


Abstract
G protein–coupled receptor (GPCR) internalization is crucial for the termination of GPCR activity, and in some cases is associated with G protein–independent signaling and endosomal receptor signaling. To date, internalization has been studied in great detail for class A GPCRs; whereas it is not well established to what extent the observations can be generalized to class C GPCRs, including the extracellular calcium-sensing receptor (CaSR). The CaSR is a prototypical class C GPCR that maintains stable blood calcium (Ca2+) levels by sensing minute changes in extracellular free Ca2+. It is thus necessary that the activity of the CaSR is tightly regulated, even while continuously being exposed to its endogenous agonist. Previous studies have used overexpression of intracellular proteins involved in GPCR trafficking, pathway inhibitors, and cell-surface expression or functional desensitization as indirect measures to investigate CaSR internalization. However, there is no general consensus on the processes involved, and the mechanism of CaSR internalization remains poorly understood. The current study provides new insights into the internalization mechanism of the CaSR. We have used a state-of-the-art time-resolved fluorescence resonance energy transfer–based internalization assay to directly measure CaSR internalization in real-time. We demonstrate that the CaSR displays both constitutive and concentration-dependent Ca2+-mediated internalization. For the first time, we conclusively show that CaSR internalization is sensitive to immediate positive and negative modulation by the CaSR-specific allosteric modulators N-(3-[2-chlorophenyl]propyl)-(R)-α-methyl-3-methoxybenzylamine (NPS R-568) and 2-chloro-6-[(2R)-2-hydroxy-3-[(2-methyl-1-naphthalen-2-ylpropan-2-yl)amino]propoxy]benzonitrile (NPS 2143), respectively. In addition, we provide compelling evidence that CaSR internalization is β-arrestin–dependent while interestingly being largely independent of Gq/11 and Gi/o protein signaling.
SIGNIFICANCE STATEMENT A novel highly efficient cell-based real-time internalization assay to show that calcium-sensing receptor (CaSR) internalization is β-arrestin-dependent and sensitive to modulation by allosteric ligands.
Introduction
The extracellular calcium-sensing receptor (CaSR) is a widely expressed class C G protein–coupled receptor (GPCR) that is essential for calcium homeostasis in the human body through regulation of the synthesis and secretion of the calcium-elevating parathyroid hormone (Brown et al., 1993; Brown, 2013). Several natural occurring CaSR mutations have been identified in patients suffering from calcium homeostasis-related disorders (e.g., familial hypocalciuric hypercalcemia and autosomal dominant hypocalcemia), thereby making the CaSR an attractive drug target in diseases related to calcium and parathyroid hormone imbalance (Brown, 2007; Hannan and Thakker, 2013). In 2004, the CaSR-specific positive allosteric modulator (PAM) cinacalcet was the first allosteric GPCR drug to be approved for clinical use. Cinacalcet effectively lowers elevated parathyroid hormone levels in disorders linked to hyperparathyroidism (Nemeth and Goodman, 2016). Recently, the CaSR PAM etelcalcetide received regulatory approval for clinical use as a more efficacious alternative for cinacalcet with a higher level of compliance (Block et al., 2017; Patel and Bridgeman, 2018; Xipell et al., 2019). CaSR expression in tissues unrelated to calcium homeostasis suggests that this receptor could also be a potential therapeutic target for cardiovascular diseases, infertility, and cancer (Ellinger, 2016; Tennakoon et al., 2016; Guo et al., 2018). Nevertheless, the precise contribution of the CaSR in these disease contexts remains uncertain.
GPCR endocytosis—commonly referred to as GPCR internalization—is a highly regulated process for removal of GPCRs from the cell membrane into intracellular compartments. Given the direct influence on GPCR cell-surface expression, understanding the mechanism(s) of internalization is of crucial importance for obtaining full comprehension of GPCR signaling and function (Drake et al., 2006; Moore et al., 2007). Internalization has been extensively studied in class A GPCRs, where the most prevalent mechanism after receptor activation includes kinase-mediated phosphorylation of the GPCR C-terminus and intracellular loops followed by β-arrestin recruitment and dynamin-driven internalization in clathrin-coated pits (Moore et al., 2007). However, the internalization mechanism(s) in class B and C GPCRs remains less well described.
In particular, the processes that underscore internalization of class C GPCRs, including the CaSR, are poorly understood and there is a lack of consensus in the current literature. Some studies conclude that the CaSR can internalize and recycle to the cell-surface independently of agonist activation (Reyes-Ibarra et al., 2007; Zhuang et al., 2012), whereas others have reported agonist-mediated internalization (Pi et al., 2005; Lorenz et al., 2007). Additionally, Grant et al. (2011) stated that constitutive (i.e., ligand-independent) CaSR internalization is primarily followed by degradation rather than recycling. Regarding the mechanism underlying CaSR internalization, Pi et al. (2005) and Lorenz et al. (2007) have conducted overexpression and inhibition studies of intracellular proteins related to GPCR trafficking, in which cell-surface expression was used as an indirect measure of internalization. Both studies found that CaSR interaction with kinases and β-arrestins was essential for functional desensitization, but not for CaSR internalization (Pi et al., 2005; Lorenz et al., 2007). Furthermore, by using nonspecific internalization inhibitors Holstein et al. (2004) have shown that a PAM may regulate CaSR internalization, although this remains to be shown via direct measurement of CaSR internalization.
In the present study, we aimed to delineate the mechanism underlying CaSR internalization and to directly measure the effects of allosteric ligands on CaSR internalization using a real-time time-resolved fluorescence resonance energy transfer (TR-FRET)–based internalization assay (Foster and Bräuner-Osborne, 2018). This assay has previously been applied to directly measure constitutive and agonist-mediated internalization for receptors across different GPCR classes, including the class A β2-adrenergic receptor (β2AR) (Roed et al., 2014), class B glucagon-like peptide 1 receptor (GLP-1R) (Roed et al., 2014, 2015), and class C GPRC6A (Jacobsen et al., 2017a). By using CaSR mutants, selective G protein inhibitors and β-arrestin1/2 and Gq/11 protein knockout (KO) cell lines, we demonstrate that CaSR internalization is β-arrestin dependent, largely Gi/o and Gq/11 protein independent, and modulated by allosteric ligands.
Materials and Methods
Materials.
Unless stated otherwise, all reagents were obtained from Sigma-Aldrich (St. Louis, MO). Dulbecco’s modified Eagle’s medium, dialyzed FBS, a mixture of 10,000 U/ml penicillin and 10,000 μg/ml streptomycin, Dulbecco’s PBS without Ca2+ and Mg2+, Opti-MEM, Hanks’ balanced salt solution (HBSS) without Ca2+, Mg2+ and phenol red, Lipofectamine 2000, and pertussis toxin (PTX) were obtained from Thermo Fisher Scientific (Waltham, MA). The allosteric modulator N-(3-[2-chlorophenyl]propyl)-(R)-α-methyl-3-methoxybenzylamine (NPS R-568) hydrochloride was purchased from Tocris Bioscience (Bristol, UK) and 2-chloro-6-[(2R)-2-hydroxy-3-[(2-methyl-1-naphthalen-2-ylpropan-2-yl)amino]propoxy]benzonitrile (NPS 2143) hydrochloride was synthesized in-house as previously published (Johansson et al., 2013). The selective Gq/11 inhibitor YM-254890 (CAS Number 568580-02-9) was purchased from Wako Chemicals GmbH (Neuss, Germany). The Gq/11 KO and β-arrestin1/2 KO cells were generated from human embryonic kidney (HEK) 293A cells using CRISPR/Cas9 technology as previously described (Schrage et al., 2015; Alvarez-Curto et al., 2016).
Plasmid DNA Constructs.
The FLAG-SNAP-tagged β2AR pEGFPN1 and FLAG-SNAP-tagged GLP-1R pEGFPN1 constructs were a kind gift from Dr. Maria Waldhoer, InterAx Biotech AG (Roed et al., 2014). These constructs contain an upstream sequence encoding a T-cell CD8A signal peptide followed by a FLAG and SNAP tag. The N-terminal FLAG and SNAP tag were inserted to allow expression analysis and real-time internalization measurements, respectively. FLAG-SNAP-tagged CaSR wild-type (WT) and mutant vectors were generated by replacing the GLP-1R sequence of the FLAG-SNAP-tagged GLP-1R pEGFPN1 construct with the CaSR WT, CaSR S170A, or CaSR E837A sequence using the MluI-NotI restriction sites. The FLAG-SNAP-tagged CaSR WT and mutant constructs were validated using an inositol monophosphate (IP1) accumulation assay (Fig. 1A) and expression analysis (Fig. 1B). Constructs containing human β-arrestin1 (GenBank accession number NM_004041.4) and β-arrestin2 (GenBank accession number NM_004313.3) were made in-house as previously published (Gabe et al., 2018).
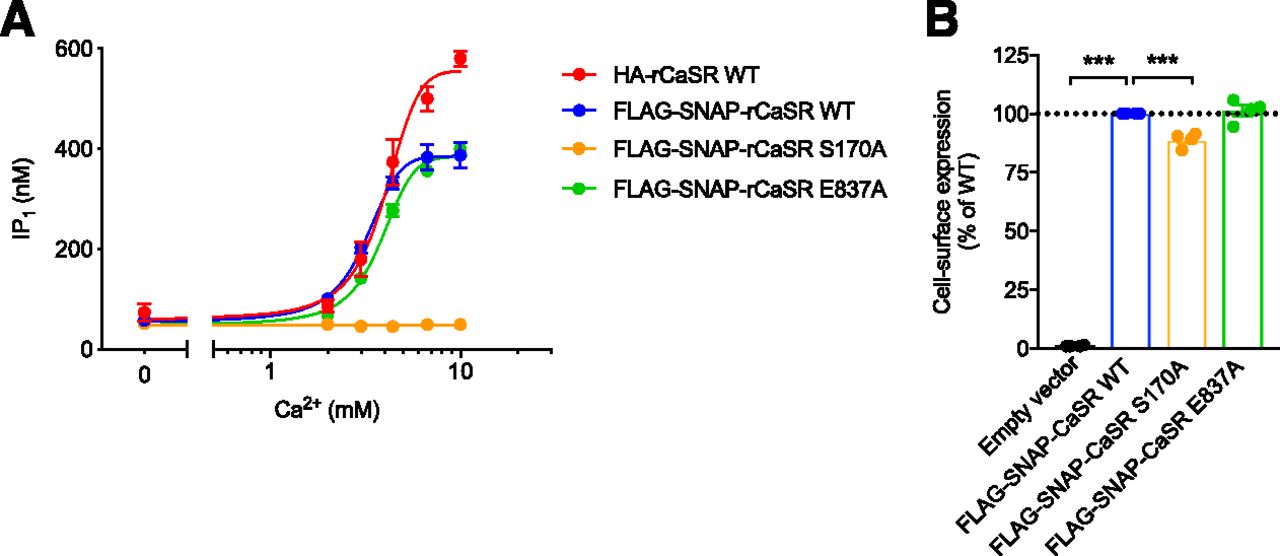
Characterization of WT and mutant CaSR constructs. WT and mutant CaSR transiently expressed in HEK293T cells were validated in an IP1 accumulation assay and by expression analysis. (A) IP1 accumulation measured after 30-minute stimulation with increasing Ca2+ concentrations. Data represent mean ± S.E.M. of three independent experiments performed in triplicate. (B) Cell-surface expression of FLAG-SNAP-tagged CaSR WT and mutant constructs obtained with anti-FLAG antibodies in ELISA. Data are mean ± S.E.M. of four experiments performed in parallel with the IP1 accumulation or internalization assay. The expression of CaSR WT was set to 100% and mutants were compared with CaSR WT using one-way ANOVA followed by Dunnett’s post-test (***P < 0.001).
Cell Culture and Transfection.
HEK293T, HEK293A, Gq/11 KO, and β-arrestin1/2 KO cells were cultured at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% dialyzed FBS and the 1% penicillin/streptomycin mixture. All cell lines were confirmed as negative for mycoplasma contamination using the LookOut Mycoplasma PCR Detection Kit (Sigma-Aldrich). Forty-eight hours prior to assays, cells were transiently transfected at a density of 17,000 cells/well in poly-D-lysine–coated 96-well plates. White opaque 96-well culture plates (PerkinElmer, Waltham, MA) were used for ELISA and clear tissue culture–treated 96-well plates (Corning, Corning, NY) for IP1 and cAMP accumulation assays. For the TR-FRET–based internalization assay, cells were transiently transfected in white 96-well culture plates (Corning). For each well, 30 ng CaSR construct diluted in 25 μl Opti-MEM was added to 0.25 µl Lipofectamine 2000 diluted in 25 μl Opti-MEM after 5-minute incubation at room temperature (RT). For β-arrestin coexpression studies, cells were transfected with a total of 60 ng/well at a CaSR:empty pEGFPN1 vector ratio of 1:1 as negative control and CaSR:β-arrestin ratios of 1:1 and 2:1:1 for coexpression with one or both β-arrestin subtypes, respectively. For the CaSR signaling and expression experiments in HEK293A and β-arrestin1/2 KO cells presented in Fig. 8, the HEK293A cells were transfected with a total of 30 ng/well at a CaSR:empty pEGFPN1 vector ratio of 1:3. The DNA:lipofectamine mixture was subsequently mixed and incubated at RT for 20 minutes prior to addition to the assay plate (50 μl/well). Next, 100 μl/well of cells diluted in culture media (17,000 cells/well) was added and the assay plate was incubated for 48 hours at 37°C and 5% CO2.
ELISA for Cell-Surface Expression.
Cell-surface expression levels of the WT and mutant CaSR constructs were examined using ELISA as described previously (Nørskov-Lauritsen et al., 2015). To ensure correct cell-surface labeling, the GABAB receptor expression system was used as a control. In brief, cells were fixed with 4% paraformaldehyde in Dulbecco’s PBS for 5 minutes at RT and washed twice using wash buffer (Dulbecco’s PBS supplemented with 1 mM CaCl2 and 1 mM MgCl2). After fixation, all wells were incubated for at least 30 minutes at RT in blocking solution (ultrapure MilliQ water, 3% skim milk, 1 mM CaCl2, and 1 mM MgCl2 50 mM Trizma hydrochloride solution, pH 7.4) followed by incubation for 45 minutes with monoclonal anti-FLAG antibody (Sigma-Aldrich) diluted 1:100,000 in blocking solution. Subsequently, cells were incubated for 45 minutes with horseradish peroxidase–conjugated anti-mouse IgG antibody (Vector Laboratories Inc., Burlingame, CA) diluted 1:2000 in blocking solution after which they were washed four times in blocking solution and four times in wash buffer. Chemiluminescence was measured on an EnSpire plate reader (PerkinElmer) immediately after addition of 10 μl/well SuperSignal ELISA Femto Substrate (Thermo Fisher Scientific).
Inositol Monophosphate Accumulation Assay.
IP1 accumulation downstream from CaSR activation was measured using the HTRF IP-One Assay Kit (Cisbio Bioassays, Codolet, France) as previously published (Jacobsen et al., 2017b). In brief, cells were washed once in 100 μl/well of assay buffer (HBSS and 20 mM HEPES, pH 7.4) prior to ligand stimulation for 30 minutes at 37°C. All ligands were prepared in ligand buffer (HBSS and 20 mM HEPES, pH 7.4, supplemented with 20 mM LiCl). Unless stated otherwise, studies with allosteric modulators or the Gq/11 inhibitor YM-254890 cells were preincubated for 30 minutes at 37°C prior to ligand stimulation in ligand buffer supplemented with allosteric modulator or inhibitor and a final DMSO concentration below 0.1%. After ligand stimulation, the cells were washed with assay buffer (100 μl/well) and subsequently lysed for 30 minutes at RT using 30 μl/well IP-One Conjugate & Lysis buffer (Cisbio Bioassays). The lysed cells were diluted with 30 μl/well assay buffer and 10 μl/well was transferred to a white 384-well Optiplate (PerkinElmer). Next, 10 μl/well of detection solution (assay buffer + 2.5% IP1-d2 conjugate + 2.5% anti-IP1 antibody Tb3+ cryptate) was added and the plate was incubated in the dark for 1 hour at RT prior to measurement on an EnVision multimode plate reader (PerkinElmer). Emissions at 615 and 665 nm were measured upon excitation at 340 nm. The resulting fluorescence resonance energy transfer ratios (615/665 nm) were converted to IP1 concentrations through standard curve interpolation of an IP1 calibrator provided with the assay kit.
cAMP Accumulation Assay.
CaSR-dependent cAMP inhibition was measured using the cAMP Dynamic 2 Assay Kit purchased from Cisbio Bioassays. For PTX treatment, cells were pretreated with 80 ng/ml PTX 24 hours prior to assay. Ligands were prepared in ligand buffer (HBSS and 20 mM HEPES, pH 7.4, supplemented with 50 μM isobutyl-1-methyl-xanthine and 0.05% DMSO) containing 1.5 μM forskolin. On the day of the assay, cells were washed once with 100 μl/well assay buffer (HBSS and 20 mM HEPES, pH 7.4) followed by ligand stimulation (50 μl/well) for 30 minutes at RT. After ligand stimulation, the cells were washed once with 100 μl/well assay buffer and then lysed for 30 minutes at RT using 30 μl/well cAMP Lysis & Detection Buffer. The lysates were diluted with 30 μl/well assay buffer and 10 μl/well was transferred to a white small-volume 384-well plate (Greiner Bio-One, Monroe). Then, 10 μl per well detection solution (assay buffer + 2.5% cAMP-d2 conjugate + 2.5% anti-cAMP antibody Eu3+ cryptate) was added and the plate was incubated in the dark for 1 hour at RT prior to measurement on an EnVision multimode plate reader (PerkinElmer). Emissions at 615 and 665 nm were measured upon excitation at 340 nm. The resulting fluorescence resonance energy transfer ratios (615/665 nm) were converted to cAMP concentrations through standard curve interpolation of a cAMP calibrator provided with the assay kit.
TR-FRET–Based Real-Time Internalization Assay.
CaSR internalization was measured using a TR-FRET–based real-time internalization assay as previously published (Roed et al., 2014, 2015; Levoye et al., 2015; Jacobsen et al., 2017a; Foster and Bräuner-Osborne, 2018; Gabe et al., 2018). For each well, cell-surface–expressed SNAP-tagged receptors were labeled for 1 hour with 50 μl 0.1 nM cell-impermeable donor fluorophore (SNAP lumi4-Tb; Cisbio Bioassays) diluted in Opti-MEM. Labeling was performed at 4°C to allow visualization of constitutive internalization, as done previously for the class C GPCR GPRC6A (Jacobsen et al., 2017a). All ligands were prepared at 2× final concentration in assay buffer (HBSS and 20 mM HEPES, pH 7.4), while the acceptor fluorophore (fluorescein-O-acetic acid) was diluted in assay buffer to a final concentration of 40 nM. The prepared ligand and acceptor fluorophore solutions were preheated to 37°C prior to addition. After labeling, cells were washed twice with 100 μl/well cold assay buffer. Subsequently, 50 μl/well ligand and 50 μl/well 40 nM fluorescein-O-acetic acid solution were added and internalization was measured immediately at 37°C on an EnVision multimode plate reader (PerkinElmer). Emissions at 615 and 520 nm were measured every 6 minutes upon excitation at 340 nm. The level of internalization depicted in the figures represents the donor over acceptor emission ratio (615/520 nm). When testing EDTA or YM-254890, the reagents were present in final concentration during both labeling and real-time measurements. When testing PTX, cells were pretreated for 24 hours with 80 ng/ml PTX prior to assay.
Data Analysis.
Curve fitting and statistical analyses were performed with Graphpad Prism 7 (Graphpad Software, San Diego, CA). Concentration-response curves were fitted using the following four-parameter nonlinear regression equation: In this equation, top and bottom are the response values obtained at the top and bottom plateau of the concentration-response curve. The EC50 value is defined as the agonist concentration necessary to elicit a half-maximal response and the n value corresponds to the Hill slope. For internalization data, each real-time trace was baseline corrected prior to area under the curve measurement. The obtained area under the curve values were normalized as described in the figure legends. Statistical analyses were performed using one-way ANOVA followed by Dunnett’s post-test or Tukey’s multiple comparisons test for comparisons against one condition or multiple conditions, respectively. Statistical significance is notated in the figures as follows: *P < 0.05, **P < 0.01, ***P < 0.001. For some data points, error bars are not visible since they are smaller than the symbol size.
In this equation, top and bottom are the response values obtained at the top and bottom plateau of the concentration-response curve. The EC50 value is defined as the agonist concentration necessary to elicit a half-maximal response and the n value corresponds to the Hill slope. For internalization data, each real-time trace was baseline corrected prior to area under the curve measurement. The obtained area under the curve values were normalized as described in the figure legends. Statistical analyses were performed using one-way ANOVA followed by Dunnett’s post-test or Tukey’s multiple comparisons test for comparisons against one condition or multiple conditions, respectively. Statistical significance is notated in the figures as follows: *P < 0.05, **P < 0.01, ***P < 0.001. For some data points, error bars are not visible since they are smaller than the symbol size.
Results
The N-Terminal SNAP-Tag Does Not Interfere with CaSR Functionality.
In this study, CaSR internalization was investigated using CaSR WT and the CaSR mutants S170A and E837A that are located in the L-amino acid and allosteric ligand binding site, respectively. CaSR internalization was assessed using a TR-FRET–based real-time internalization assay that requires the addition of an N-terminal SNAP tag to the receptor of interest. Prior to internalization, the FLAG-SNAP-tagged constructs were validated for their agonist response using an IP1 accumulation assay (Fig. 1A) and for their cell-surface expression using an ELISA (Fig. 1B). To investigate whether the SNAP tag interfered with the signaling capacity of the CaSR, FLAG-SNAP-tagged CaSR WT was compared with hemagglutinin (HA)-tagged CaSR WT, which we have previously characterized in detail (Jacobsen et al., 2017b). In a functional IP1 accumulation assay there was no difference in agonist potency of Ca2+ for the two receptor constructs (Fig. 1A; pEC50 values of 2.50 ± 0.02 and 2.39 ± 0.03 for FLAG-SNAP-CaSR WT and HA-CaSR WT, respectively), thereby confirming that the SNAP tag did not prevent Ca2+-mediated activation of the Gq/11 pathway. We next used the FLAG-SNAP-CaSR construct to study Ca2+-mediated CaSR internalization.
The CaSR Displays Constitutive and Ca2+-Mediated Internalization.
CaSR internalization was measured in the absence and presence of its endogenous agonist Ca2+ and internalization was observed under both conditions (Fig. 2A). Increasing Ca2+ concentrations induced internalization in a concentration-dependent manner (pEC50 of 2.48 ± 0.04) (Fig. 2B). Direct comparison with the IP1 accumulation data (Fig. 1A) revealed that Ca2+ triggers internalization and Gq/11-mediated activity of the CaSR with equal potency.

The CaSR displays constitutive and concentration-dependent Ca2+-mediated internalization. HEK293T cells were transiently transfected with FLAG-SNAP-CaSR WT, orthosteric loss-of-function CaSR mutant (S170A), or FLAG-SNAP-β2-adrenergic receptor (β2AR). (A) Representative real-time traces showing CaSR WT internalization in response to buffer and increasing Ca2+ concentrations. (B) Area under the curve (AUC) plotted against the Ca2+ concentration. Data are mean ± S.E.M. of four independent internalization experiments performed in triplicate. (C) Representative real-time traces and (D) baseline-corrected AUC data grouped from three independent experiments of β2AR internalization upon stimulation with 1 μM isoproterenol in the absence and presence of 0.5 or 1 mM EDTA. (E–H) The effect of 0.5 mM EDTA on CaSR WT (E and F) as well as CaSR S170A (G and H) internalization was measured in the absence (open symbols, constitutive internalization) or presence (closed symbols, Ca2+-mediated internalization) of 10 mM Ca2+. (E and G) Representative real-time traces and (F and H) normalized baseline-corrected AUC data grouped from three independent experiments performed in triplicate are shown. Real-time internalization traces are presented as mean ± S.D. of a single representative experiment performed in triplicate. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s post-test (D, F and H). Significance is notated as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
The physiologic ligands of the CaSR include essential components of cell culture media and assay buffers such as divalent cations (e.g., Ca2+ and Mg2+) and L-amino acids (e.g., L-phenylalanine and L-tryptophan). Additionally, cells are able to release such ligands either through passive or active membrane transport (Meier et al., 2002; Brini and Carafoli, 2011). To determine whether the constitutive internalization was mediated by divalent cations and/or L-amino acids released from the cells during the real-time assay, we employed two strategies. First, EDTA was used to chelate extracellular cations present during the internalization assay. Since L-amino acids require the presence of Ca2+ to activate the CaSR, chelation of Ca2+ by EDTA also indirectly eliminates the amino acid–induced effects (Conigrave et al., 2000; Geng et al., 2016). To test if EDTA had nonspecific activity on the cells or assay, we used the FLAG-SNAP-β2AR as a control. As shown in Fig. 2, C and D, 1 mM EDTA significantly reduced the level of isoproterenol-mediated internalization (P < 0.01, one-way ANOVA followed by Dunnett's post-test), while 0.5 mM EDTA had no effect. Hence, 0.5 mM EDTA was used during labeling and real-time internalization measurements of the CaSR. The usage of 0.5 mM EDTA did not affect constitutive internalization of the CaSR (Fig. 2, E and F). These results indicate that constitutive internalization is not caused by ambient divalent cations and L-amino acids but rather is an inherent property of the CaSR.
Second, constitutive internalization was investigated using a CaSR loss-of-function mutant, S170A. The S170 residue is located in the highly conserved L-amino acid orthosteric binding site, where it directly interacts with L-Trp according to the crystal structures of the CaSR extracellular domain (Geng et al., 2016; Zhang et al., 2016). Mutating this residue has been shown to prevent Ca2+-mediated signaling of the CaSR (Bräuner-Osborne et al., 1999; Geng et al., 2016). In agreement with the literature, FLAG-SNAP-CaSR S170A abolished Ca2+-mediated IP1 accumulation (Fig. 1A). Cell-surface expression analysis by ELISA verified that the loss of Ca2+-mediated activity was not caused by a lack of cell-surface expression. The surface expression of CaSR S170A was marginally reduced relative to CaSR WT; however, this alone could not explain the loss of function (Fig. 1B, P < 0.001, one-way ANOVA followed by Dunnett’s post-test). In the internalization assay, CaSR S170A internalized constitutively while no Ca2+-mediated internalization was observed (Fig. 2, G and H). Moreover, constitutive internalization of CaSR S170A was unaffected by the presence of 0.5 mM EDTA. Thus, the constitutive internalization was not eliminated by mutating the orthosteric ligand binding site or chelation of extracellular ambient Ca2+. These results strongly suggest that the CaSR displays constitutive internalization.
Ca2+-Mediated CaSR Internalization Is Modulated by Allosteric Ligands.
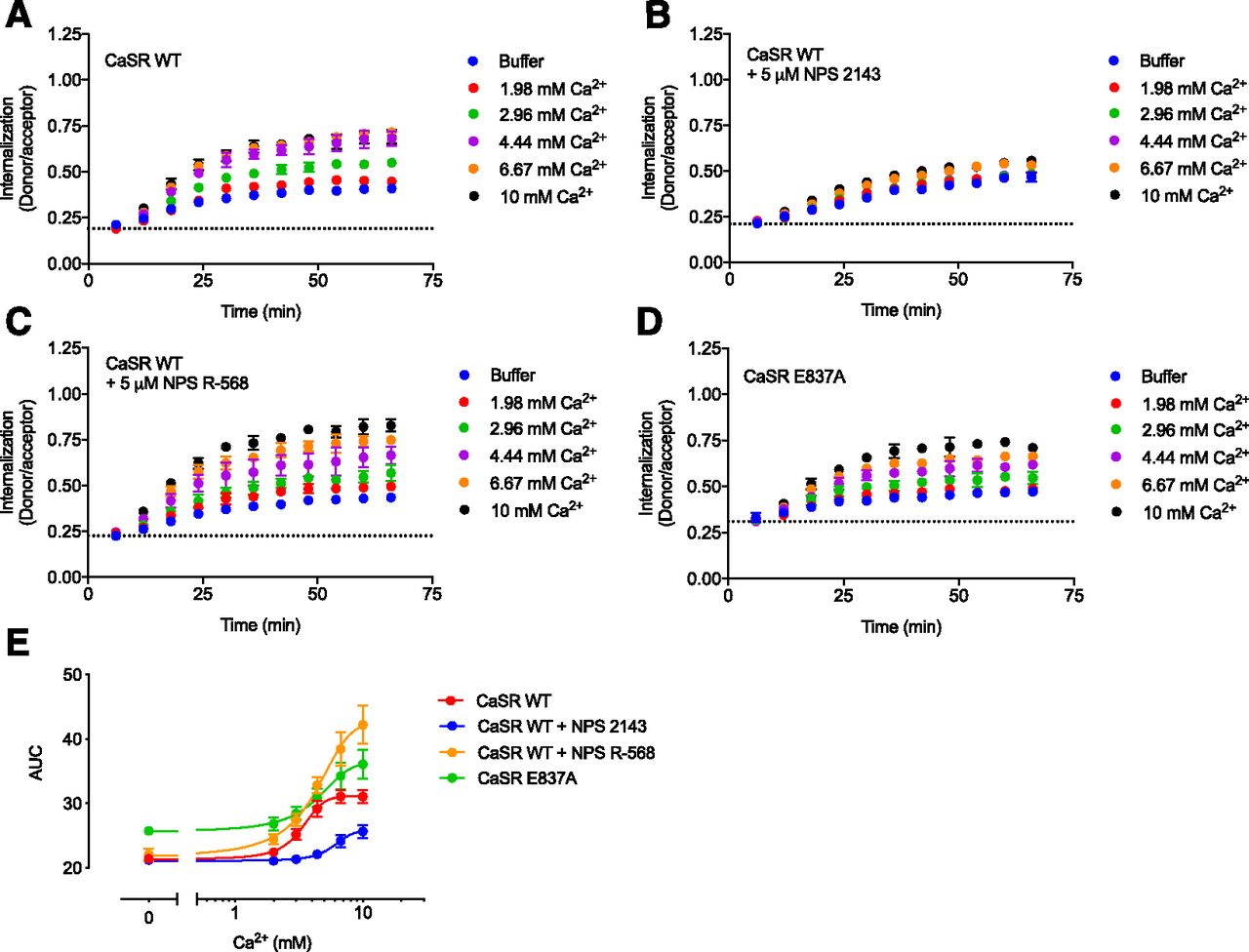
To study the effect of allosteric ligands on CaSR internalization, we measured internalization in the presence of the representative negative allosteric modulator (NAM) NPS 2143 or PAM NPS R-568 (Fig. 3). Ca2+-mediated internalization of the CaSR was reduced in the presence of 5 μM NPS 2143 and enhanced in the presence of 5 μM NPS R-568. This concentration of NPS 2143 and NPS R-568 was found to fully inhibit or potentiate the Ca2+-induced CaSR response in the IP1 accumulation assay, respectively (Figs. 4A and 5A).
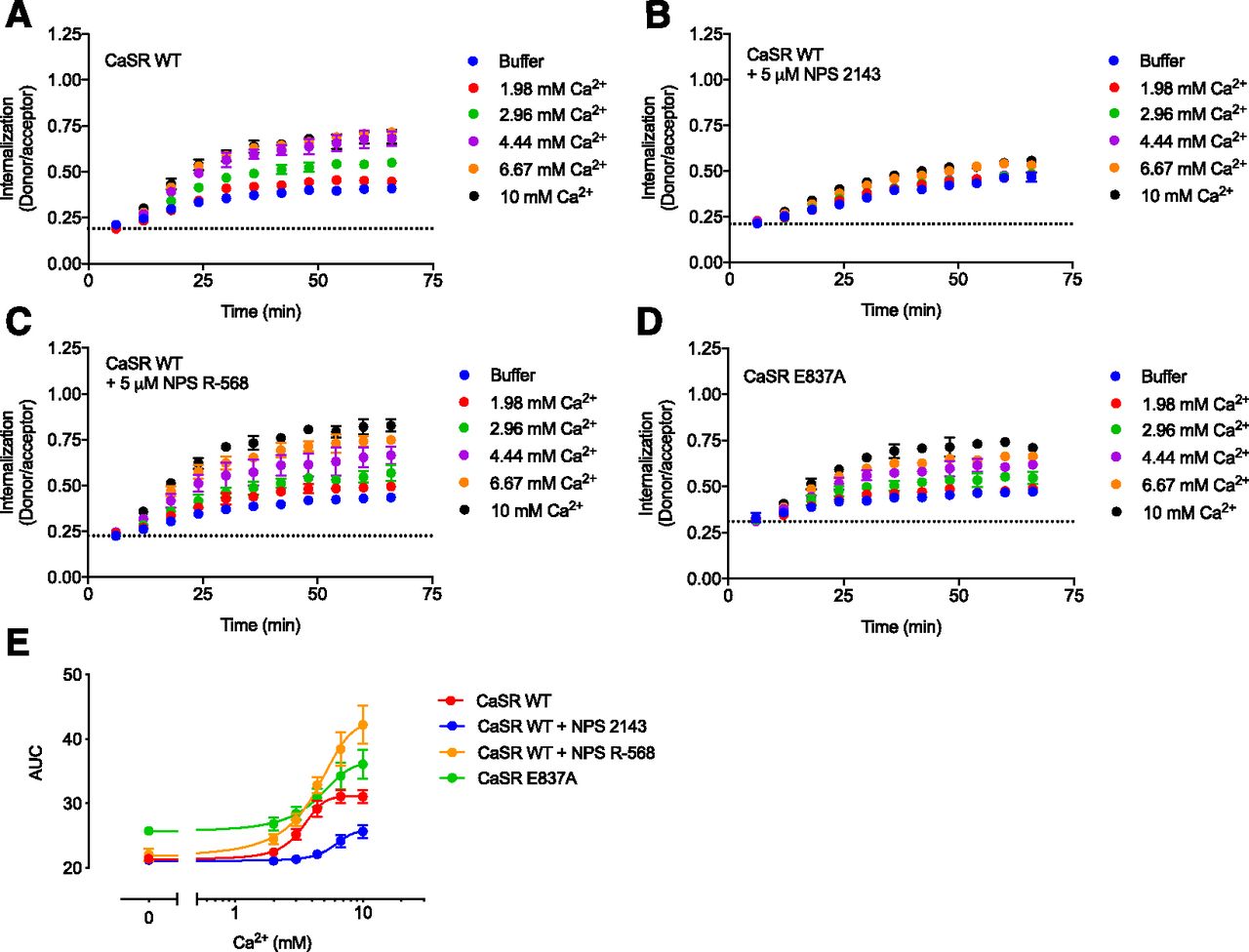
CaSR internalization is allosterically modulated. Real-time internalization traces of HEK293T cells transiently transfected with FLAG-SNAP-CaSR WT in response to increasing Ca2+ concentrations without allosteric modulators (A), in the presence of 5 μM NPS 2143 (B), and in the presence of 5 μM NPS R-568 (C). (D) Real-time internalization traces for increasing Ca2+ concentrations without allosteric modulators recorded from HEK293T cells transiently transfected with FLAG-SNAP-CaSR E837A. Data are mean ± S.D. of a single representative experiment performed in triplicate. (E) Area under the curve (AUC) plotted against the Ca2+ concentration. Data are mean ± S.E.M. of three or four independent internalization experiments performed in triplicate.
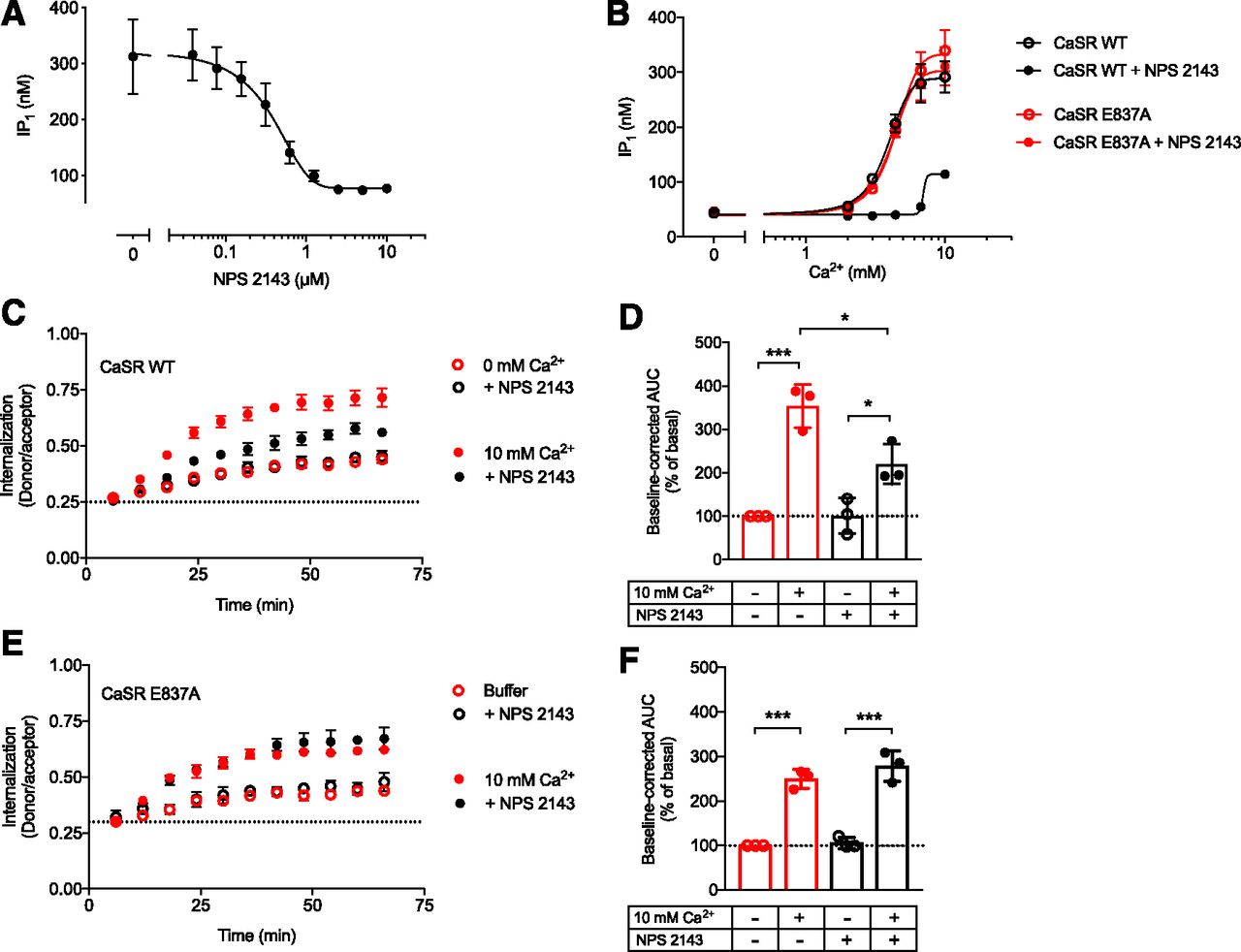
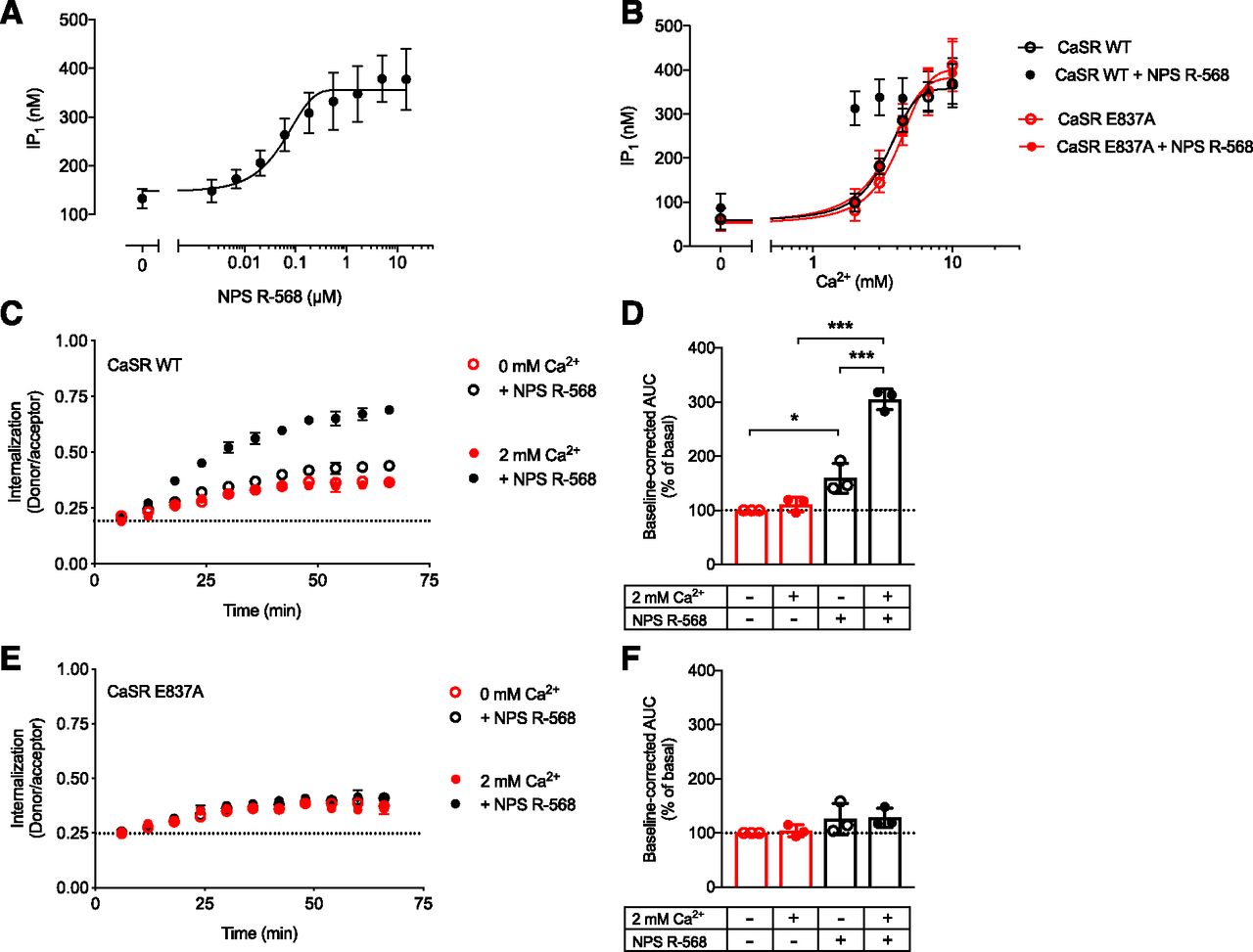
The CaSR allosteric site E837A mutant abolishes NPS 2143–induced inhibition of Ca2+-mediated CaSR internalization. (A) IP1 accumulation of CaSR WT upon stimulation with 4 mM Ca2+ and increasing NPS 2143 concentrations. (B) IP1 accumulation for CaSR WT and CaSR E837A upon stimulation with increasing Ca2+ concentrations in the absence or presence of 5 μM NPS 2143. (A and B) Data are mean ± S.E.M. of three independent experiments performed in triplicate. (C–F) The effect of NPS 2143 on CaSR WT (C and D) as well as CaSR E837A (E and F) internalization was measured in the absence (open symbols, constitutive internalization) or presence (closed symbols, Ca2+-mediated internalization) of 10 mM Ca2+. (C and E) Real-time traces and (D and F) normalized baseline-corrected area under the curve (AUC) data grouped from three independent experiments performed in triplicate. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test. Real-time internalization traces are presented as mean ± S.D. of a single representative experiment performed in triplicate. Significance is notated as follows: *P < 0.05; ***P < 0.001.

The CaSR allosteric site E837A mutant abolishes NPS R-568–induced potentiation of constitutive and Ca2+-mediated CaSR internalization. (A) IP1 accumulation of CaSR WT upon stimulation with 2 mM Ca2+ and increasing NPS R-568 concentrations. (B) IP1 accumulation for CaSR WT and CaSR E837A upon stimulation with increasing Ca2+ concentrations in the absence or presence of 5 μM NPS R-568. Data are mean ± S.E.M. of three independent experiments performed in triplicate. (C–F) The effect of NPS R-568 on CaSR WT (C and D) as well as CaSR E837A internalization (E and F) was measured in the absence (open symbols, constitutive internalization) or presence (closed symbols, Ca2+-mediated internalization) of 2 mM Ca2+. (C and E) Real-time traces and (D and F) normalized baseline-corrected area under the curve (AUC) data grouped from three independent experiments performed in triplicate. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test. Real-time internalization traces are presented as mean ± S.D. of a single representative experiment performed in triplicate. Significance is notated as follows: *P < 0.05; ***P < 0.001.
CaSR-specific NAMs and PAMs generally share an overlapping binding site in the seven-transmembrane domain. Previous publications have revealed that the modulating effects from both NPS 2143 and NPS R-568 can be eliminated by mutating the E837 residue situated within this overlapping binding site (Hu et al., 2002, 2006; Petrel et al., 2004; Jacobsen et al., 2017b). To determine if the observed modulation of CaSR internalization was directly mediated by the NAM and PAM, internalization and functional testing was performed with FLAG-SNAP-CaSR E837A.
CaSR E837A cell-surface expression was equivalent to CaSR WT as demonstrated by ELISA (Fig. 1B). Moreover, the Ca2+-induced effect on CaSR E837A was similar to CaSR WT in both the IP1 accumulation assay (Fig. 1A) and the real-time internalization assay (Fig. 3, D and E). Thus, mutating E837 did not affect surface expression or Ca2+-mediated receptor activation and internalization.
In accordance with previously published studies, the E837A mutant did prevent NPS 2143–mediated inhibition and NPS R-568–mediated potentiation of Ca2+-induced IP1 accumulation (Figs. 4B and 5B). When tested in the real-time internalization assay, the modulating effects of NPS 2143 and NPS R-568 were likewise abolished for the Ca2+-mediated internalization of CaSR E837A (Figs. 4, E and F and 5, E and F), while the Ca2+-mediated internalization of CaSR WT was subjected to NPS 2143-induced inhibition and NPS R-568-induced potentiation as expected (Figs. 4, C and D and 5, C and D). Furthermore, a small yet statistically significant increase in constitutive internalization from NPS R-568 (Fig. 5D, P < 0.05, one-way ANOVA followed by Tukey’s multiple comparisons test) was abrogated in CaSR E837A (Fig. 5, D and F). These data demonstrate that the CaSR-specific NAM NPS 2143 and PAM NPS R-568 are directly responsible for the modulation of CaSR internalization.
Constitutive and Ca2+-Mediated CaSR Internalization Is Independent of G Protein Activation.
It is widely accepted that the CaSR interacts with the Gq/11 and Gi/o protein families, although interaction with other G proteins has also been reported (Thomsen et al., 2012). To elucidate the mechanism of CaSR internalization, we investigated whether activation of these G proteins drive CaSR internalization. Commercially available natural products YM-254890 and PTX were used to inhibit Gq/11- and Gi/o-mediated activity, respectively (Campbell and Smrcka, 2018). As seen in Fig. 6A, 30-minute preincubation with 1 μM YM-254890 to block Gq/11-dependent signaling reduced Ca2+-mediated IP1 generation via CaSR WT to the basal IP1 level. Similarly, the Ca2+-mediated decrease in cAMP accumulation was abrogated by 24-hour preincubation with 80 ng/ml PTX (Fig. 6B).
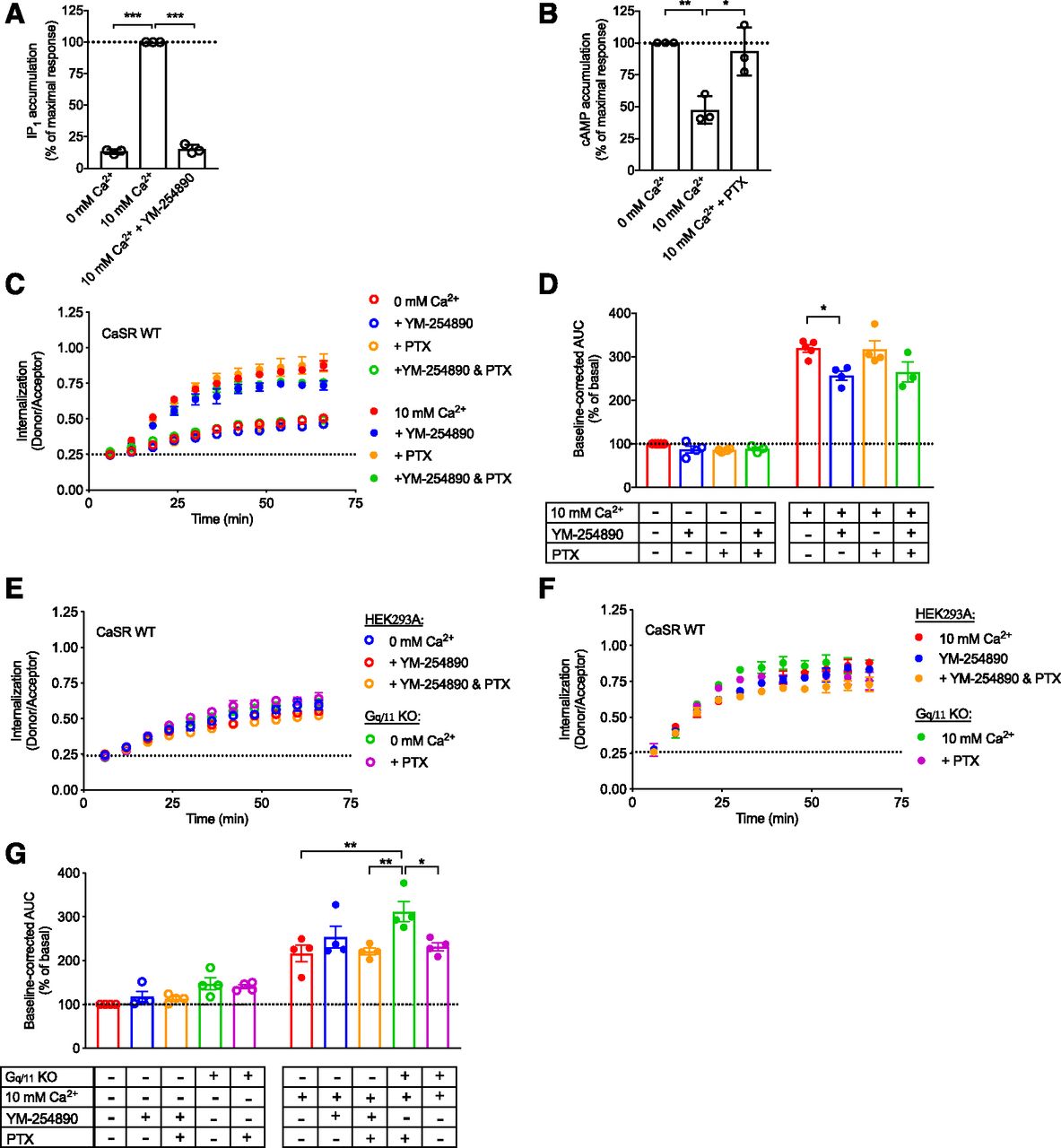
CaSR internalization is largely independent of G protein activation. HEK293T cells transiently transfected with FLAG-SNAP-CaSR WT were pretreated for 30 minutes with 1 μM Gq/11 of inhibitor YM-254890 or 24 hours with 80 ng/ml of Gi/o inhibitor PTX. (A) Normalized IP1 accumulation upon stimulation with 10 mM Ca2+ in the absence or presence of YM-254890. (B) Normalized cAMP accumulation upon stimulation with 10 mM Ca2+ in the absence or presence of PTX. (C) Real-time traces in the absence or presence of the G protein inhibitors for constitutive internalization (open symbols, 0 mM Ca2+) and Ca2+-mediated internalization (closed symbols, 10 mM Ca2+). (D) Normalized grouped area under the curve (AUC) data for constitutive and Ca2+-mediated internalization. Constitutive (E) and Ca2+-mediated (F) real-time internalization traces of FLAG-SNAP-CaSR WT transiently transfected in HEK293A or Gq/11 KO cells. (G) Normalized grouped AUC data after donor signal correction for constitutive and Ca2+-mediated internalization. (A, B, D, and G) Data are mean ± S.E.M. from three to five independent experiments performed in triplicate. (C, E, and F) Real-time data are mean ± S.D. of a single representative experiment performed in triplicate. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test (A, B, and G) and one-way ANOVA followed by Dunnett’s post-test (D) (*P < 0.05; **P < 0.01; ***P < 0.001).
Both G protein inhibitors did not affect constitutive CaSR internalization (Fig. 6, C and D, open symbols). Preincubation with PTX had no effect on Ca2+-mediated internalization, while a modest 20% decrease was observed in the presence of YM-254890 (Fig. 6, C and D, closed symbols, P < 0.05, one-way ANOVA followed by Dunnett’s post-test). Preincubation with PTX and YM-254890 did not further suppress Ca2+-mediated internalization, demonstrating that the two G protein families do not compensate for each other.
As presented in Fig. 6, E–G, similar results were obtained in CRISPR/Cas9 edited HEK293A cells lacking the Gq/11 protein (Gq/11 KO cells). In particular, both the constitutive and Ca2+-mediated internalizations were maintained in the Gq/11 KO cells, consistent with the pharmacologically blocked Gq/11 in YM-254890–treated HEK293A cells. Overnight PTX treatment in the Gq/11 KO cells led to a slight reduction in Ca2+-mediated internalization, but did not affect constitutive internalization (Fig. 6, E–G). Collectively, these results suggest that constitutive and Ca2+-mediated CaSR internalization is largely independent of Gq/11 or Gi/o activation.
CaSR Internalization Is Predominantly Driven by β-Arrestin.
To investigate the involvement of β-arrestin, CaSR internalization was measured in a CRISPR/Cas9 edited cell line lacking β-arrestin1 and β-arrestin2 (β-arrestin1/2 KO) (O’Hayre et al., 2017). By expressing the CaSR in both the β-arrestin1/2 KO cell line and its parental cell line HEK293A, it was demonstrated that constitutive internalization of the CaSR was reduced, while the Ca2+-mediated internalization of the CaSR was abolished in the absence of β-arrestins (Fig. 7A) despite having equivalent surface labeling of the CaSR in both cell lines (data not shown).
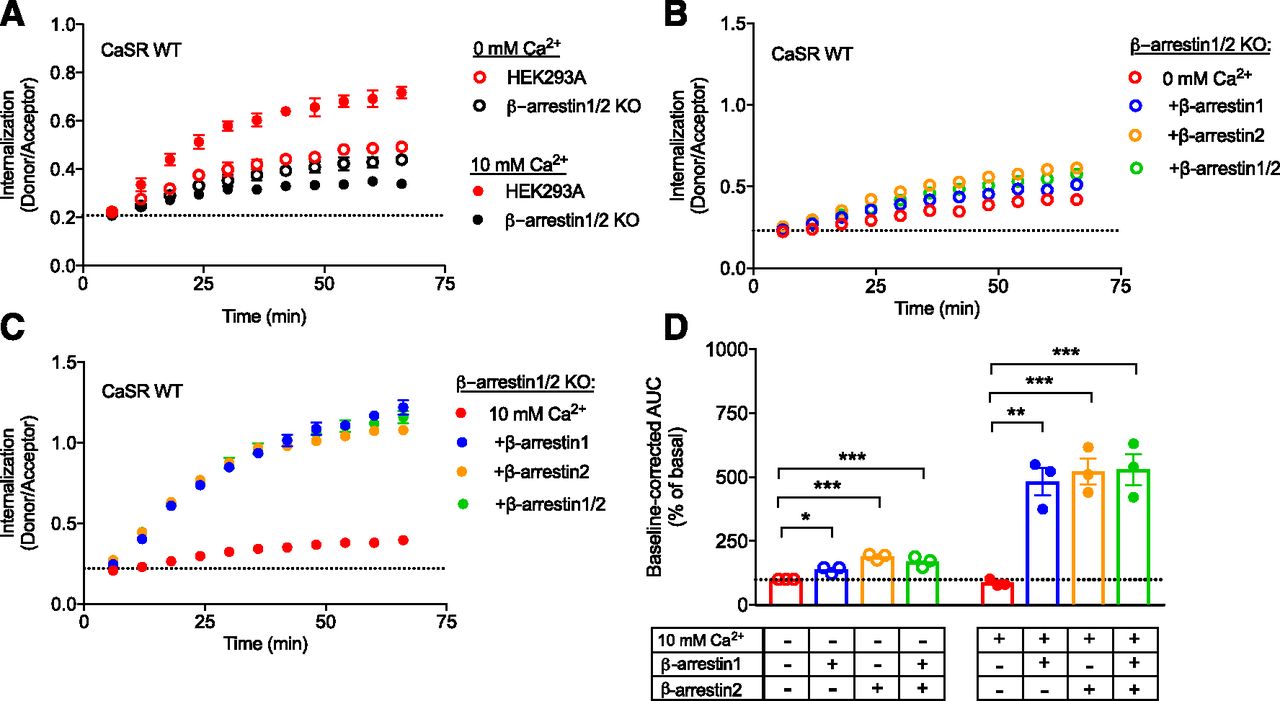
Constitutive and Ca2+-mediated CaSR internalizations are β-arrestin1/2 dependent. The β-arrestin1/2 KO cells and HEK239A cells were transiently transfected with FLAG-SNAP-CaSR WT. (A) Constitutive (open symbols, 0 mM Ca2+) and Ca2+-mediated (closed symbols, 10 mM Ca2+) real-time traces of β-arrestin1/2 KO cells and HEK293A cells. Similar results were generated in at least two additional experiments. (B) Constitutive (open symbols, 0 mM Ca2+) and (C) Ca2+-mediated (closed symbols, 10 mM Ca2+) real-time internalization traces of β-arrestin1/2 KO cells transiently cotransfected with empty vector, β-arrestin1, β-arrestin2, or a combination of both subtypes (β-arrestin1/2). (D) Normalized grouped area under the curve (AUC) data for constitutive and Ca2+-mediated internalization analyzed using one-way ANOVA followed by Dunnett’s post-test (*P < 0.05; **P < 0.01; ***P < 0.001). Real-time data presented in (A–C) are mean ± S.D. of a single representative experiment performed in triplicate, while AUC data in (D) are shown as mean ± S.E.M. of three independent experiments performed in triplicate.
To validate that the loss of Ca2+-mediated internalization was caused by the lack of β-arrestin expression, the two β-arrestin subtypes were transiently expressed in the β-arrestin1/2 KO cell line and CaSR internalization was measured. The introduction of each β-arrestin subtype did not affect CaSR surface labeling (data not shown). Constitutive internalization was significantly enhanced by both β-arrestin1 and β-arrestin2 (Fig. 7, B and D, one-way ANOVA followed by Dunnett’s post-test). Furthermore, the reintroduction of either β-arrestin1, β-arrestin2, or both β-arrestins restored Ca2+-mediated internalization in the β-arrestin1/2 KO cells (Fig. 7, C and D). These results demonstrate that CaSR internalization is largely β-arrestin dependent.
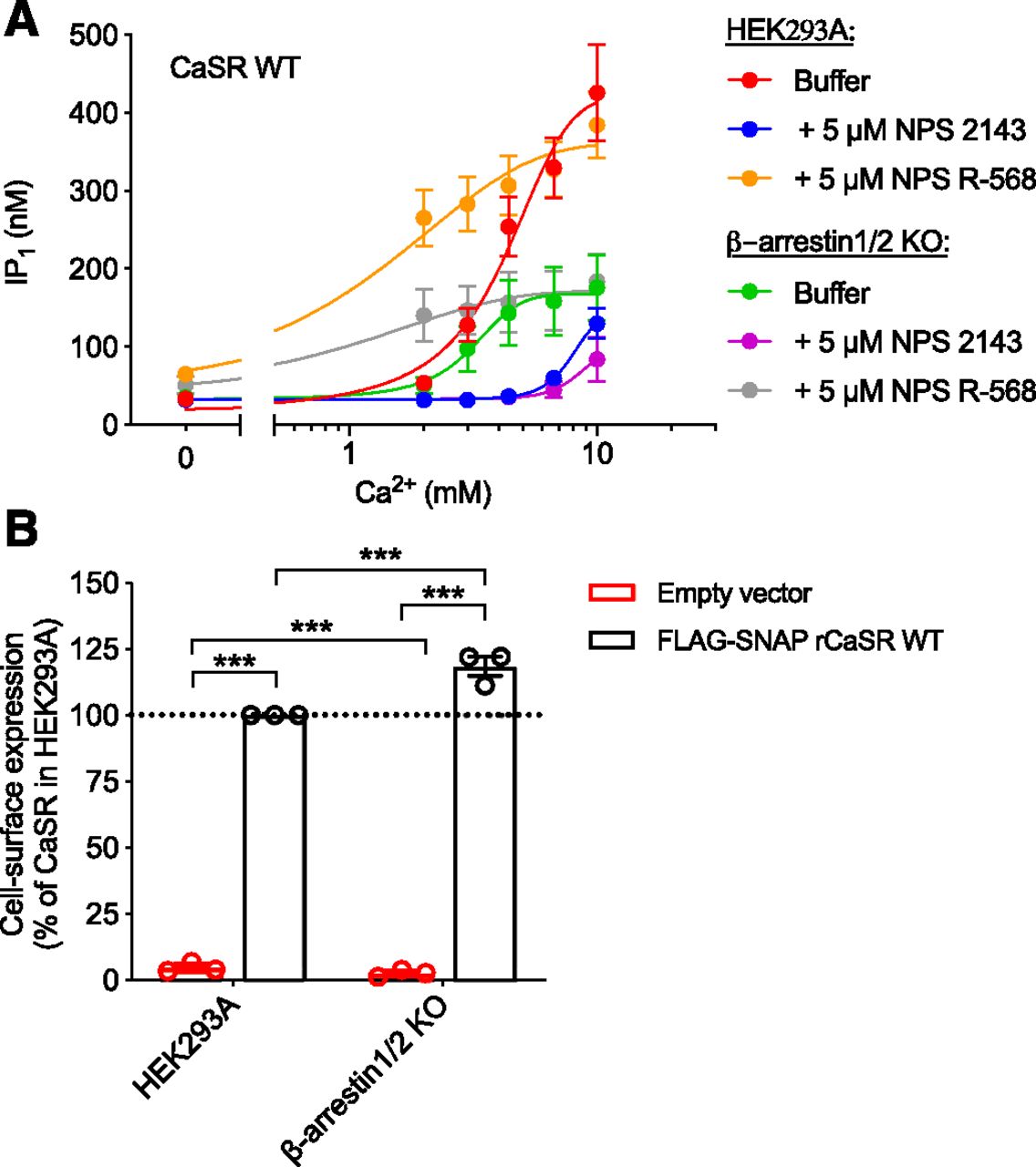
In this study, NPS 2143 and NPS R-568 have been shown to affect not only CaSR signaling but also internalization. The question remains whether these allosteric ligands modulate Ca2+-mediated CaSR signaling because they modulate internalization or vice versa. To investigate this, we measured the immediate effects of NPS 2143 and NPS R-568 on CaSR signaling in β-arrestin1/2 KO cells, where Ca2+-mediated internalization was shown to be abolished (Fig. 8A). Despite slightly higher cell-surface expression (Fig. 8B), the maximum Ca2+-induced IP1 accumulation response in the β-arrestin1/2 KO cells was lower than that observed with the HEK293A cells. However, NPS 2143 and NPS R-568 were still able to qualitatively modulate CaSR signaling in the β-arrestin1/2 KO cell line to the same extent as in the parental HEK293A cell line. Thus, removal of β-arrestin1/2, and thereby Ca2+-mediated internalization, does not impair modulation of CaSR signaling by allosteric ligands.

CaSR signaling in β-arrestin1/2 KO cells is still sensitive to allosteric modulation. The β-arrestin1/2 KO cells and HEK239A cells were transiently transfected with FLAG-SNAP-CaSR WT. (A) IP1 accumulation of CaSR WT after 30-minute stimulation with increasing Ca2+ concentrations in the absence or presence of 5 μM NPS 2143 or NPS R-568. (B) Cell-surface expression of CaSR WT measured using anti-FLAG antibodies in ELISA. Data are mean ± S.E.M. of three experiments performed in parallel with the IP1 accumulation assay. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test. Significance is notated as follows: ***P < 0.001.
Discussion
In the present study, we demonstrate that the CaSR displays constitutive and Ca2+-mediated internalization, which is β-arrestin dependent and sensitive to allosteric modulators. In accordance with our results, several studies have previously reported that the CaSR is constitutively internalized (Reyes-Ibarra et al., 2007; Grant et al., 2011; Zhuang et al., 2012). However, the potential impact of ambient CaSR ligands such as extracellular cations or L-amino acids was not addressed in these studies. In the current study, we have shown that constitutive internalization of the CaSR occurs independently of extracellular cations and L-amino acids, since constitutive internalization was not abolished by the chelating agent EDTA or the CaSR loss-of-function mutant S170A.
To gain these new insights into CaSR internalization, we have used a TR-FRET–based assay, which allows high-throughput, sensitive, and readily quantified detection of receptor internalization in real time (Foster and Bräuner-Osborne, 2018). This assay provides a useful platform to study the role of β-arrestin and allosteric modulation of CaSR internalization in greater detail than was previously possible. Indeed, there are inconsistencies in the literature regarding the key processes and proteins involved in CaSR internalization. Reyes-Ibarra et al. (2007) and Zhuang et al. (2012) showed constitutive CaSR internalization followed by recycling, while Grant et al. (2011) reported merely lysosomal degradation. In other studies, the CaSR has been reported to undergo Ca2+-mediated internalization (Pi et al., 2005; Lorenz et al., 2007). These data have been generated using a variety of approaches including receptor truncation/mutations, trafficking and signaling inhibitors (e.g., tunicamycin and gallein), and overexpression or small interfering RNA knockdown of proteins (such as β-arrestin1/2 or GPCR kinases) (Pi et al., 2005; Lorenz et al., 2007; Reyes-Ibarra et al., 2007; Grant et al., 2011, 2015; Zhuang et al., 2012), which likely explain the differences in conclusions drawn. Importantly, the majority of studies have used static measures of CaSR localization, including immunoprecipitation (Reyes-Ibarra et al., 2007) or immunofluorescence-based approaches (Zhuang et al., 2012), or have indirectly tracked CaSR internalization by measuring changes in functional desensitization and/or cell-surface expression (Pi et al., 2005; Lorenz et al., 2007; Grant et al., 2011, 2015). These approaches typically only allow a qualitative measure and not, as in the current study, a detailed quantitative measure of receptor internalization.
NPS R-568 and NPS 2143 are CaSR allosteric modulators with well-characterized pharmacological profiles (Nemeth et al., 1996; Nemeth, 2002), but their effect on CaSR internalization has not been systematically investigated. In the present study, we show that CaSR internalization is negatively and positively modulated by NPS 2143 and NPS R-568, respectively. These results are in agreement with previous findings on other receptors reporting allosteric modulation of ligand-mediated internalization for the class A M1 and M4 muscarinic acetylcholine receptors (Leach et al., 2010; Yeatman et al., 2014), D2 dopamine receptor (Basu et al., 2013), cannabinoid receptor type 1 (Ahn et al., 2012; Laprairie et al., 2015), and the class C metabotropic glutamate receptor 7 (Pelkey et al., 2007). Furthermore, NPS R-568 triggered a small but statistically significant increase in constitutive CaSR internalization. We propose that this could either be caused by positive modulation of the small amounts of ambient agonist present during the assay or direct activation of the CaSR internalization pathway.
Additionally, CaSR modulators have been reported to affect the biosynthesis and forward trafficking of WT and mutant CaSR. For NPS 2143 and NPS R-568, a respective decrease or increase in CaSR WT surface expression was obtained following overnight exposure with the allosteric modulators (Huang and Breitwieser, 2007; White et al., 2009; Cavanaugh et al., 2010; Leach et al., 2013). Here, we further demonstrate that these modulators affect CaSR internalization in a similar manner, although immediately after agonist stimulation.
CaSR interaction with β-arrestin1 and 2 has previously been shown to be crucial for functional desensitization, but not for Ca2+-mediated internalization (Pi et al., 2005; Lorenz et al., 2007). Pi et al. (2005) and Lorenz et al. (2007) used a flow cytometry method in which internalization was indirectly measured as a loss in cell-surface expression. In their studies, the potential involvement of β-arrestin was determined by β-arrestin overexpression, which could mask their importance since endogenously expressed β-arrestin in the cells could already saturate the internalization pathway. In the present study, we clearly demonstrate the requirement for β-arrestin in Ca2+-mediated CaSR internalization in cells lacking both β-arrestin subtypes. In reciprocal experiments, transfection with either β-arrestin1 or β-arrestin2 recovered internalization of the CaSR. Interestingly, both constitutive and Ca2+-mediated internalizations are largely independent of the Gq/11 and Gi/o signaling pathways, indicating that the β-arrestins are recruited independently of the major CaSR G protein pathways.
The CaSR PAM cinacalcet was the first allosteric GPCR modulator to be approved for clinical use. As a result, drug discovery for CaSR-related diseases has shifted interest toward the development of allosteric drugs. To date, a great number of allosteric ligands acting on the CaSR have been reported with well-known pharmacological and efficacy and safety profiles in clinical trials, but there is still limited knowledge on the clinical importance of CaSR internalization. For some GPCRs, constitutive internalization and recycling ensure a constant pool of functional receptors at the cellular surface (Hein et al., 1994; Mcdaniel et al., 2012; Basagiannis and Christoforidis, 2016). The CaSR is characterized by its constant or prolonged exposure to agonist, and thus a persistent cell-surface expression of functional receptors is required to retain responsiveness. Accordingly, constitutive internalization and recycling has been observed for the CaSR (Reyes-Ibarra et al., 2007; Zhuang et al., 2012) as well as for the related class C metabotropic glutamate receptor subtype 5 receptor (Trivedi and Bhattacharyya, 2012), GABAB receptor (Grampp et al., 2007; Vargas et al., 2008), and GPRC6A (Jacobsen et al., 2017a). Grant et al. (2011) propose an alternative strategy for the CaSR to remain responsive, in which prolonged Ca2+ exposure triggers constitutive internalization followed by degradation rather than recycling. Instead, the CaSR from a persistent intracellularly available pool is directed to the cell surface (Grant et al., 2011). Although we cannot investigate the anterograde trafficking of the CaSR since our TR-FRET assay requires labeling of cell-surface–expressed receptors, our principal findings on CaSR modulators agree with this hypothesis.
Interestingly, we find that agonist-induced IP1 signaling is reduced in the β-arrestin1/2 KO cell line compared with the parental HEK293A cell line, despite slightly increased receptor cell-surface expression in the former. This is in accordance with recent studies investigating disease-causing mutations of the adaptor protein-2σ subunit, which impair CaSR internalization, increase CaSR cell-surface expression, and reduce CaSR signaling (Gorvin et al., 2018). These results suggest that receptor internalization is required to obtain a full IP1 response, which could be driven by sustained signaling from internalized receptors (Gorvin et al., 2018), or alternatively point to the requirement for receptor internalization, dephosphorylation, and recycling to resensitize the receptor for sustained effects (Ward and Riccardi, 2012). However, more studies are needed to delineate the mechanism of this observation. Moreover, we find that the NAM and PAM qualitatively modulate the IP1 response to the same level in both cell lines, demonstrating that PAM/NAM effects on signaling are not downstream from modulating receptor internalization or vice versa.
In conclusion, we demonstrate that the CaSR undergoes both constitutive and Ca2+-mediated internalization using a TR-FRET–based real-time internalization assay. Ca2+-mediated CaSR internalization was shown to be concentration dependent with a Ca2+ potency equal to that determined for Gq/11-mediated IP1 accumulation. In addition, this study reveals that Ca2+-mediated CaSR internalization is sensitive to allosteric modulation, G protein independent, and largely β-arrestin1/2 dependent. More studies are needed to determine whether β-arrestin and/or G protein involvement is a universal phenomenon or specific to the HEK293 cellular background (i.e., the dependence on expression levels of receptor and intracellular signaling and scaffolding proteins). Future experiments investigating CaSR internalization upon stimulation with other CaSR-specific PAMs and NAMs could determine whether differences in pharmacological profiles can be related to biased signaling and distinct effects on internalization. Overall, these findings contribute to the full delineation of the mechanisms regulating CaSR signaling.
Acknowledgments
We thank Mie F. Pedersen for providing the β-arrestin cDNA constructs and for fruitful discussions regarding the use of β-arrestin1/2 knockout cells to study GPCR internalization. We thank Dr. Asuka Inoue (Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Japan, Japan) for the generous gift of the Gq/11 and β-arrestin1/2 knockout cell lines.
Authorship Contributions
Participated in research design: Mos, Jacobsen, Foster, Bräuner-Osborne.
Conducted experiments: Mos, Jacobsen.
Performed data analysis: Mos, Jacobsen.
Wrote or contributed to the writing of the manuscript: Mos, Jacobsen, Foster, Bräuner-Osborne.
Footnotes
- Received March 30, 2019.
- Accepted July 30, 2019.
↵1 Current affiliation: Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology, Monash University, Clayton, Australia.
↵2 I.M. and S.E.J. contributed equally to this work.
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under [Grant 675228], Lundbeck Foundation, Carlsberg Foundation, Augustinus Foundation, and Toyota Foundation.
Abbreviations
- β2AR
- β2-adrenergic receptor
- CaSR
- calcium-sensing receptor
- GLP-1R
- glucagon-like peptide 1 receptor
- GPCR
- G protein–coupled receptor
- HA
- hemagglutinin
- HBSS
- Hanks’ balanced salt solution
- HEK
- human embryonic kidney
- IP1
- inositol monophosphate
- KO
- knockout
- NAM
- negative allosteric modulator
- NPS 2143
- 2-chloro-6-[(2R)-2-hydroxy-3-[(2-methyl-1-naphthalen-2-ylpropan-2-yl)amino]propoxy]benzonitrile
- NPS R-568
- N-(3-[2-chlorophenyl]propyl)-(R)-α-methyl-3-methoxybenzylamine
- PAM
- positive allosteric modulator
- PTX
- pertussis toxin
- RT
- room temperature
- TR-FRET
- time-resolved fluorescence resonance energy transfer
- WT
- wild type
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}