



Abstract
G protein–coupled receptors are the most important drug targets for human diseases. An important number of them remain devoid of confirmed ligands. GPR27 is one of these orphan receptors, characterized by a high level of conservation among vertebrates and a predominant expression in the central nervous system. In addition, it has recently been linked to insulin secretion. However, the absence of endogenous or surrogate ligands for GPR27 complicates the examination of its biologic function. Our aim was to validate GPR27 signaling pathways, and therefore we sought to screen a diversity-oriented synthesis library to identify GPR27-specific surrogate agonists. To select an optimal screening assay, we investigated GPR27 ligand-independent activity. Both in G protein–mediated pathways and in β-arrestin 2 recruitment, no ligand-independent activity could be measured. However, we observed a recruitment of β-arrestin 2 to a GPR27V2 chimera in the presence of membrane-anchored G protein-coupled receptor kinase-2. Therefore, we optimized a firefly luciferase complementation assay to screen against this chimeric receptor. We identified two compounds [2,4-dichloro-N-(4-(N-phenylsulfamoyl)phenyl)benzamide (ChemBridge, San Diego, CA; ID5128535) and 2,4-dichloro-N-{4-[(1,3-thiazol-2-ylamino)sulfonyl]phenyl}benzamide (ChemBridge ID5217941)] sharing a N-sulfamoylphenyl-2,4-dichlorobenzamide scaffold, which were selective for GPR27 over its closely related family members GPR85 and GPR173. The specificity of the activity was confirmed with a NanoLuc Binary Technology β-arrestin 2 assay, imaging of green fluorescent protein–tagged β-arrestin 2, and PathHunter β-arrestin 2 assay. Interestingly, no G protein activation was detected upon activation of GPR27 by these compounds. Our study provides the first selective surrogate agonists for the orphan GPR27.
Introduction
With more than 800 members in the human genome, the G protein–coupled receptors (GPCRs) represent the largest family of membrane receptors and are the most commonly exploited class of drug targets. Indeed, GPCRs are targeted directly by more than 30% of current medicines (Rask-Andersen et al., 2011).
According to the current paradigm, ligand binding to GPCRs stabilizes conformations with increased affinity for intracellular signaling partners such as heterotrimeric G proteins (Wettschureck and Offermanns, 2005), GPCR kinases (GRKs), or β-arrestins (Lefkowitz and Shenoy, 2005). This process, described as receptor activation, induces various events such as second-messenger modulation by several families of G proteins, phosphorylation of intracellular receptor residues by GRKs, or desensitization and internalization mediated by β-arrestins. Depending on the affinity for the different β-arrestin isoforms (β-arrestin 1 or β-arrestin 2) and the stability of the GPCR–β-arrestin complex during internalization, GPCRs are referred to as class A (transient interaction) or class B (sustained interaction) (Oakley et al., 2000). In addition, β-arrestins can act as important scaffolds and promote G protein–independent signaling such as extracellular signal–regulated kinase (ERK)1/2 phosphorylation (Defea, 2008; Luttrell and Gesty-Palmer, 2010).
According to the most recent International Union of Basic and Clinical Pharmacology list, 87 (excluding pseudogenes) human GPCRs of the rhodopsin family are still considered orphan, being unmatched with a physiologic ligand or being paired only with nonconfirmed ligands (Davenport et al., 2013). Given the past successes of the GPCR family in drug discovery, some of these receptors may represent innovative therapeutic targets. Ligands that modulate a receptor activity are an invaluable tool to precisely understand the physiology of a given GPCR (Roth and Kroeze, 2015). Consequently, many efforts focus on the identification of endogenous ligands for orphan GPCRs. However, the deorphanization rate has decreased during the past few years, and surrogate ligands (small molecules, antibodies, peptides, etc.) have been proposed as a complementary approach to investigate the most challenging orphans (Ngo et al., 2016).
Many GPCRs have been shown to be able to spontaneously adopt active conformations even in the absence of ligands. This concept of constitutive activity (Bond and Ijzerman, 2006) was introduced in 1989 together with the notion of inverse agonists when Costa and Herz (1989) demonstrated negative intrinsic activity for antagonists of the opioid δ receptors. Constitutive activity is considered today as a central paradigm in GPCR pharmacology. In the field of orphan GPCRs, constitutive signaling is an important receptor property because it sheds light on the signaling pathways and hence facilitates the detection of ligands.
The three receptors of the superconserved receptors expressed in the brain (SREB) family, GPR27 (SREB1), GPR85 (SREB2), and GPR173 (SREB3), compose a small cluster inside the rhodopsin-like GPCRs (Fredriksson et al., 2003). They share 52–63% amino acid identity and have the distinctive characteristic of being highly conserved (from 70 to 100%) throughout vertebrate evolution (Matsumoto et al., 2000). Although they are classified within the α-group of the rhodopsin family (Fredriksson et al., 2003), they lack some of the features usually found in the members of this family, such as the DRY motif at the junction of transmembrane domain III and intracellular loop 2 (replaced by a TRY motif) or the NPxxY motif in transmembrane domain VII (replaced by NPxxC) (Matsumoto et al., 2000). SREB are principally expressed in the central nervous system but have also been found in several peripheral organs (Regard et al., 2008). To date, no endogenous ligand has been described for GPR27, but it has been suggested that it is involved in insulin promoter activity and secretion (Ku et al., 2012). The precise signaling events linking GPR27 and its effects on insulin are still elusive, and its function in the central nervous system remains unknown.
Given the putative role of GPR27 in metabolism and its high rate of conservation, we sought to identify surrogate ligands as pharmacological tools to decipher its signaling pathways and function. To find an optimal pathway to screen against, we investigated the receptor coupling to G proteins (Gs, Gi, and Gq) and arrestins. In a split firefly luciferase complementation assay, we demonstrated the ability of a chimeric GPR27V2 to recruit β-arrestin 2 in a ligand-independent fashion. We optimized this assay for screening and identified the first specific and selective surrogate agonists for GPR27 in a diversity-oriented synthesis library. Using these tools together with various orthogonal assays, we validated the coupling of the receptor to β-arrestin 2, but not to G proteins.
Materials and Methods
Materials.
All chemicals used were from Sigma-Aldrich (St. Louis, MO), unless stated otherwise. Chemical library DIVERSet (50,000 compounds), purchased hits, and analogs were from ChemBridge. PHAR169522 and PHAR034416 were purchased from Pharmeks (Moscow, Russia), and CXCL12 from PeproTech (Rocky Hill, NJ). The following commercially available antibodies were used for several applications: monoclonal anti-Flag clone M2 (F3165) from Sigma-Aldrich; anti-mouse IgG (H + L), F(ab′)2 fragment (4408, Alexa Fluor1 488 conjugate), anti-mouse IgG, horseradish peroxidase–linked (7076), anti–phospho-p44/42 mitogen-activated protein kinase (MAPK; ERK1/2) (Thyr202/Thyr204, D13.14.4E, rabbit, 4370), anti-p44/42 MAPK (ERK1/2) (rabbit, 4695), anti-rabbit IgG, and horseradish peroxidase–linked (7074) from Cell Signaling Technology (Danvers, MA). D-luciferin (sodium salt) was from Synchem (Felsberg/Altenburg, Germany), and coelenterazine h was from Regis Technologies (Grove, IL). Poly-D-lysine (P7280) was from Sigma-Aldrich.
Plasmids.
Human GPR27 (SREB1) and GPR85 (SREB2) were amplified from pcDNA3.1+ GPR27 or GPR85 (cDNA Resource Center, Bloomsburg, PA); GPR173 (SREB3) was amplified from human embryonic kidney 293 (HEK293) cell genomic DNA. Vasopressin V2 receptor (V2R) was amplified from human ORFeome (version 7.1, http://horfdb.dfci.harvard.edu/hv7/), β 2-adrenoceptor (β 2AR) from HEK293 cDNA, human GPR151 from peripheral blood mononuclear cell genomic DNA, and ACKR3 (CXCR7) from pBABE-ACKR3 (Chevigne et al., 2014). All receptors were cloned into the pIRESpuro vector (Clontech Laboratories, Mountain View, CA) (for stable transfections) and/or pcDNA3.1 (Invitrogen, Carlsbad, CA) (for transient transfections) after addition of the Flag epitope (DYKDDDDK) at the N terminus, preceded by the signal sequence KTIIALSYIFCLVFA (Guan et al., 1992) for SREB receptors, unless specified otherwise. GPR151, V2R, and ACKR3 were cloned directly into the firefly luciferase assay vector (described below). β-adrenergic receptor kinase 1 (GRK2) was amplified from cDNA from HEK293 and inserted into the pcDNA3.1 vector. Membrane-anchored GRK2 (GRK2m) was obtained through modification by polymerase chain reaction of the C terminus by a CAAX motif (Inglese et al., 1992; Oakley et al., 2006). SREB-V2 chimeras were obtained by replacement of the last 20 (GPR27), 14 (GPR85), or 19 (GPR173) amino acids by the C terminus of V2R (last 29 amino acids). Human β-arrestin 1 (Arrb1) was amplified from HEK293 cDNA, and rat β-arrestin 2 (Arrb2) from β-arrestin 2 green fluorescence protein (GFP; 35411; Addgene, Cambridge, MA). Fusions with firefly luciferase for complementation assay were made in a two-step cloning. First, linkers and firefly luciferase moieties [amino acid 1–415 (Fn)/413–549 (Fc)] described in (Takakura et al., 2012) were inserted in pIREShygro3/pIRESpuro vectors. In a second step, the GPCR/Arrb1/Arrb2 was added in frame to obtain pIREShygroFnLArrb1/Arrb2 and pIRESpuroGPCRLFc vectors. The pGloSensor-22F cAMP (cAMP GloSensor) plasmid was obtained from Promega (Madison, WI). Plasmids for alkaline phosphatase (AP)–transforming growth factor (TGF)-α shedding assay (pCAGGS/AP-TGF-α), chimeric/promiscuous Gα subunits (Gα q/s, Gα q/i1, Gα q/i3, Gα q/o, Gα q/z, Gα q/12, Gα q/13, and Gα 16 in pCAGGS), and dopamine D2 receptor (pCAGGS/D2) were a gift of A. Inoue (Graduate School of Pharmaceutical Science, Tohoku University, Sendai, Japan). For the NanoLuc Binary Technology (NanoBiT) system, receptors and β-arrestin 2 were cloned, respectively, into pNBe2 and pNBe3 vectors (Promega). All constructs were verified by sequencing.
Cell Culture and Transfection.
HEK293 cells were from American Type Culture Collection (Manassas, VA) and grown in Dulbecco’s modified Eagle medium (Lonza, Verviers, Belgium) containing 10% fetal bovine serum (Biochrom AG, Berlin, Germany), 1% penicillin and streptomycin (Lonza), and 1% L-glutamine (Lonza) at 37°C with 5% CO2. Cells were transfected at 70–80% confluency with XtremeGene 9 (Basel, Roche, Switzerland) in a 3:1 (reagent:DNA) ratio following manufacturer’s recommendations, unless stated otherwise. Stably transfected clones were obtained through limited dilution and selection of clones following epitope (Flag) labeling and analysis in flow cytometry.
Cell Surface Receptor Expression by Flow Cytometry.
Polyclonal HEK293.FlagGPR27 or HEK293.ssFGPR27 cells (1.4 × 105 cells/tube) were incubated with monoclonal anti-Flag M2 (1:1000) for 45 minutes at 4°C in fluorescence-activated cell sorter buffer [1% bovine serum albumin and 0.1% NaN3 in phosphate-buffered saline (PBS)]. After washing, cells were incubated with anti-mouse IgG (H + L), F(ab′)2 fragment (Alexa Fluor 488 conjugate; 1:1000) for 45 minutes at 4°C in the dark. Data were acquired on BD FACSCalibur (two lasers) and analyzed with BD CellQuest Pro (BD Biosciences, Franklin Lakes, NJ).
Visualization of Receptor Expression by Confocal Microscopy.
HEK293 cells grown on poly-D-lysine–coated coverslips in a 24-well plate were transfected with pcDNA3.1FlagGPR27 or pcDNA3.1ssFlagGPR27 (250 ng/well). Forty-eight hours post-transfection, cells were starved for 30 minutes in PBS (37°C). They were then placed on ice, washed once with ice-cold PBS, and blocked for 30 minutes with 2% bovine serum albumin in PBS on ice. After 1-hour incubation on ice with monoclonal anti-Flag M2 antibody (1:1000), cells were washed three times with ice-cold PBS and fixed in 4% formaldehyde for 15 minutes at room temperature. All subsequent steps were conducted at room temperature. Autofluorescence was quenched for 15 minutes with NH4Cl (50 mM in PBS), and cells were incubated with anti-mouse IgG (H + L), F(ab′)2 fragment (Alexa Fluor 488 conjugate; 1:1000), and 4′,6-diamidino-2-phenylindole (1 µg/ml) for 1 hour in the dark. After four washing steps with PBS, slides were mounted using Aqua Poly/Mount (Polysciences, Warrington, PA). Image acquisition was performed on a confocal microscope NIKON A1R (60× oil immersion objective; Tokyo, Japan).
Quantification of Inositol Monophosphate.
Changes in second-messenger inositol monophosphate were quantified on cell lysates using the IP-One enzyme-linked immunosorbent assay (ELISA) kit (Cisbio, Codolet, France), according to the manufacturer’s instructions. Briefly, HEK293 cells were seeded into a 24-well plate (100,000 cells/well) and, after overnight incubation, transfected with 250 ng pcDNA3.1ssFlagGPR27 or empty vector. Twenty-four hours post-transfection, culture medium was replaced by 200 µl provided stimulation buffer (without additional compounds when monitoring constitutive activity) and returned to the incubator for 1 hour. Cells were then lysed by adding 50 µl lysis reagent (2.5%) and further incubated for 30 minutes at 37°C. Finally, 50 µl cell lysate was transferred into the ELISA plate, and the assay was conducted according to the manufacturer’s instructions. Absorbance was read at 450 and 620 nm on an Infinite M200 Pro reader (TECAN, Männedorf, Switzerland).
Determination of cAMP Levels.
Variations of cAMP levels were measured with the GloSensor technique (Promega). For constitutive activity, HEK293 cells stably expressing the cAMP-sensitive luciferase, pGlo cells (Gilissen et al., 2015), were transiently transfected in poly-D-lysine–coated 96-well plates (50 ng/well). Twenty-four hours later, the medium was replaced by the assay buffer [Hanks’ balanced salt solution (HBSS; 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 10 mM HEPES, pH 7.4) containing 3-isobutyl-1-methylxanthin (300 µM) and D-luciferin (833 µM)], and luminescence was directly recorded for 30 minutes on a Centro XS3 LB 960 reader (Berthold Technologies, Bad Wildbad, Germany). For ligand stimulation assays, pGlo cells stably transfected with GPR27 were resuspended in the assay buffer and incubated 20 minutes at room temperature in the dark. They were then seeded in white 96-well plates (655074; Greiner Bio-One, Kremsmünster, Austria), and basal luminescence was recorded for 5 minutes. After adding the ligands, luminescence was recorded for 30 minutes before and after injecting forskolin (1 µM). Isoproterenol and CXCL12 acting on their endogenous receptors β 2AR and CXCR4 were used as controls for Gs- and Gi-mediated cAMP modulation.
Determination of Receptor Expression by ELISA.
Cells for the receptor quantification by ELISA were prepared in parallel to those for the β-arrestin 2 recruitment assay (constitutive activity). Cells were seeded in poly-D-lysine–coated 96-well plates, and 24 hours later transfected with various amounts of receptor plasmids (0–50 ng). The next day, cells were tested in both assays. For the ELISA, cells were washed and fixed for 15 minutes in 4% formaldehyde at room temperature. After blocking and permeabilization for 30 minutes in PBS containing 2% bovine serum albumin and 0.12% Triton X-100, cells were incubated with anti-Flag M2 antibody (1:3000) for 1 hour. After extensive washing with PBS, cells were incubated for 1 hour with anti-mouse horseradish peroxidase–linked antibody (1:3000). After several washing steps, cells were left in 30 µl PBS. Chemiluminescence was recorded for 15 minutes after addition of 30 µl Pierce ECL Western blot substrate (Thermo-Fisher Scientific, Waltham, MA) on a Centro XS3 LB 960 reader (Berthold Technologies). Results are presented as the ratio of the area under the curve divided by that of untransfected cells.
Measurements of Intracellular Ca2+ Mobilization.
Intracellular calcium was monitored with a calcium-sensitive bioluminescent fusion protein consisting of aequorin and GFP (G5A) (Baubet et al., 2000). The assay has been conducted according to previous description (Hanson et al., 2013; Gilissen et al., 2015). Briefly, cells overexpressing G5A were seeded in poly-D-lysine–coated transparent-bottom 96-well plates. After an overnight incubation, cells (70% confluent) were transfected with equal amounts (25 ng receptor- and 25 ng Gα subunit-encoding plasmids) of either pcDNA3.1ssFlagGPR27 or pCAGGS.DRD2 and empty vector or chimeric/promiscuous Gα subunits (Gα q/s, Gα q/i1, Gα q/i3, Gα q/o, Gα q/z, Gα q/12, Gα q/13, and Gα 16 in pCAGGS). Twenty-four hours later, medium was replaced by 50 µl assay buffer (HBSS supplemented with 10 mM glucose) containing 5 µM coelenterazine h (Regis Technologies, Grove, IL) and plates were incubated for 1 hour in the dark at 37°C. After replacing this buffer by 100 µl assay buffer supplemented with 1.8 mM CaCl2, plates were transferred to a Centro XS3 LB 960 reader (equipped with two dispensers; Berthold Technologies). After following the basal luminescence for 3 seconds (200-ms integration), ligands were added and luminescence was measured for an additional 25 seconds (200-ms integration).
Measurement of TGF-α Shedding.
HEK293 cells grown in poly-D-lysine–coated transparent-bottom 96-well plates (80% confluent) were transfected with pCAGGS/AP-TGF-α, receptor (pcDNA3.1ssFlagGPR27 or pCAGGS.DRD2), and empty vector or chimeric/promiscuous Gα subunits (Gα q/s, Gα q/i1, Gα q/i3, Gα q/o, Gα q/z, Gα q/12, Gα q/13, and Gα 16 in pCAGGS) in a 5:2:1 ratio (AP-TGF-α: receptor: Gα subunit; total DNA amount/well: 50 ng). Twenty-four hours later, medium was replaced with 100 µl HBSS (37°C). After incubating 10 minutes at room temperature, buffer was replaced by 50 µl HBSS and plates were returned to the incubator for 30 minutes. After stimulating the cells (by adding 50 µl 2× ligands in HBSS), plates were further incubated 1 hour at 37°C. Then, 80 µl conditioned medium was transferred into a new flat-bottom 96-well plate. When both plates had cooled down to room temperature, 80 µl freshly prepared AP solution (1.2 ml Tris-HCl 2 M, pH 9.5, 0.2 ml NaCl 4 M, 0.2 ml MgCl2 1 M, 18.4 ml H20, 200 µl pNPP 1 M) was added into each well. Optical density at 405 nm was measured directly (measure 1) and after incubation for 45 minutes (measure 2) on an Infinite M200 Pro reader (TECAN). After subtracting measure 1 from measure 2, and calculating the total AP activity by adding the obtained optical density(cells) and optical density(conditioned medium), shedding efficiency was presented as the percentage of AP activity in the conditioned medium.
Measurement of β-Arrestin Recruitment by Firefly Luciferase Complementation.
For constitutive β-arrestin 2 binding, HEK293 cells stably expressing β-arrestin 2 fused with the N-terminal part of the firefly luciferase (HEK293.FnLArrb2) were seeded in poly-D-lysine–coated 96-well plates (30,000 cells/well) and transfected with different amounts (0–50 ng) of the plasmid DNA coding for the receptor fused with the C-terminal part of the luciferase (GPCRLFc). After overnight incubation, medium was replaced with phenol red–free Dulbecco’s modified Eagle medium (Lonza), and, after addition of D-luciferin (500 µM), luminescence was measured on a Centro XS3 LB960 plate reader (Berthold Technologies). The same protocol was used to assess the influence of GRKs, where stable cell lines (overexpressing GPR27 or GPR27V2) were transfected with 50 ng plasmid DNA containing the different GRK coding sequence.
Cells for screening were prepared by seeding HEK293.FnLArrb2.GPR27V2LFc cells into white 96-well plates (655083; Greiner Bio-One) (35,000 cells/100 µl/well) and incubating the plates overnight at 37°C with 5% CO2 atmosphere. Cells (at 95% confluence) were stimulated for 10 minutes at room temperature by adding 1 µl 100× concentrated dimethyl sulfoxide compound stock solutions (using the liquid handling platform Freedom Evo 150 and 200; TECAN) to reach a final concentration of 15.5 ± 3.1 µM (mean ± S.D.; mol. wt. of compounds: 334.6 ± 64.2 g/mol). Luminescence was read after addition of D-luciferin (500 µM) on a Centro XS3 LB960 plate reader (Berthold Technologies). For concentration–response curves, cells resuspended in HBSS (130,000 cells/well) were stimulated by ligands at indicated concentrations. The same protocol was used for β-arrestin 1 binding after a transient transfection of both the β-arrestin 1– and receptor-encoding plasmids into HEK293 cells in a 1:1 ratio (total DNA amount = 4 µg/60 cm2).
β-Arrestin 2 Recruitment by NanoLuc Luciferase Complementation.
We followed the manufacturer instructions and tested several plasmid DNA dilutions for the NanoBiT assay (Promega). The optimal DNA mix for transfection in this assay was constituted of 5% pNBe2.GPR27V2, 5% pNBe3.Arrb2, and 90% empty pcDNA3.1. For competition experiments with wild-type β-arrestin 2, the proportion of empty vector was reduced to include 15% or 50% pcDNA.Arrb2 (300% or 1000% compared with pNBe3.Arrb2). Twenty-four hours post-transfection, cells were detached and incubated in HBSS supplemented with the substrate for 45 minutes at 37°C. After seeding in a white 96-well plate (100,000 cells/well), cells were stimulated by adding 1 µl 100× ligand solutions (in dimethyl sulfoxide) and luminescence was recorded for 40 minutes (Centro XS3 LB960 or Mithras LB940; Berthold Technologies). Results are expressed as the ratio of the areas under the curve.
Measurement of β-Arrestin 2 Recruitment by β-Galactosidase Complementation.
The PathHunter eXpress GPR27 CHO-K1 β-arrestin assay (DiscoverX, Fremont, CA) uses split β-galactosidase to monitor the recruitment of β-arrestin 2 to GPR27. CHO-K1 cells stably expressing β-arrestin 2 fused to enzyme acceptor of β-galactosidase and GPR27 fused to the ProLink donor peptide were seeded 48 hours before the experiment in 96-well plates. After stimulating the cells by adding 1 µl 100× concentrated dimethyl sulfoxide compound stock solutions, plates were incubated 90 minutes at 37°C. PathHunter detection reagent was added, and chemiluminescence was recorded on a Centro XS3 LB960 plate reader (Berthold Technologies).
β-Arrestin 2–GFP Translocation Assay.
HEK293 cells were transfected in a 24-well plate with 200 ng pcDNA3.1GPR27V2 or pNBe2.GPR85V2 and 40 ng β-arrestin 2-GFP (5:1). Twenty-four hours post-transfection, cells were seeded into poly-D-lysine–coated CELLview cell culture dishes (four compartments; Greiner Bio-One) to reach 40–50% confluency after overnight incubation. Prior to the experiment, medium was replaced by HBSS. Stimulation and imaging were performed on a confocal microscope NIKON A1R at room temperature using a 60× oil immersion objective.
Determination of ERK Phosphorylation.
HEK293 cells seeded in 20-cm2 culture dishes were transiently transfected with pcDNA3.1ssFlagGPR27 or empty vector (MOCK). Twenty-four hours later, cells were starved with medium containing 1% fetal bovine serum overnight. After incubating with the indicated ligands for 10 minutes at 37°C, cells were immediately put on ice and lysed with ice-cold radioimmunoprecipitation assay buffer (25 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS; pH 7.6) supplemented with protease inhibitors and phosphatase inhibitors (Roche, Basel, Switzerland). Cell lysates were analyzed by SDS-PAGE electrophoresis, followed by immunoblotting. ERK1/2 phosphorylation was detected with a rabbit monoclonal phospho-p44/42 MAPK antibody (1:2000), and total ERK1/2 with a rabbit monoclonal p44/42 MAPK antibody (1:1000). The membranes were then probed with corresponding horseradish peroxidase–conjugated secondary antibody (1:2000).
Data Analysis and Hit Selection Criteria.
Concentration–response curves were fitted to the four-parameter Hill equation using the least-squares method (GraphPad Prism, version 5.0 for Windows; GraphPad, La Jolla, CA).
Compounds were considered as hits in primary or secondary screening when fulfilling these two criteria: 1) active on GPR27V2 and inactive on MOCK (GPR151), in which activity is defined as a ratio > (mean ratio(VEH) + 3 S.D.(VEH)); 2) ratio(GPR27V2) > (ratio(MOCK) + 3 S.D.(VEH)). Ratio represents relative light units(compound)/relative light units(VEH).
Results
GPR27 Shows No Detectable Constitutive Activity in G Protein Pathways upon Heterologous Expression in HEK293 Cells.
To establish a cellular model for GPR27, a Flag epitope–tagged version of human GPR27 (Fig. 1A) was transfected and overexpressed in HEK293 cells. Analysis by flow cytometry (Fig. 1B) and immunofluorescence of transiently transfected cells (Fig. 1C) revealed only modest cell surface expression. To increase receptor export to the membrane compartment, a cleavable signal sequence (Guan et al., 1992) was added at the N terminus of the receptor (Fig. 1A). Flag-tagged GPR27 preceded by this signal sequence showed marked increase of surface expression (Fig. 1, B and C) and is referred to in this study as GPR27.

Cellular expression of heterologously expressed GPR27 and constitutive signaling through Gi, Gs, and Gq proteins. (A) DNA constructs for expression of GPR27 preceded at the N terminus by a Flag-tag or a cleavable signal sequence (ss) and the Flag-tag. (B) Flow cytometry analysis of surface Flag-tagged receptor expression in polyclonal HEK293.FlagGPR27 (red) and HEK293.ssFlagGPR27 (blue) compared with cells labeled with secondary antibody only (black). (C) Visualization of tagged receptors on cell surface 48 hours after transfection of Flag-tagged GPR27 with or without the signal sequence (ss) by immunofluorescence on nonpermeabilized cells. Scale bars, 15 µm. (D and E) Effect of the transfection of GPR27 (compared with MOCK, empty plasmid) on basal cAMP (living cells; GloSensor technology) and inositol monophosphate (IP1) levels (cell lysates; ELISA). Data represent the mean ± 95% confidence interval (n ≥ 5).
Information about coupling of orphan GPCR to intracellular partners is critical for choosing the right screening assay/pathway. Therefore, we aimed at measuring constitutive activity of GPR27 in G protein pathways (Gs, Gi, and Gq). The overexpression of GPR27 had no significant effect on either cAMP levels (Fig. 1D) or inositol monophosphate accumulation (Fig. 1E), suggesting that there is no or low constitutive coupling to these signaling pathways in HEK293 cells.
GPR27 Does Not Markedly Recruit β-Arrestin 2 Either in Basal Condition or in the Presence of Membrane-Anchored GRK2, in Contrast to the Chimeric GPR27V2.
Next, we used an in-house firefly luciferase complementation assay to evaluate the basal recruitment of β-arrestin 2 to GPR27 (Fig. 2A). The signal is represented as a function of receptor expression because luciferase activity increases with the relative expression of luciferase moieties, even in the absence of specific association between two labeled proteins. In this assay, no marked ligand-independent interaction between GPR27 and β-arrestin 2 could be observed (Fig. 2B), when compared with V2R and ACKR3, used as controls with respectively low and high constitutive β-arrestin 2 binding (Barak et al., 2001; Luker et al., 2010).

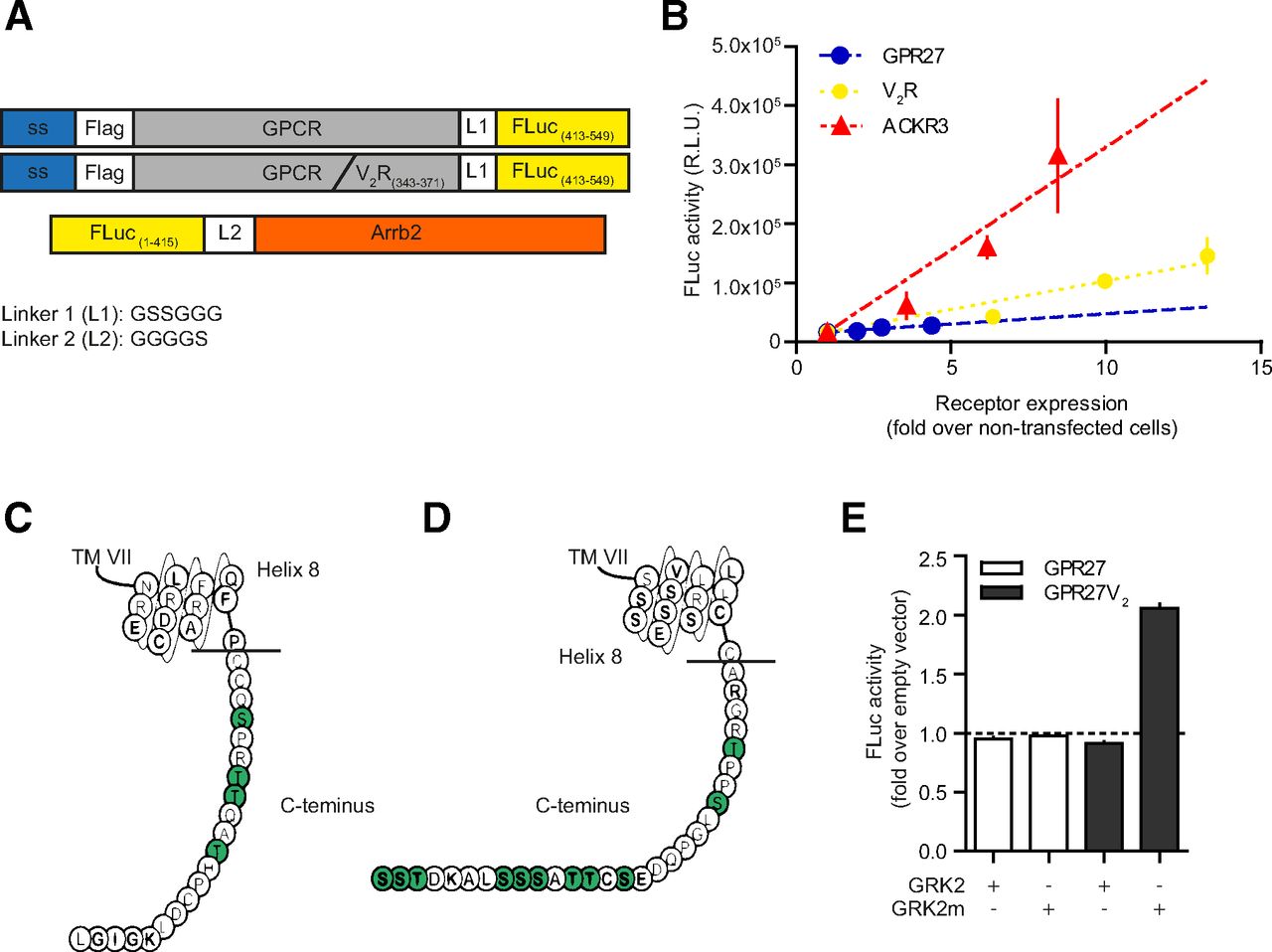
β-arrestin 2 binding to GPR27 and the chimeric GPR27V2. (A) DNA constructs for the firefly luciferase (FLuc) complementation assay for wild-type receptors or GPCR-V2 chimera. (B) Constitutive (ligand-independent) β-arrestin 2 binding. V2R and ACKR3 were used as control receptors with, respectively, low and high constitutive activity. Parental HEK293 cells stably expressing the β-arrestin 2 construct for firefly luciferase assay were transfected with the receptor construct and assayed in the firefly luciferase complementation assay. On duplicate plates, receptor expression level was quantified by ELISA on permeabilized cells. Results are presented as a function of receptor expression. Data represent the mean ± S.E.M. (n = 3). (C and D) Helix 8 and C terminus of GPR27 (C) and V2R (D), adapted from www.gpcrdb.org (Munk et al., 2016). Ser/Thr residues of the C terminus are colored in green; the horizontal line indicates the cleaving position for C terminus exchange and chimera generation. (E) Effect of GRK2 and membrane-anchored GRK2m overexpression on β-arrestin 2 recruitment to GPR27 and GPR27V2 by firefly luciferase complementation. Data represent the mean ± S.E.M. (n = 5).
We reasoned that the absence of signal in both G protein and arrestin pathway could be the consequence of absent or low constitutive activity. To examine whether β-arrestin 2 would bind to an activated receptor, we cotransfected a GRK2m that is thought to increase receptor phosphorylation (Inglese et al., 1992; Oakley et al., 2006). No increased β-arrestin 2 binding was observed for GPR27 in our assay (Fig. 2E). We therefore hypothesized that GPR27 could be a class A receptor with regard to β-arrestin recruitment due to the limited number of putative Ser/Thr phosphorylation sites (4 Ser/Thr in the C terminus) (Fig. 2C). To obtain a more sustained interaction between the receptor and β-arrestin 2, we replaced the C terminus of GPR27 with that of the archetypical arrestin class B V2R that contains several Ser/Thr clusters (Fig. 2D). We repeated the experiment with and without GRK2m on the GPR27V2 chimera and observed a robust increase in β-arrestin 2 binding for the chimeric GPR27V2 (Fig. 2E).
A Luciferase-Based Screening of a Small-Molecule Library on GPR27V2 Unveils Two Active Compounds with a Common Chemical Scaffold.
Having confirmed the ability of GPR27V2 to interact with β-arrestin 2, we implemented a screening-compatible protocol for the firefly luciferase complementation assay (Supplemental Fig. 1). The validation of the assay was not possible on GPR27 due to the absence of positive control; therefore, the assay performance was determined on the β 2AR, and a Z′ factor (Zhang et al., 1999) of 0.79 was obtained (Supplemental Fig. 1). With our semiautomated protocol using the firefly luciferase complementation assay, we screened on GPR27V2 6160 compounds from a diversity-oriented synthesis library (DIVERSet; ChemBridge). Final compound concentration and mol. wt. were, respectively, 15.5 ± 3.1 µM and 334.6 ± 64.2 g/mol (mean ± S.D.). The screening was performed in parallel with an unrelated receptor (GPR151) used as MOCK condition. Compounds modifying the signal on both cell lines were discarded. After the primary screening, 141 compounds met our criteria (active only on GPR27V2 with a threshold based on mean + 3× S.D. of vehicle-treated wells) and were tested in a secondary screening performed with the same assay in triplicate (Fig. 3A). Two hits were identified, ChemBridge ID 5128535 and 5217941. Interestingly, they shared a N-sulfamoylphenyl-2,4-dichlorobenzamide scaffold (Fig. 3B). Response curves over a wide range of concentrations revealed concentration-dependent activity on GPR27V2 while being inactive on MOCK-transfected cells (Fig. 3C).

Identification of two agonists for GPR27V2. (A) Following the initial screening using the β-arrestin 2–based firefly luciferase complementation assay, 141 compounds were retested in triplicate at the same concentration (15 µM) on both GPR27V2 and MOCK cells. (B) Chemical structure, PubChem CID, and (C) concentration–response curves of the two active compounds. Results are expressed as the ratio over vehicle-treated cells. Data represent the mean ± S.E.M. (n = 3).
N-Phenyl-2,4-Dichlorobenzamide Agonist Activity on GPR27V2-Mediated β-Arrestin 2 Recruitment Is Structure-Dependent.
Sixty-five commercially available two- and three-dimensional analogs of 5128535 (notably diversely substituted benzamides) were tested at different concentrations on GPR27V2 to assess structure dependency of the activity toward the target (Supplemental Table 1). Concentration–response curves were performed on a subset of analogs sharing the N-phenyl-2,4-dichlorobenzamide scaffold of the screening hits (Fig. 4A). None of them was more potent than 5128535 (pEC50: 6.38 ± 0.13) in the firefly luciferase complementation assay, but close derivatives displayed varying potencies and efficacies on GPR27V2, suggesting specific activity for this family of compounds (Fig. 4A; Supplemental Table 1).

Structure-related activity and selectivity of the identified compounds. (A) Common N-phenyl-2,4-dichlorobenzamide scaffold and effect of diverse substituents (R) on compound activity in the β-arrestin 2–based firefly luciferase complementation assay. Data points represent the mean ± S.E.M. of three independent experiments. Emax is expressed as the percentage of 5128535 maximal activity. (B) Selectivity of the identified compounds over the other SREB members in the firefly luciferase complementation assay. Results are normalized as a ratio over vehicle-treated cells. Data points represent the mean ± S.E.M. of three independent experiments performed in quadruplicate.
5128535 and 5217941 Are Selective for GPR27V2 in the SREB Family.
We assessed the selectivity of our compounds by examining their ability to activate the closely related GPR85 and GPR173 in the same assay. We generated chimeric receptors fused with the V2R tail and tested 5128535 and 5217941. Both were unable to induce recruitment of β-arrestin 2 to GPR85V2 or GPR173V2 even at the highest concentration tested (Fig. 4B).
Orthogonal Confirmation of N-SulfamoylpPhenyl-2,4-Dichlorobenzamide–Induced β-Arrestin 2 Recruitment by NanoBiT Technology and Confocal Microscopy.
To corroborate the validity of our β-arrestin recruitment results obtained with the firefly luciferase complementation assay, we tested both ligands in two alternative systems for the detection of β-arrestin recruitment (orthogonal confirmation). The first one was the NanoBiT protein:protein interaction system from Promega (Cannaert et al., 2016; Dixon et al., 2016). This assay is based on the complementation of two subunits, Large BiT (LgBiT; 17.6 kDa) and Small BiT (SmBiT, 11 amino acids), fused to the two proteins of interest. Upon interaction, the bright signal of NanoLuc luciferase is emitted, and, due to the reversibility of the weak interaction between LgBiT and SmBiT, rapid associations/dissociations can be detected. By fusing GPR27V2 with SmBiT and β-arrestin 2 with LgBiT (Fig. 5A), we confirmed the activity of both ligands in the low micromolar range (Fig. 5B; EC50 of 834 nM and 1.3 µM for 5128535 and 5217941, respectively). In this assay, in contrast to the firefly luciferase complementation system, 5217941 showed a reduced maximal efficacy compared with 5128535 (Fig. 5B). The difference in maximal efficacy was probably due to different luciferases, the Nanobit system being more reversible than the split Firefly luciferase.

Orthogonal confirmation of the β-arrestin 2 recruitment by NanoLuc complementation. (A) DNA constructs for the NanoBiT technology. (B) Concentration–response curves on GPR27V2 in the NanoBiT assay. (C) Competition of cotransfected wild-type Arrb2 in the NanoBiT assay on GPR27V2. The amount of wild-type (untagged) β-arrestin 2 is expressed as a percentage of the functionalized β-arrestin 2 (tagged with LgBiT). Results are normalized as a ratio over vehicle-treated cells. Data points represent the mean ± 95% confidence interval (n ≥ 4).
To further validate the specificity of the β-arrestin–mediated increase of the complementation signal, increasing amounts of plasmid DNA containing the untagged β-arrestin 2 sequence were cotransfected as an in-assay control, which competed with LgBiT-functionalized β-arrestin for receptor binding (Fig. 5C).
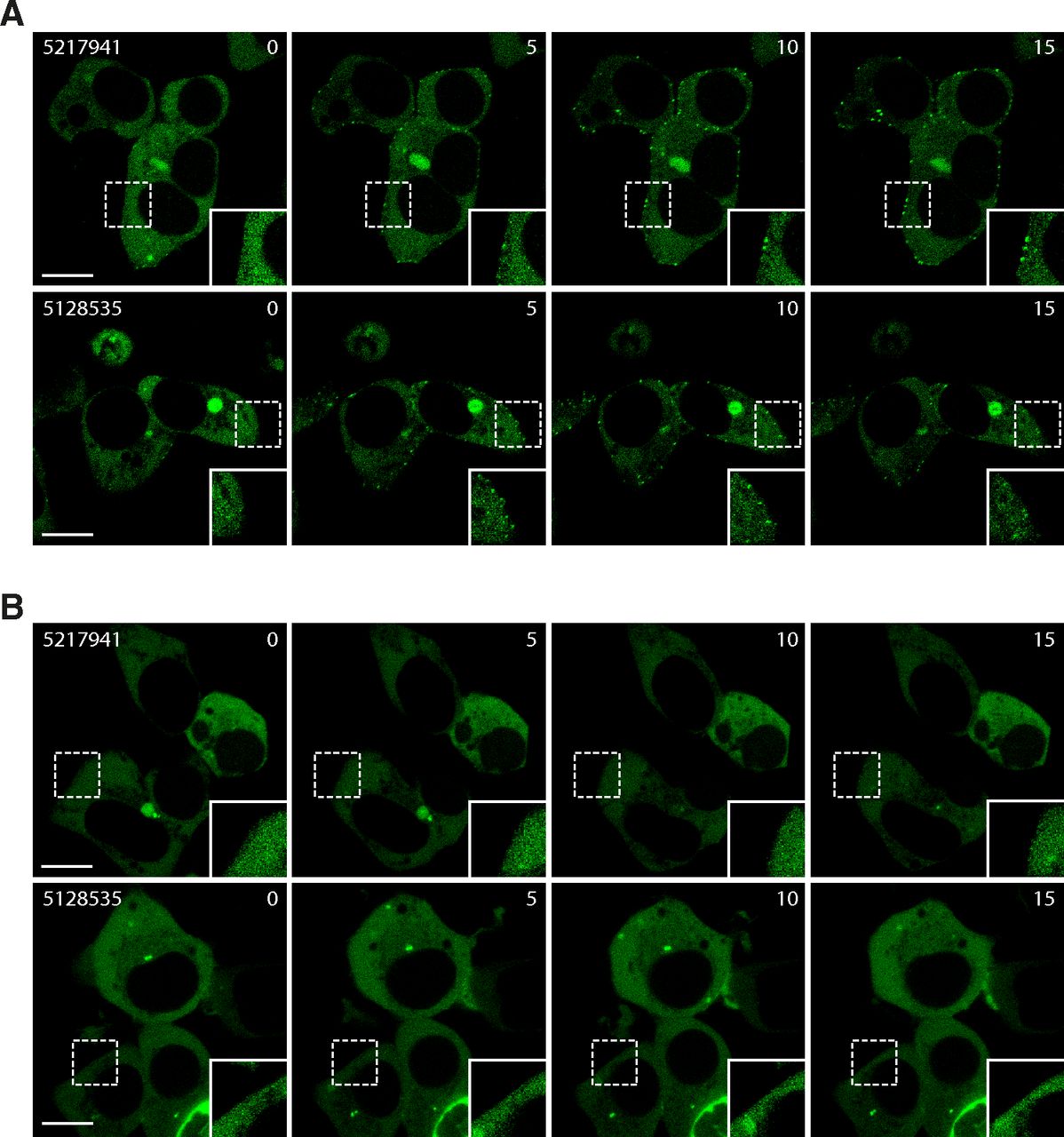
Furthermore, the β-arrestin 2 recruitment was evaluated by confocal microscopy on living cells using a β-arrestin 2–GFP fusion (Shenoy and Lefkowitz, 2005), translocating from the cytoplasm to the membrane/endosomes upon receptor stimulation (Barak et al., 1997). We observed the formation of small endosome-like clusters near the membrane in the presence of 5217941 (50 μM) or 5128535 (46 µM) and the receptor GPR27V2 (Fig. 6A). No translocation was observed for cells transfected with GPR85V2 (Fig. 6B).

Orthogonal confirmation of the β-arrestin 2 recruitment by confocal microscopy. Translocation of β-arrestin 2-GFP to GPR27V2 (A) or GPR85V2 (B) upon stimulation with 5217941 (50 μM) or 5128535 (46 µM) for 0–15 minutes. Images are representative of at least three experiments. Scale bar, 10 µm.
Pharmacological Activation of GPR27 Does Not Initiate G Protein–Mediated Signaling in the Range of Tested Concentrations, but Recruits β-Arrestin 2.
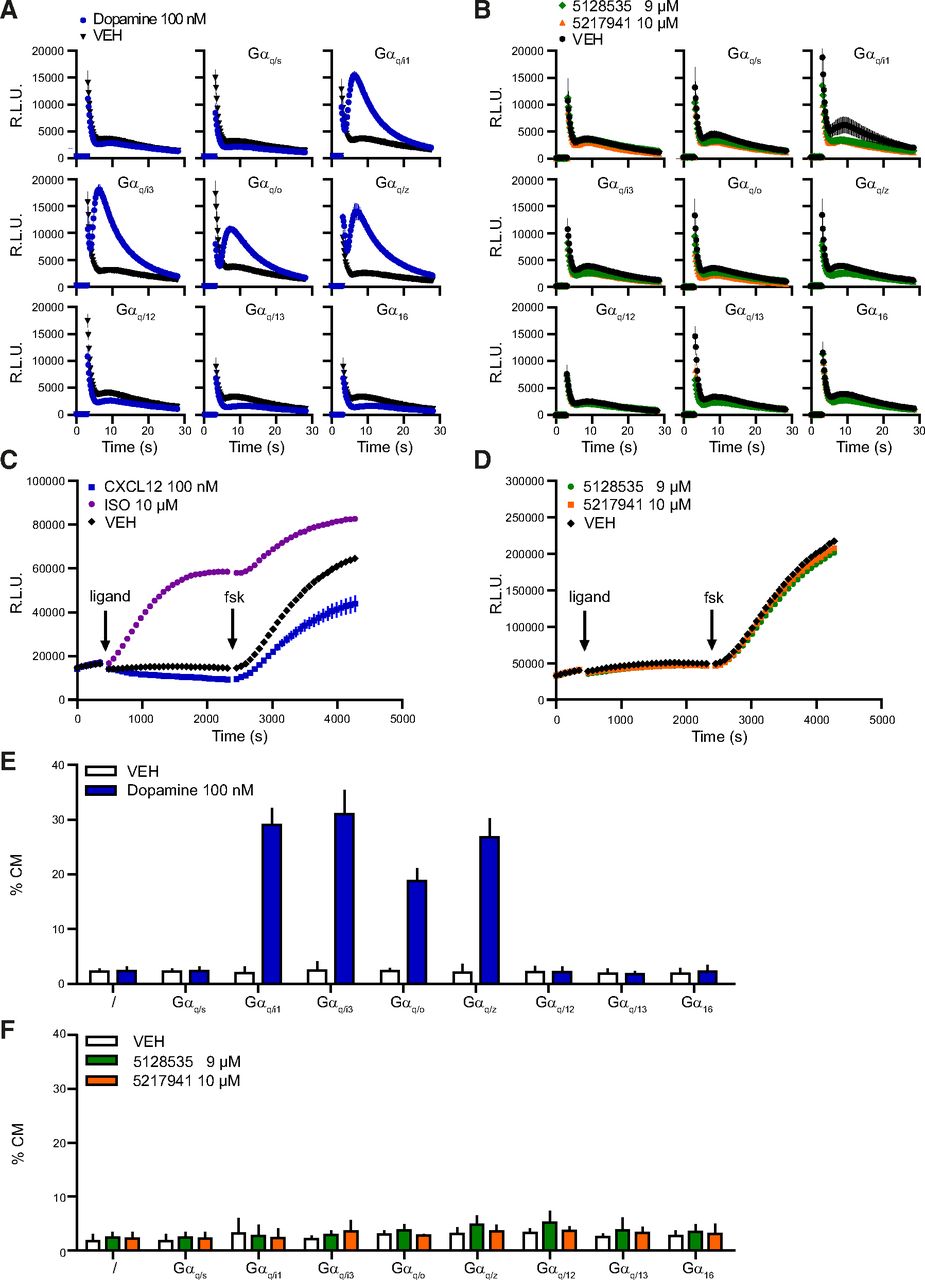
We examined whether the compounds were able to induce signaling through GPR27 with unmodified C terminus. In an aequorin-based Ca2+ assay, monitoring directly the Gq-related signaling pathway and indirectly other G protein pathways by using corresponding chimeric G proteins, no intracellular calcium release could be detected upon stimulation of HEK293 cells overexpressing GPR27 with 5128535 and 5217941 (Fig. 7B). We confirmed the validity of the assay with the D2 receptor used as a positive control. As expected, HEK293 cells transiently transfected with D2 released intracellular Ca2+ upon dopamine treatment in presence of the chimeric G proteins of the pathway it couples to (Fig. 7A) (Masuho et al., 2015). The experiments on GPR27 and D2 were performed in parallel, the same day, with the same batch of HEK293 cells. Expression of GPR27 at the membrane following transient transfection with our protocol was verified by fluorescence microscopy following FLAG epitope labeling.

GPR27 G protein pathway analysis in response to ChemBridge ID 5128535 and 5217941. (A and B) Calcium release in the absence or presence of different chimeric/promiscuous Gα subunits in the aequorin–GFP assay for the control receptor D2 (A) or GPR27 (B). Compounds were injected after 3 seconds. Data represent the mean ± S.E.M. (n = 3). (C and D) Effect on cAMP levels in cells stably expressing the cAMP GloSensor and GPR27 of control compounds acting on their respective endogenous receptors (C) or 5128535 and 5217941 (D). Data represent the mean ± S.E.M. [(C) n = 4, or (D) 8]. (E and F) TGF-α shedding assay in the absence or presence of eight chimeric/promiscuous Gα subunits for the control receptor D2 (E) and GPR27 (F). Results are expressed as the percentage of alkaline phosphatase activity in the conditioned medium (CM). Data points represent the mean ± 95% confidence interval (n ≥ 3). %CM, percentage of alkaline phosphatase activity in the conditioned medium; fsk, forskolin; R.L.U., relative luminescence units; VEH, vehicle.
Moreover, no effect of 5128535 and 5217941 was observed on cells overexpressing GPR27 in a cAMP assay to examine the Gs- or Gi-related pathway without using chimeric or promiscuous G proteins (Fig. 7D), in contrast to CXCL12 and isoproterenol used as controls for Gi- and Gs-mediated cAMP modulation by acting on their endogenously expressed receptors (Fig. 7E).
Furthermore, no effect of 5128535 or 5217941 could be observed in the Gq/G12/13-related AP-TGF-α shedding assay (Inoue et al., 2012), in the absence or presence of eight chimeric/promiscuous G proteins (Fig. 7F). Similar to the calcium assay, the dopamine D2 receptor was used as a control (Fig. 7E). D2 and GPR27 experiments were performed the same day with the same batch of HEK293 cells.
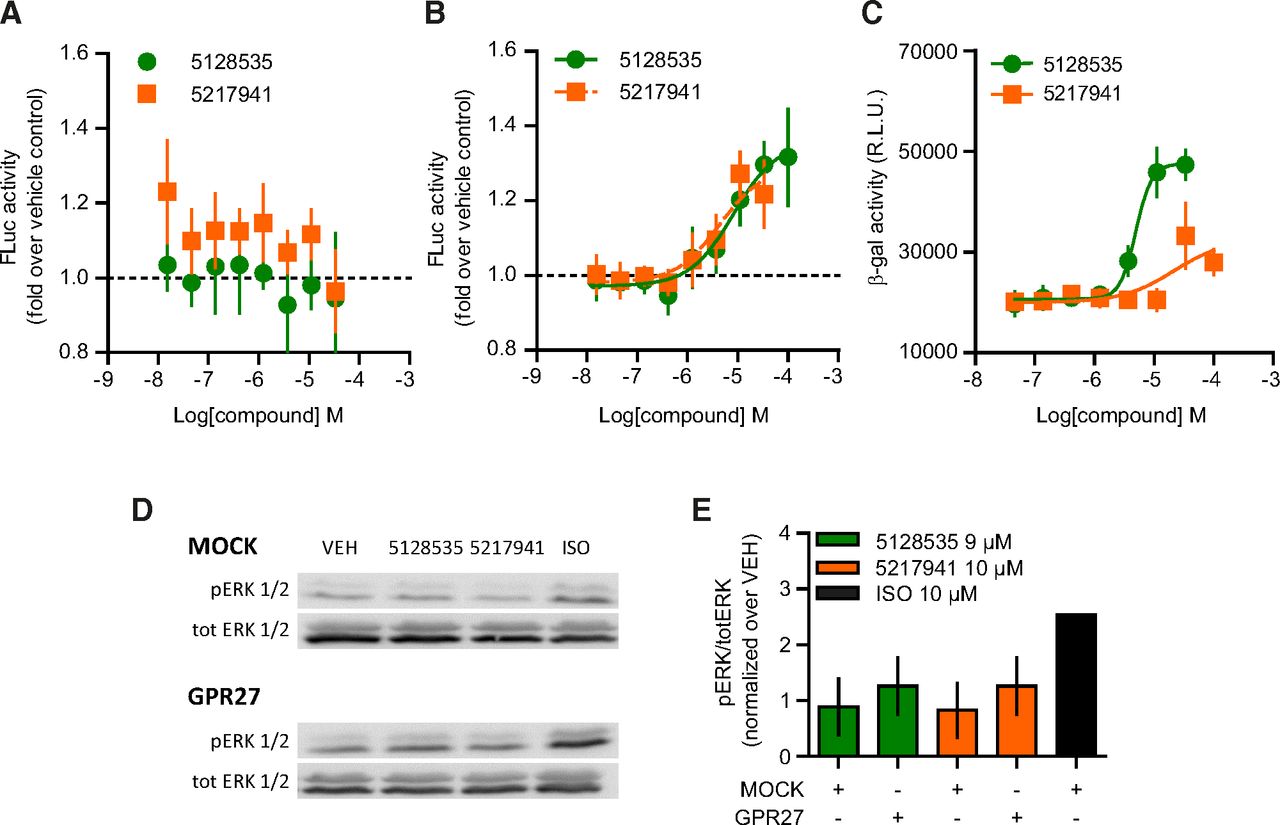
We investigated the β-arrestin recruitment to the unmodified wild-type GPR27 in the firefly luciferase complementation assay. Whereas no recruitment of β-arrestin 1 could be detected (Fig. 8A), the two compounds were able to recruit β-arrestin 2 with a similar potency (Fig. 8B; pEC50: 5.10 ± 0.16 and 5.40 ± 0.20 for 5128535 and 5217941, respectively) but with lower amplitude compared with GPR27V2 (Fig. 4B). In an alternative assay based on β-galactosidase complementation upon recruitment of β-arrestin 2 to GPR27 in CHO-K1 cells, both ligands concentration-dependently increased the signal (Fig. 8C), although the maximal efficacy and potency were more important for 5128535 (pEC50: 5.34 ± 0.03) compared with 5217941.
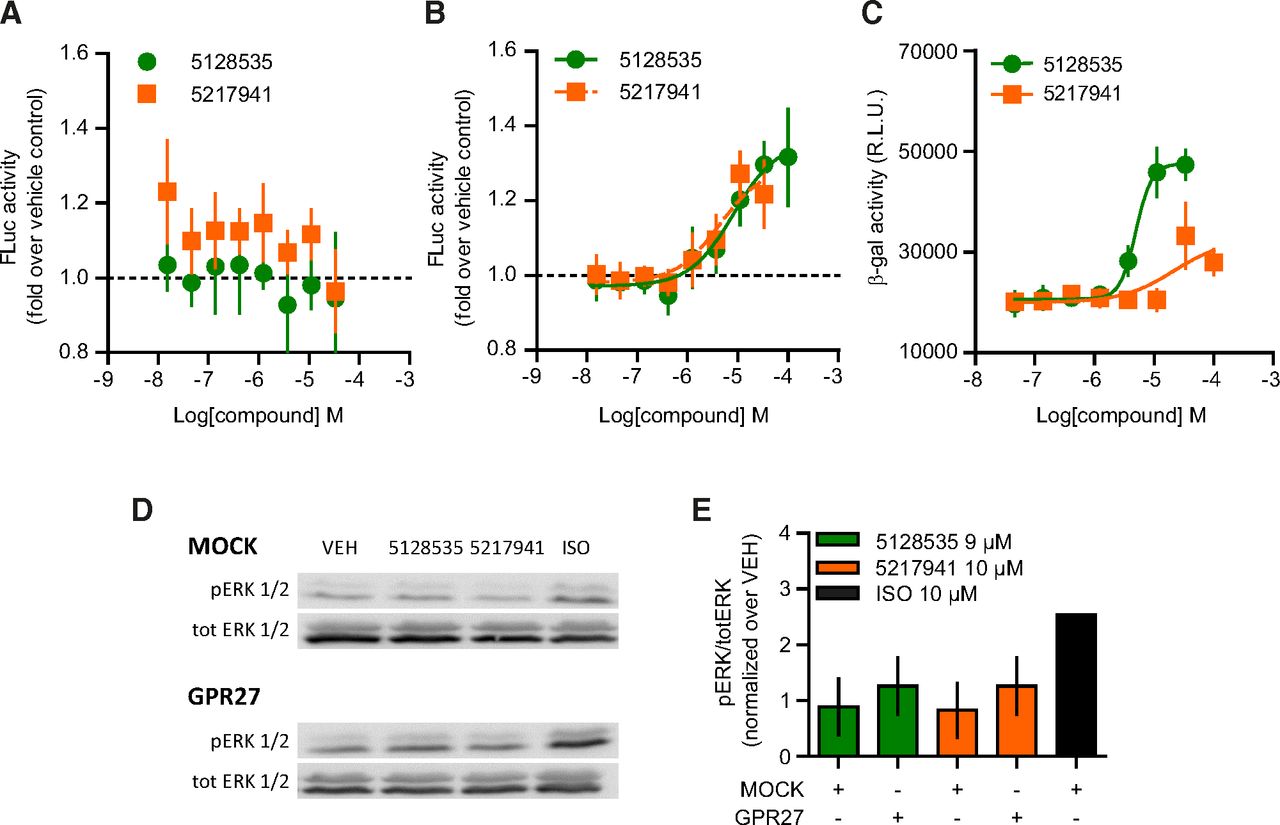
GPR27-mediated β-arrestin 2 recruitment and ERK1/2 phosphorylation. (A and B) β-arrestin 1 (A) and β-arrestin 2 (B) recruitment to GPR27 by firefly luciferase complementation. Results are normalized as the ratio over vehicle-treated cells. Data points represent the mean ± 95% confidence interval (n ≥ 4). (C) β-arrestin 2 recruitment to GPR27 by β-galactosidase complementation. Data points represent the mean ± 95% confidence interval (n ≥ 4). (D and E) Phosphorylation of ERK1/2 by Western blot on lysates of HEK293 cells transiently transfected with empty vector (MOCK) or GPR27 and stimulated for 10 minutes at 37°C (10 µM and 9 µM for 5217941 and 5128535, respectively). Isoproterenol (ISO, 10 µM) acting on endogenous receptors was used as a positive control. Data represent the mean ± 95% confidence interval (n = 3). pERK, phospho-ERK; VEH, vehicle.
β-arrestin binding to GPCR has been reported to promote the activation of diverse intracellular kinases, notably the MAPK pathway (Shenoy et al., 2006). We investigated the phosphorylation of ERK1/2 upon exposure to 5128535 and 5217941 (Fig. 8, D and E). No increase of ERK1/2 phosphorylation above the background noise could be detected (Fig. 8E).
Discussion
The present work focuses on GPR27, an orphan GPCR hypothesized to have important functions due to its high level of conservation through vertebrate evolution. The published literature on this receptor is scarce, and attempts to identify ligand for GPR27 have failed to date (Matsumoto et al., 2000). Several reasons can be postulated to explain the absence of known endogenous ligands for the remaining orphan GPCRs (Levoye and Jockers, 2008; Davenport et al., 2013). First, the isolation of the endogenous ligands from tissue extracts can be challenging due to their tightly regulated and transient production or high instability. Secondly, screening campaigns of tissue extracts may have failed due to an incomplete receptor system in cell-based assays, such as missing interacting partners like other receptors (obligatory heterodimers) or the use of assays based on signaling pathways irrelevant for the scrutinized receptor. Thirdly, the orphan receptor may have ligand-independent functions due to high level of constitutive activity or act as a scaffold protein.
Currently, this shortage of endogenous ligands precludes research that could be undertaken on many orphan GPCRs. One attractive and validated option to overcome this issue is to identify and use surrogate ligands as pharmacological tools to gain information on the receptor function, which facilitates the quest for endogenous ligands (Davenport et al., 2013; Roth and Kroeze, 2015; Ngo et al., 2016). This strategy has recently been successfully applied, for example, to the orphan receptors GPR17, GPR65, and GPR68 (Hennen et al., 2013; Huang et al., 2015). Therapeutic arsenal can also benefit from surrogate ligands if they become experimental drugs, as exemplified by the administration to humans of the bombesin receptor BB3 agonist MK-5046 for the treatment of obesity (Reitman et al., 2012; Civelli et al., 2013).
The first step in the identification of a ligand for a receptor is to select an optimal assay. One of the current trends is to apply universal detection techniques that would be suitable regardless of the signaling pathways a receptor is coupled to (Oakley et al., 2002; Verkaar et al., 2008; Zhu et al., 2008). These generic assays are based for instance on promiscuous G proteins (e.g., G16) or label-free technologies, and have proven highly successful for some receptors (Hennen et al., 2013). However, many exceptions and atypical GPCRs are regularly described, such as receptors being unable to couple to promiscuous G proteins (Kostenis, 2001). In addition, label-free technologies such as the measurement of cell impedance are more challenging for the screening of large libraries due to the important rate of false-positive compounds that require many follow-up assays. Because the assay choice and development have a critical impact on screening campaigns (Kenakin, 2009), we reasoned that an assay specifically optimized and validated for GPR27 would increase the odds of successful screening. Choosing an adequate assay for orphan receptors is particularly challenging due to the lack of knowledge on its signaling pathways. Therefore, we conducted a thorough characterization of the molecular pharmacology of GPR27.
First, we analyzed the G protein signaling pathways downstream of GPR27 by focusing on its ligand-independent activity in a heterologous expression system. We were not able to detect constitutive activity in Gq, Gi, or Gs protein pathways. This absence of constitutive G protein signaling is in contradiction with previously reported results (Ku et al., 2012; Martin et al., 2015). Although these discrepancies could be the consequence of slightly different approaches and models, we noticed that key controls such as normalization to cell number or quantity of expressed receptor were not disclosed in those earlier reports. Consistent with our results, no ligand-independent β-arrestin 2 recruitment could be detected in a firefly luciferase complementation assay (Fig. 2B). Although the recruitment of β-arrestins to stimulated GPCRs is sometimes qualified as ubiquitous in the GPCR family (Kroeze et al., 2015), several exceptions have been described. Prominent examples are the gastric inhibitory peptide receptor (Al-Sabah et al., 2014), β 3-adrenoceptor (Nantel et al., 1993; Cao et al., 2000), α 2A-adrenoceptor (DeGraff et al., 1999, 2002), or the human gonadotropin-releasing hormone receptor (Hislop et al., 2005) that do not bind to β-arrestins. Therefore, we questioned the ability of activated GPR27 to bind to β-arrestins. One approach has been described to verify whether the translocation of β-arrestin would be a suitable readout for a given orphan GPCR (Oakley et al., 2006). It is based on the use of GRK mutants to trigger receptor phosphorylation in nonstimulated conditions. Applying this strategy on a chimeric GPR27V2 receptor, in which the C terminus of GPR27 was exchanged with that of V2R, we could confirm the recruitment of β-arrestin 2 to the receptor when cotransfected with GRK2m (Fig. 2E). The modification of the C terminus is a commonly used approach to increase the affinity for β-arrestins (Kroeze et al., 2015) without affecting the pharmacology of the receptor itself (Oakley et al., 2000). Therefore, we postulated that the GPR27V2 would be able to recruit β-arrestin 2 when stimulated by an agonist and that the firefly luciferase complementation was an adequate system to perform a screening.
We identified two synthetic ligands in the ChemBridge DIVERSet library able to specifically and selectively induce β-arrestin 2 recruitment to GPR27. Interestingly, none of these ligands could give rise to detectable activation of the G protein pathways. Besides an insufficient sensitivity of the G protein assays, several hypotheses can be made for the absence of measurable activation in cAMP, intracellular Ca2+, and TGF-α shedding assays. On the one hand, the ligands that we have identified could be strongly biased toward the β-arrestin pathway. Such compounds have been described for other receptors (Kenakin and Christopoulos, 2013; Kenakin, 2015a). On the other hand, GPR27 could belong to an atypical, non-G protein–coupled class of receptors that are well described among rhodopsin-like GPCRs. For example, the atypical chemokine receptor ACKR3/CXCR7 is thought to function as a scavenger receptor through an exclusive binding to β-arrestins (Rajagopal et al., 2010; Hanson and Chevigne, 2016; Szpakowska et al., 2016). Another prominent example of atypical GPCR that is thought to be devoid of G protein signaling is the LGR5 (Snyder et al., 2013). Moreover, the absence of G protein pathway activation in the presence of the GPR27 agonists is consistent with the lack of G protein constitutive activity in a controlled experimental setting. It is unclear whether the TRY and NPxxC that replace the conserved DRY and NPxxY motifs in the SREB receptor family could confer an atypical activation mechanism toward G proteins or have other impacts on receptor signaling. Of important note, these motifs have been identified as critical for G protein binding and/or activation in several rhodopsin-like GPCRs (Wess, 1998). At present, it is impracticable to test either the biased ligand or the atypical GPCR hypotheses. This would require additional, preferably endogenous, ligands that would go against our observations. Recently, Yanai et al. (2016) described a series of nonselective surrogate inverse agonists for the SREB family, using a GTP-γ–S assay on SREB-Gs fusion proteins in Spodoptera frugiperda Sf9 cells. However, we were not able to detect GPR27-specific signal in our assays with these compounds (Supplemental Fig. 2).
In the absence of an endogenous ligand, it is currently impossible to predict whether our small molecules bind in the orthosteric or an allosteric site, or how the observed β-arrestin recruitment relates to a physiologically relevant signaling pathway. Synthetic surrogate ligands should be seen as complement rather than substitutes of the endogenous ligands (Ahmad et al., 2015). They represent key research tools to gain information about receptor properties (Kenakin, 2015b), especially in the field of orphan GPCRs. For example, these tools may be used in future investigations for insulin secretion assays or in various neuronal models to study GPR27 signaling in a context in which it is expressed endogenously. Additionally, availability of agonists opens the door to ligand-guided optimization of homology models (Ngo et al., 2016) or pharmacophore-based prediction of the structure of endogenous ligands (Isberg et al., 2014). These ligands should also facilitate the search for the endogenous ligand because they can be used as positive controls to develop more sensitive assays or serve as displaced probes in binding experiments (Loison et al., 2012).
In conclusion, to the best of our knowledge, we provide the first molecular analysis of the signaling pathways of the highly conserved orphan receptor GPR27. We described the first selective compounds with agonist properties for this receptor. These small molecules are commercially available and readily accessible to the scientific community. They should promote further studies focused on the physiologic relevance of this elusive receptor. Moreover, they constitute interesting chemical scaffolds that may serve as starting points in the design of more potent analogs to be developed by pharmacomodulation.
Acknowledgments
The authors thank the GIGA Imaging and Flow Cytometry Platform for technical support. The authors gratefully acknowledge the technical assistance of Céline Piron. The plasmids used for the shedding assay were a gift of Dr. Asuka Inoue.
Authorship Contributions
Participated in research design: Dupuis, Twizere, Hanson.
Conducted experiments: Dupuis, Laschet, Franssen, Szpakowska.
Contributed new reagents or analytic tools: Dupuis, Laschet, Gilissen, Soni.
Performed data analysis: Dupuis, Laschet, Geubelle, Parent, Pirotte, Hanson.
Wrote or contributed to the writing of the manuscript: Dupuis, Szpakowska, Gilissen, Parent, Pirotte, Chevigné, Hanson.
Footnotes
- Received December 6, 2016.
- Accepted March 16, 2017.
This work was supported by the Fonds pour la Recherche Scientifique (F.R.S.-FNRS) Incentive Grant for Scientific Research [F.4510.14]; University of Liège [Crédit de Démarrage-Fonds Spéciaux]; Luxembourg Institute of Health [Ministère de l'enseignement supérieur et de la recherche (MESR) Grant 20160116]; Fonds National de la Recherche (FNR) Luxembourg [AFR-3004509]; and Léon Fredericq Foundation. J.H. and C.L. are F.R.S.-FNRS Research Associate and PhD fellow, respectively. N.D. is a Fonds pour la Recherche dans l'Industrie et l'Agriculture (FRIA) PhD fellow. M.S. is an Aides à la Formation-Recherche FNR-Luxembourg PhD fellow.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AP
- alkaline phosphatase
- Arrb1
- β-arrestin 1
- Arrb2
- β-arrestin 2
- β2AR
- β2-adrenoceptor
- ELISA
- enzyme-linked immunosorbent assay
- ERK
- extracellular signal–regulated kinase
- GFP
- green fluorescence protein
- GPCR
- G protein–coupled receptor
- GRK
- GPCR kinase
- GRK2m
- membrane-anchored GRK2
- HBSS
- Hanks’ balanced salt solution
- HEK293
- human embryonic kidney 293
- MAPK
- mitogen-activated protein kinase
- NanoBiT
- NanoLuc Binary Technology
- PBS
- phosphate-buffered saline
- SREB
- superconserved receptors expressed in the brain
- TGF
- transforming growth factor
- V2R
- vasopressin receptor V2
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}