist in an EFC-based β-arrestin2 recruitment assay. Moreover, JNJ 7777120 recruited β-arrestin2 in a Gαi protein-independent manner, which subsequently led to the phosphorylation of ERK1/2 (Rosethorne and Charlton, 2011).
In this study we investigated whether JNJ 7777120 is the only ligand displaying this biased agonism or whether other hH4R ligands exhibit this thus far hidden efficacy as well. Therefore, 31 hH4R ligands of nine different structural classes were examined for their ability to modulate Gαi protein-independent β-arrestin2 recruitment and Gαi protein-dependent CRE-luciferase activity. We compared ligand potency and efficacy in both pathways and determined whether hH4R ligands were biased toward one of the tested signaling pathways. On the basis of distinct efficacy values between the two assays, we identified hH4R ligands that are biased toward the Gαi-dependent pathway: two isothioureas (IV, clobenpropit and its analog VUF5222) (Istyastono et al., 2011), one aminopyrimidine (VI, VUF10778), and one triazole (II, VUF10185) (Wijtmans et al., 2011). Because the β-arrestin2 recruitment assay is novel for these hH4R ligands, no comparison can be made with literature data. The potency of clobenpropit (pEC50 = 8.3; efficacy = 132%) in the CRE assay was higher than previously reported by us in a SK-N-MC cell line that stably expressed hH4R and CRE-β-galactosidase (pEC50 = 7.7; efficacy = 80%) (Lim et al., 2005). On the other hand, the clobenpropit data do fit to the literature in which histamine and clobenpropit have comparable efficacies (Buckland et al., 2003; Ling et al., 2004) or clobenpropit has a higher efficacy than histamine (Gutzmer et al., 2009). The potency and efficacy of VUF10185 were comparable to previous results in HEK293T cells (Wijtmans et al., 2011). Of interest, VUF10185 was identified as a competitive antagonist for the histamine-induced β-arrestin2 recruitment, with a pA2 value of 6.8 that almost equals its affinity (pKi = 6.4). Some hH4R ligands are known to interact with the other histamine receptor subtypes, especially with the Gαi-coupled hH3R (Lim et al., 2005). We therefore performed our CRE assay in the presence of the competitive H4R antagonist JNJ 7777120 (Lim et al., 2005, 2006; Rosethorne and Charlton, 2011). Saturating concentrations (10 μM) of JNJ 777120 inhibited agonist-induced Gαi-mediated signaling, confirming that signaling was mediated by hH4R. Moreover, hH4R modulation of CRE activity can be fully inhibited by preincubation with PTx, a known inhibitor of Gαi proteins.
The hH4R ligands that display a clear bias toward β-arrestin2 recruitment can be divided into three structural groups: one quinoxaline (VIII, VUF10214) (Smits et al., 2008), one isothiourea (IV, VUF5223) (Istyastono et al., 2011), and four indolecarboxamides (VII, VUF10056, VUF6002, VUF11273, and VUF11012). These compounds are neutral antagonists or inverse agonists for Gαi protein-dependent signaling but act as agonists in the β-arrestin2 recruitment assay. The 26% higher efficacy of VUF10214 compared with that of the partial agonist JNJ 7777120 in this assay makes VUF10214 the most effective hH4R biased agonist. hH4R signal specificity in β-arrestin2 recruitment was previously shown via competitive antagonism experiments using the hH3R/hH4R antagonist thioperamide (Rosethorne and Charlton, 2011). Furthermore, this EFC-based assay relies on the specific interaction of two enzyme parts and thereby assures signal specificity through the tagged proteins.
The majority of hH4R ligands were more potent in the CRE assay than in the β-arrestin2 recruitment assay. This discrepancy is most likely the consequence of a stoichiometry difference between the two assays. Whereas the β-arrestin2 recruitment assay depends on enzyme fragment complementation, CRE-luciferase is a reporter gene downstream of the second messenger cAMP. Thus, the EFC assay has a 1:1 interaction stoichiometry between hH4R and the β-arrestin2 protein and is characterized by a lack of stimulus amplification, and in this study we found a linear correlation between hH4R affinities and potencies. In contrast, the CRE assay is subjected to downstream signal amplification and can therefore yield higher potencies compared with the affinity values. Nonetheless, potency values of the ligands tested in both assays correlate in a linear way, indicating that the magnitude of signal amplification in the CRE assay at least is comparable for each ligand. The potential amplification in the CRE assay can cause an increase in efficacy compared with the β-arrestin2 recruitment; however, the lack of correlation between ligand efficacies in β-arrestin2 recruitment and CRE activity suggests that signal amplification in the latter is not causing the discrepancy in efficacies between the two functional readouts.
The hH4R ligands that did not exhibit a positive efficacy in either the Gαi protein-dependent or -independent assay were the isothioureas (IV) VUF9107 and thioperamide, the aminopyrimidines (VI) VUF11422, VUF10460, and VUF10777, and the quinazoline sulfonamides (IX) VUF10558 and VUF10519. These ligands are currently considered to be hH4R antagonists/inverse agonists. However, this is obviously only true for all pathways that have been investigated so far.
The hH4R compounds that were effective in both assays, but did not show distinct efficacy differences, were further analyzed using the “operational model of pharmacological agonism” (Black and Leff, 1983) and calculation of σ (Rajagopal et al., 2011; Rivero et al., 2012). On the basis of our definition that histamine is a full agonist in both assays, histamine and 4-MeHA are nonbiased ligands (i.e., situated on unity line). All other compounds tested tended to show a preference for one of the two pathways. Subsequent calculation and analysis of the bias factor (Rajagopal et al., 2011; Rivero et al., 2012) revealed that VUF10192 (II), UR-PI376 (III), VUF5222 (IV), VUF10306 (IV), VUF4656 (IV), clozapine (V), and VUF10778 (VI) are Gαi-biased ligands, whereas VUF5228 (IV) is a β-arrestin2-biased ligand. Intriguingly, this ligand classification based on the operational model is impossible to identify from the individual concentration-response curves. However, clobenpropit and VUF10185, which were first classified as Gαi-biased ligands on the basis of the large efficacy difference between both pathways, show up as unbiased ligands on the basis of the bias factor calculation using the operational model. This discrepancy for VUF10185 is probably caused by the relatively large error of one of its τ values, which is the consequence of the marginal efficacy in the β-arrestin2 recruitment assay. The reason for the different outcome observed for clobenpropit is not very clear and again emphasizes the caution that should be taken when interpreting ligand bias. A similar observation was reported for the β2-adrenergic receptor and the angiotensin II type 1A receptor. Three different methods for the determination of biased agonism resulted in three different outcomes (Rajagopal et al., 2011). This result demonstrates the limitation of the current analysis methods and shows that the ultimate method to plot and calculate biased agonism still has to be identified.
In conclusion, we have tested a series of structurally diverse hH4R ligands in a Gαi protein-dependent CRE assay and in a Gαi protein-independent β-arrestin2 recruitment assay. The previously reported β-arrestin2 biased agonism of JNJ 7777120 is clearly not limited to one hH4R ligand or even one chemical class. We have been able to identify several other β-arrestin2-biased H4R ligands that cover diverse chemical structures. In addition, we have demonstrated for the first time that Gαi protein bias also exists for the hH4R, with several ligands preferentially activating this pathway over β-arrestin2. This observed bias was not caused by assay differences (e.g., signal amplification and hH4R expression levels) because potency values between both assays correlated linearly and receptor levels were comparable. Moreover, after fitting our data to the operational model of agonism, we found additional biased hH4R ligands. A general tendency in pathway preference could not be observed within the nine different ligand classes, except for the indole-carboxamides (VII, i.e., JNJ 7777120 analogs) that all induced β-arrestin2 recruitment, but did not stimulate Gαi signaling, and the quinazoline sulfonamides (IX) that were antagonists in both assays. In particular, the isothioureas (IV) were able to give a broad spectrum of efficacies upon minor structural changes, from Gαi protein-biased (VUF5222) to β-arrestin2-biased (VUF5223) to nonbiased antagonist (VUF9107). The consequences of biased GPCR signaling have been studied extensively but are not completely understood for all receptors. In the case of the hH4R, preclinical data should be analyzed with care because biased hH4R signaling is still an unexplored area that urgently needs clarification. The identified compounds may prove to be valuable tools for further investigations to unravel the contributions of these distinct pathways in H4R (patho)physiology. Moreover, in light of the ongoing clinical trials and the high therapeutic interest in hH4R ligands, the discovery of ligand-specific signaling bias might have important therapeutic implications. β-Arrestin-biased ligands could be beneficial over a neutral antagonist for long-term treatments. Such a β-arrestin-biased ligand would potentially cause H4R internalization and long-term desensitization without initiating traditional G protein-dependent signaling events.
Authorship Contributions
Participated in research design: Nijmeijer, Vischer, Rosethorne, Charlton, and Leurs.
Conducted experiments: Nijmeijer.
Performed data analysis: Nijmeijer and Rosethorne.
Wrote or contributed to the writing of the manuscript: Nijmeijer, Vischer, Rosethorne, Charlton, and Leurs.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.S.N., H.F.V., and R.L. participate in the European COST BM0806.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
ABBREVIATIONS:
- H4R
- histamine H4 receptor
- hH4R
- human histamine H4 receptor
- GPCR
- G protein-coupled receptor
- ERK
- extracellular signal-regulated kinase
- PTx
- pertussis toxin
- EFC
- enzyme fragment complementation
- JNJ 7777120
- 1-[(5-chloro-1H-indol-2-yl)carbonyl]-4-methyl-piperazine
- HEK293T
- human embryonic kidney cells expressing the large T-antigen of simian virus 40
- CRE
- cAMP response element
- 4-MeHA
- 4-methylhistamine.

- Received June 29, 2012.
- Accepted September 12, 2012.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}