


Abstract
The ability of agonists to selectively activate some but not all signaling pathways linked to pleiotropically signaling receptors has opened the possibility of obtaining molecules that emphasize beneficial signals, de-emphasize harmful signals, and concomitantly deemphasize harmful signals while blocking the harmful signals produced by endogenous agonists. The detection and quantification of biased effects is straightforward, but two important factors should be considered in the evaluation of biased effects in drug discovery. The first is that efficacy, and not bias, determines whether a given agonist signal will be observed; bias only dictates the relative concentrations at which agonist signals will appear when they do appear. Therefore, a Cartesian coordinate system plotting relative efficacy (on a scale of Log relative Intrinsic Activities) as the ordinates and Log(bias) as the abscissae is proposed as a useful tool in evaluating possible biased molecules for progression in discovery programs. Second, it should be considered that the current scales quantifying bias limit this property to the allosteric vector (ligand/receptor/coupling protein complex) and that whole-cell processing of this signal can completely change measured bias from in vitro predictions.
Introduction
Over the last 20 years in pharmacology evidence has accumulated to indicate that agonists need not activate all signaling pathways linked to pleiotropically coupled receptors but in fact may emphasize signaling to some pathways more than, and at the expense of, others; this is referred to as biased receptor signaling. The key to the appreciation of this concept was the introduction of new functional assays in pharmacology that allowed the separate observance of the various processes mediated by seven transmembrane receptors (7TMRs). As these assays were employed in recombinant and natural systems, literature reports began to emerge to suggest that not all systems had a monotonic stimulus-response linkage that made agonist potency ratios cell-type independent—that receptor activation was uniform. These effects augured the concept of bias and became appreciated as a therapeutic way forward: “The possibility is raised that selective agonists and antagonists might be developed which have specific effects on a particular receptor-linked effector system” (Roth and Chuang, 1987). The first molecular description of this phenomenon ascribed this effect to the stabilization of different receptor active states by different agonists (Kenakin and Morgan, 1989; Kenakin, 1995). The ability of agonists to choose signaling pathways has been given a number of names (“stimulus trafficking,” Kenakin, 1995; “functional selectivity,” Lawler et al., 1999; Kilts et al., 2002; Shapiro et al., 2003; “functional dissociation,” Whistler and von Zastrow, 1999; “biased inhibition,” Kudlacek et al., 2002; “differential engagement,” Manning, 2002); the term “biased signaling” was introduced by Jarpe et al. (1998) and is favored as a general descriptor of the effect. It is worth considering the present place of this idea in new drug discovery.
Therapeutic Applications of Signaling Bias
Although there are quantitative scales to characterize biased signaling (vide infra), the effect can readily be seen with no arithmetic manipulation of data through a bias plot; this expresses the response to an agonist in one signaling pathway as a function of the observed agonist response in another pathway. When signaling is seen with this tool, almost all ligands are biased because of the varying sensitivities of different functional assays and varying efficiency of coupling of receptor populations to different signaling proteins in the cell; this is system bias. This type of cellular bias will be imposed on all agonists operating within the two systems being studied and thus would not be useful therapeutically.
However, within this system bias there may be unique ligand-selective signaling bias in the form of diverging bias plot curves seen for any two pathways. This is the ligand bias that has potential for production of selective therapeutic effect. This is illustrated in Fig. 1 where the G-protein and β-arrestin activating properties of κ-opioid ligands are shown on a bias plot, and a clear differentiation is shown between agonists (White et al., 2014). Ligand-mediated receptor biased signaling must be explicitly defined and differentiated from simple differences in ligand effect for different functional assays; if this can be done, then a potentially therapeutically applicable ligand property can be associated with a chemical scaffold and subsequently optimized.

Bias plot for three κ-opioid agonists. Ordinates are agonist-mediated fractional recruitment of β-arrestin measured in the Tango assay, and abscissae are fractional activation of Gαi protein through a firefly luciferase cyclic AMP assay. A relatively unbiased response is produced by salvanorin A, a response biased toward β-arrestin is produced by GR89696 [10ΔΔLog(τ/KA) relative to salvanorin A = 4.3], and a response biased toward Gαi protein is produced by RB-64 [10ΔΔLog(τ/KA) relative to salvanorin A = 26.4]. Data from White et al. (2014).
In drug discovery, there are generally three reasons for seeking biased ligands:
The emphasis of a favorable cellular signal, such as β-arrestin-2 activation for parathyroid agonists in osteoporosis (Ferrari et al., 2005; Gesty-Palmer et al., 2006, 2009) and β-arrestin-1 activation for glucagon-like peptide 1 (GLP-1) effect in diabetes (Sonoda et al., 2008).
The deemphasis of a debilitating cellular signal, such as elimination of β-arrestin signaling for opioid analgesics (Raehal et al., 2005; DeWire et al., 2013; Charfi et al., 2015) and the loss of β-arrestin–mediated dysphoric effects of κ-opioid agonists (White et al., 2014).
The de-emphasis of a debilitating cellular signal and the blockade of the ability of the natural agonist to produce the same signal, such as TRV120027 [Sar-Arg-Val-Tyr-Ile-His-Pro-D-Ala-OH] blockade of angiotensin-mediated vasoconstriction in heart failure with concomitant preservation of angiotensin-mediated β-arrestin signaling (Violin et al., 2006, 2010).
Quantifying Bias
Although a bias plot can be used to detect unique ligand-mediated signaling bias, it cannot quantify such effects. Within any structure-activity relationship for bias this latter step is important in furnishing a scale by which medicinal chemists can gauge progress toward or away from a defined signaling pathway. Such a scale can be derived from the Black/Leff operational model of agonism (Black and Leff, 1983), which furnishes estimates of affinity in the form of KA values (equilibrium dissociation constants of agonist-receptor complexes) and agonist efficacy (in the form of τ values for a given signaling pathway). A null method to derive such a factor (termed RAi denoted as receptor activity) has been published (Ehlert et al., 1999) and subsequently applied to agonist bias (Griffin et al., 2007; Figueroa et al., 2009; Tran et al., 2009; Ehlert et al., 2011).
A modification and extension of this approach amenable to the statistical comparison of multiple agonists to yield ΔΔLog(τ/KA) values (denoted as transducer coefficients) has been published to furnish logarithms of bias factors between signaling pathways (Kenakin et al., 2012). Transducer coefficients are based on allosteric constants among ligands, receptors, and signaling proteins and thus yield bias within the allosteric vector defined by this ternary complex (Kenakin and Christopoulos, 2013). This makes the values independent of cell type and useful for quantification of biased effects. Calculation of transducer coefficients for the κ-opioid agonists shown in Fig. 1 indicate bias values of 4.3-fold toward G-protein for GR89696 [methyl 4-[2-(3,4-dichlorophenyl)acetyl]-3-(pyrrolidin-1-ylmethyl)piperazine-1-carboxylate] and 26.4-fold toward β-arrestin for RB-64 [methyl (2S,4aR,6aR,7R,9S,10aS,10bR)-2-(furan-3-yl)-6a,10b-dimethyl-4,10-dioxo-9-(2-thiocyanatoacetyl)oxy-2,4a,5,6,7,8,9,10a-octahydro-1H-benzo[f]isochromene-7-carboxylate] when compared with salvinorin A [methyl (2S,4aR,6aR,7R,9S,10aS,10bR)-9-acetyloxy-2-(furan-3-yl)-6a,10b-dimethyl-4,10-dioxo-2,4a,5,6,7,8,9,10a-octahydro-1H-benzo[f]isochromene-7-carboxylate].
These values are certainly useful to identify unique molecular modes of action and for visualizing the relative concentration-dependence of biased signals when the efficacy of the ligand is sufficient to demonstrate agonism, but they do not in themselves assist in the prediction of actual biased response. This still lies within the realm of relative intrinsic efficacy. Because of this fact, it is useful to assess biased ligands in terms of a Cartesian coordinate system of relative efficacy on an ordinate scale as a function of bias on the abscissal scale. Figure 2 shows such a presentation of the relative ability of 30 κ-opioid agonists to produce G-protein versus β-arrestin response (on a scale of Log(Intrinsic ActivityG-protein/Intrinsic Activityβ-arrestin), where Intrinsic Activity is the maximal response to the agonist in the assay (Ariens, 1954) as a function of the Log(Bias) for these responses (ΔΔLog(τ/KA) values). It can be seen that a considerable scatter results reflecting variance in the efficacy and affinity of these ligands for the receptor as it interacts with different signaling systems. Such an array is useful for choosing ligands that are more prone to produce desired responses versus those more prone to block undesired responses. For instance, it can be seen that relatively similar values of bias can yield compounds of differing ability to produce response as in the case of RB-65 [22-methoxysalvinorin A] and RB-64 and separately for 6ʹ-GNTI [6′-guanidino-17-(cyclopropylmethyl)-6,7-didehydro-4,5α-epoxy-3,14-dihydroxyindolo[2′,3′:6,7]morphinan] and salvinorin B [(2S,4aR,6aR,7R,9S,10aS,10bR)-2-(3-furanyl)dodecahydro-9-hydroxy-6a,10b-dimethyl-4,10-dioxo-methyl ester-2H-naphtho[2,1-c]pyran-7-carboxylic acid].

Activity of 30 agonists for κ-opioid receptors. Ordinates are logarithms of the relative intrinsic activity (maximal response) of the agonists for G-protein/β-arrestin. Abscissae are ΔΔLog(Log (τ/KA) values depicting bias relative to salvinorin A. Inset concentration curves are for two sets of agonists with similar bias but differing agonist characteristics (solid line: G-protein response; dotted line: β-arrestin response). Rel. I.A., relative Intrinsic Activities. Data from White et al. (2014).
Because 7TMRs are allosteric proteins, their affinity for ligands depends on the nature and concentration of cobinding species. Different cobinding ligands have been shown to affect the receptor affinity of ligands in accordance with this allosteric reciprocity (i.e., salvanorin affinity for κ-opioid receptors with Gα16 versus Gαi2; Yan et al., 2008 and changes in ghrelin receptors with addition of β-arrestin to nanodiscs, Mary et al., 2012). In view of the fact that functional response depends on the ternary complex of agonist/receptor/signaling protein (Onaran and Costa, 2012), it is conceivable that the affinity of the receptor-signaling complex shows variable affinity for agonists depending on the nature of the signaling protein interacting with the receptor. This raises the possibility that agonists may have different affinities for the receptor depending on which signaling pathway is being activated. In fact, significantly different functional affinities for partial agonists have been shown for 5-HT2A (5-hydroxytryptamine 2A) receptor agonists activating G-proteins versus producing extracellular signal-regulated kinase phosphorylation (Strachan et al., 2010).
Very different EC50 values for partial agonists also have been reported for μ-opioid receptors (McPherson et al., 2010; Nickolls et al., 2011), histamine H4 receptors (Nijmeijer et al., 2012), and β1-adrenoceptors (Casella et al., 2011). This suggests that effective bias may be obtained through combinations of efficacies and affinities for the receptor as it interacts with different pathways. Figure 3 shows two hypothetical agonists both designed to produce G-protein agonism (solid lines) and little β-arrestin activation (dotted lines). Interestingly, two very different values for bias could still yield favorable effects in terms of de-emphasizing β-arrestin signaling; agonist A (ΔΔLog(τ/KA) = −1.8) produces G-protein activation but little β-arrestin activation. However, agonist A would be a relatively poor antagonist of the natural agonist activation of β-arrestin. In contrast, agonist B (ΔΔLog(τ/KA) = 2.2) produces G-protein activation, no β-arrestin activation, and also would be a selective antagonist of the natural agonist in activating β-arrestin.

Profiles of two hypothetical agonists that would be predicted to produce selective G-protein (solid line: concentration curves) over β-arrestin (dotted line: concentration response curves). Gray open circle symbols represent the relative location of the agonists on a Log (relative Intrinsic Activities, Rel. I.A.) versus Log(Bias) plot (see Fig 2). Agonist A would produce selective G-protein agonism, but it would not selectively block natural agonist β-arrestin signaling. Agonist B would produce selective G-protein signaling, not signal through β-arrestin, and would selectively prevent activation of β-arrestin by the natural agonist system.
The consideration of possible biased affinity raises another condition seen in discovery programs aimed at biased molecules, namely, those compounds that may not have sufficient efficacy to generate a response in one of the signaling pathways. This is frequently observed for β-arrestin assays and has erroneously been described as “perfect bias.” Usually there are no independent data to conclude that such molecules may not produce a response in more sensitive tissues, so it is important to estimate a possible bias value for these molecules.
A lower limit for bias can be estimated by using the molecule as an antagonist for a more efficacious agonist and determining the affinity of the test molecule; this will be the EC50 of that molecule in more sensitive assays for the signaling pathway of the molecule when it produces partial agonism. Then an estimate of bias can be obtained by assuming a 5% error on the ability of the assay to detect an agonist response, setting the maximal response to be 0.05 in calculation of Log(τ/KA) where KB is the antagonist value for blockade by the molecule of the signaling pathway. The resulting ΔΔLog(RA) value is a lower limit for bias; that is, bias will be at least this value or more. This procedure is illustrated in Fig. 4. A useful and rigorous procedure for this estimation also has been recently been published by Stahl et al. (2015).

Estimation of minimal bias for a ligand that produces no response. If a ligand has low efficacy for a given pathway and the assay for that pathway is of insufficient sensitivity to demonstrate agonist response, the minimal bias for that compound can still be assessed. Specifically, the ligand can be used as an antagonist, and a measure of the KA can be made through antagonism. It then is assumed that the ligand produced a 5% response (possibly within error of the assay measurement), and an estimate of the limiting value of the Log(τ/KA) is made. This is used for the ΔLog(τ/KA) and subsequently the ΔΔLog(τ/KA) measurement to yield an estimate of the minimal bias. This is the best-case scenario that the ligand has efficacy for that pathway. It could have less activity, in which case the bias will be greater than the minimal estimation.
The System Independence of Transducer Coefficients
Agonist values of ΔΔLog(τ/KA) are dependent on the allosteric cooperativity constants controlling the interaction of agonists, receptors, and individual signaling proteins (Kenakin, 2013). Thus the agonist KA reflects the change in the natural affinity of the receptor for the signaling protein in the absence versus the presence of an agonist in the form of the allosteric parameter α (Stockton et al., 1983; Ehlert, 1988) and the change in the efficacy of interaction between the receptor and signaling protein in the absence or presence of the agonist (denoted β in the functional allosteric model in Ehlert, 2005; Kenakin, 2005; and Price et al., 2005; denoted as B in Ehlert, 1988). Under these circumstances, the transducer coefficient is unique to the allosteric vector made up of agonist/receptor/signaling protein and is thus cell type and system independent. Therefore, full agonist potency ratios [ΔΔLog(τ/KA) for full agonists] should not vary when measured in different cell types if the response is solely dependent on a signal emanating from the allosteric vector with no modification by the cell.
However, cell type and system variation of agonist potency ratios have long been documented, thereby indicating that receptor stimulus may, in some cases, be considerably modified by the cell (i.e., calcium entry, extracellular signal-regulated kinase phosphorylation, label-free drug response such as dynamic mass redistribution or cell layer electrical impedance). For instance, the relative potency ratios of the calcitonin agonists human and porcine calcitonin and human calcitonin gene–related peptide (CGRP) vary by orders of magnitude when the receptor is transfected into COS (monkey kidney cells) versus CHO (Chinese hamster ovary) cells (Christmanson et al., 1994).
Agonist-mediated β-adrenoceptor agonist potency ratios measured at the allosteric vector (cyclic AMP) and from the whole-cell response (cellular impedance) have been shown to vary, thereby indicating a modification by the cell (Peters et al., 2007). Even modification of host cell background such as coexpression of Gαs protein in human embryonic kidney 293 cells has been shown to reverse calcitonin receptor agonist potency ratios for eel and porcine calcitonin (Watson et al., 2000). These effects strongly suggest that bias values measured in vitro may not always predict therapeutic biased signaling in vivo and bring into question how measured bias can be used to progress drug candidate molecules.
It is useful to consider how bias numbers can effectively be applied to drug discovery in light of their sometimes dubious predicting power for therapeutic effect. Before biased signaling was considered as a pharmacologic mechanism, new molecules detected in high-throughput screens were sorted on the basis of potency and advanced into more complex and resource demanding models. With the advent of bias has come the ability to retest the active molecules found in a high-throughput screen in a functional test for another signaling pathway to identify biased molecules. This practice allows the detection of signaling bias resulting in the advancement of molecules that are known to stabilize different receptor active states. These molecules would be known to be different on a molecular level, and progression to the next pharmacologic model and in vivo testing would therefore increase the likelihood of detecting truly different in vivo phenotypic therapeutic activity. In addition, if a favorable in vivo phenotype is discovered, then the linking of that phenotype to in vitro bias assays furnishes pharmacologists and medicinal chemists with the tools to optimize the activity. It is within this realm that ΔΔLog(τ/KA) values yield the scale to constructively track changes in signaling bias with chemical structure.
There are other possible applications of transducer ratios that actually take advantage of the cell-type variability effect. One is for the screening of biased molecules. This is because cell-based variance in agonist potency ratios occurs only in cases where the agonists produce a biased signal from the allosteric receptor-based vectors. Therefore, if a high-throughput screen is carried out in two separate assays of cellular response (i.e., label-free screening in two cellular backgrounds) then the relative potencies of only the biased ligands will diverge between the two cell backgrounds.
A second possible application would be to use transducer coefficients to identify unique cell-based activity. For example, Fig. 5 shows a bias plot of the agonist activity of five dopamine agonists for natural dopamine D1 receptors in U-2 and SK-N-MC cells (Peters and Scott, 2009). It can be seen that while four of the agonists have comparable activity in the two cell types, A77636 [(1R,3S)-3-(1-adamantyl)-1-(aminomethyl)-3,4-dihydro-1H-isochromene-5,6-diol] diverges to have an 11-fold bias toward U-2 cells. In this case it is difficult to ascribe a physiologic significance to these data, but the same effect seen in a comparison of two physiologically meaningful cell types (i.e., tumor versus healthy cells, normal cells versus cells from a heart failure model) could possibly identify pathologically selective ligands with unique activity. This modification of allosteric vector signals can extend beyond cell type to cell state (e.g., uterine smooth muscle in pregnancy or state of estrous, or cell viability in disease states).
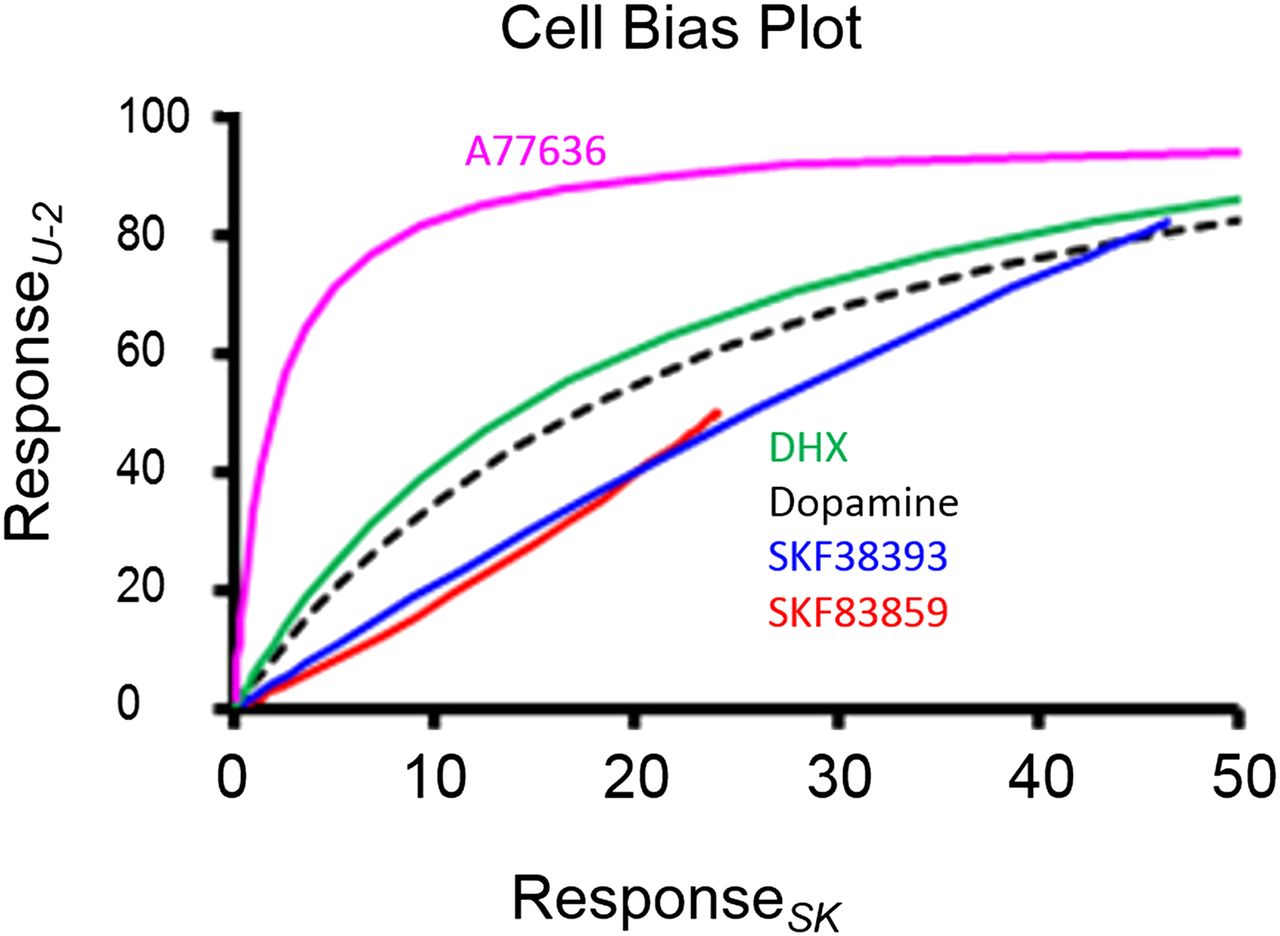
Bias plot for the dopamine receptor–mediated label-free assay response to five agonists for endogenous dopamine D1 receptors in U-2 cells (ordinates) and SK-N-MC cells (abscissae). The agonist A77636 is selectively 11-fold biased toward production of responses in U-2 cells. Data from Peters and Scott (2009). DHX, 10,11-dihydroxyhexahydrobenzo(a)phenanthridine.
Bias Induced by Negative Allosteric Molecules and Positive Allosteric Modulators
Agonists for pleiotropic receptors show an array of efficacies—pluridimensional efficacy (Galandrin and Bouvier, 2006)—as different signaling patterns are activated in varying degrees. Thus, agonist efficacy toward the whole cell has a quality as well as a quantity. Representations of these different qualities of efficacy can be shown on a multiaxis plot (referred to as web plots, radar plots, or spider plots); for example, ligand-specific webs of efficacy have been shown for β-adrenoceptors (Evans et al., 2010) and κ-opioid receptors (Zhou et al., 2013). The questions of efficacy quality and biased signaling become relevant to the interaction of 7TMR ligands and allosteric molecules.
The increase in functional, versus binding, screens in discovery programs has increased the likelihood of obtaining an allosteric molecule (Rees et al., 2002). Allosteric molecules for 7TMRs that can reduce signaling (negative allosteric molecules) or potentiate signaling (positive allosteric modulators) are becoming increasingly important as potential therapeutic entities. Because allosteric molecules are by nature permissive in that they may allow the interaction of the receptor with the natural agonist, there is a probability that the allosteric ligand will change the quality of the natural agonist signaling—that is, it will produce a bias for the natural agonist effect. Such bias has been noted for negative allosteric molecules in the selective blockade of NK2 receptors (Maillet et al., 2007), prostaglandin D2 receptors (Mathiesen et al., 2005), and calcium-sensing receptors (Davey et al., 2012; Cook et al., 2015) and positive allosteric modulators for glucagon-like peptide 1 (GLP-1) receptors (Koole et al., 2010) and mGlutamic acid 5 receptors (Bradley et al., 2011). The possibility of producing induced-bias in natural signaling can be viewed as a potential positive aspect of allosteric modulation of drug effect but also should be seen as an added consideration in the development of allosteric molecules.
Bias as a Perspective in Discovery
The ability of ligands to produce portions of a given receptor’s signaling portfolio has increased the scope for selective 7TMR-based new drugs. It might be asked whether biased signaling is a rare or common phenomenon and whether it will impact 7TMR therapeutics. In light of a molecular dynamics view of 7TMR function, the expectation of biased signaling would be predicted to be a common, not rare, phenomenon. Specifically, molecular dynamics describes receptor systems as composed of ensembles of numerous receptor states, the interconversion of which can be modeled as the receptor rolling on an energy landscape of conformations (Frauenfelder et al., 1991; Freire, 1998; Hilser et al., 1998, 2006; Hilser and Thompson, 2007).
Seen in terms of this hypothesis, the cell interacts with a spontaneous ensemble in the absence of a ligand and a ligand-formed ensemble in the presence of the ligand. This ensemble is composed of the stabilized conformations dictated by the individual affinities that the ligand has for the various receptor states (Onaran and Costa, 1997; Onaran et al., 2000; Kenakin, 2002). For identical ligand-stabilized ensembles to be created would mean that two ligands would have to have identical affinities for every conformation in the ensemble, a statistically unlikely event. Therefore, it would be predicted that different ligands would form at least slightly different ensembles which, in turn, would lead to different bias for signaling proteins.
However, this expectation would need to be tempered by the fact that only a few conformations are associated with cell signaling, so the chance that these would vary with ligands may not be as high as for total ensembles. In general, however, molecular dynamics predicts that biased signaling might be a fairly common and inherent property of 7TMR-ligand systems.
Conclusion
The existing data on agonist signaling suggest that bias is a prevalent pharmacologic phenomenon that should be considered in all new drug discovery programs. Bias can readily be measured and quantified through simple in vitro experiments using scales such as ΔΔLog(τ/KA), but it should be recognized that cell type and physiologic context may change these numbers and accordingly the predictions that bias measurements make.
In addition, the realization that bias is an amalgam of efficacy and affinity differences introduces concepts of affinity- versus efficacy-dominant bias ligands. This opens the possibility of biased antagonism as a viable therapeutic option. Finally, it can no longer be assumed that a synthetic agonist or antagonist or an allosteric modulator for a receptor will not alter the quality of efficacy produced by the natural agonist(s).
This makes it incumbent upon drug discovery efforts to pharmacologically characterize new drug activity with multiple functional assays and with quantitative scales. Although there are simple tools available to do this, the modification of these measured differences in signaling by cell type and other factors in vivo presently still make direct prediction of biased signaling to therapeutic systems a challenging prospect.
Acknowledgments
The author thanks the ever-stimulating Bryan Roth laboratory at the University of North Carolina for discussion and inspiration, Arthur Christopoulos for thoughts bounced back a little differently, and Kate White for an excellent set of data.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Kenakin.
Footnotes
- Received May 3, 2015.
- Accepted July 2, 2015.
Abbreviations
- A77636
- (1R,3S)-3-(1-adamantyl)-1-(aminomethyl)-3,4-dihydro-1H-isochromene-5,6-diol
- 6′-GNTI
- 6′-guanidino-17-(cyclopropylmethyl)-6,7-didehydro-4,5α-epoxy-3,14-dihydroxyindolo[2′,3′:6,7]morphinan
- GR89696
- methyl 4-[2-(3,4-dichlorophenyl)acetyl]-3-(pyrrolidin-1-ylmethyl)piperazine-1-carboxylate
- RB-64
- methyl (2S,4aR,6aR,7R,9S,10aS,10bR)-2-(furan-3-yl)-6a,10b-dimethyl-4,10-dioxo-9-(2-thiocyanatoacetyl)oxy-2,4a,5,6,7,8,9,10a-octahydro-1H-benzo[f]isochromene-7-carboxylate
- RB-65
- 22-methoxysalvinorin A
- TRV120027
- Sar-Arg-Val-Tyr-Ile-His-Pro-D-Ala-OH
- 7TMR
- seven transmembrane receptor
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Therapeutic Applications of Signaling Bias
- Quantifying Bias
- The System Independence of Transducer Coefficients
- Bias Induced by Negative Allosteric Molecules and Positive Allosteric Modulators
- Bias as a Perspective in Discovery
- Conclusion
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters