


Abstract
The two G protein-coupled receptors (GPCRs) C-X-C chemokine receptor type 4 (CXCR4) and atypical chemokine receptor 3 (ACKR3) are part of the class A chemokine GPCR family and represent important drug targets for human immunodeficiency virus (HIV) infection, cancer, and inflammation diseases. CXCR4 is one of only three chemokine receptors with a US Food and Drug Administration approved therapeutic agent, the small-molecule modulator AMD3100. In this review, known modulators of the two receptors are discussed in detail. Initially, the structural relationship between receptors and ligands is reviewed on the basis of common structural motifs and available crystal structures. To date, no atypical chemokine receptor has been crystallized, which makes ligand design and predictions for these receptors more difficult. Next, the selectivity, receptor activation, and the resulting ligand-induced signaling output of chemokines and other peptide ligands are reviewed. Binding of pepducins, a class of lipid-peptides whose basis is the internal loop of a GPCR, to CXCR4 is also discussed. Finally, small-molecule modulators of CXCR4 and ACKR3 are reviewed. These modulators have led to the development of radio- and fluorescently labeled tool compounds, enabling the visualization of ligand binding and receptor characterization both in vitro and in vivo.
SIGNIFICANCE STATEMENT To investigate the pharmacological modulation of CXCR4 and ACKR3, significant effort has been focused on the discovery and development of a range of ligands, including small-molecule modulators, pepducins, and synthetic peptides. Imaging tools, such as fluorescent probes, also play a pivotal role in the field of drug discovery. This review aims to provide an overview of the aforementioned modulators that facilitate the study of CXCR4 and ACKR3 receptors.
Introduction
Chemokine receptors are a family of 24 seven-transmembrane (7TM) domain G protein-coupled receptors (GPCRs) that respond to chemokines, a class of 52 chemotactic cytokines (Bachelerie et al., 2013). The chemokine-chemokine receptor system is involved in the regulation of immune response, inflammation, and cancer (Scholten et al., 2012). In this review we focus on two related chemokine receptors, C-X-C chemokine receptor type 4 (CXCR4) and atypical chemokine receptor 3 (ACKR3), both binding the chemokine C-X-C chemokine ligand 12 (CXCL12). CXCR4 function is critical for the localization of hematopoietic stem cells, for binding of HIV-1 to T-cell-tropic strains, and cancer cell development (Feng et al., 1996; Horuk, 1999; Chatterjee et al., 2014; Pozzobon et al., 2016; Teixidó et al., 2018; Neves et al., 2019). ACKR3, also known as C-X-C chemokine receptor type 7 (CXCR7), is not a classic GPCR and signals primarily through β-arrestin recruitment (Rajagopal et al., 2010) and therefore belongs to the class of atypical chemokine receptors (ACKRs). The properties of the ACKR3-CXCL12 and ACKR3-C-X-C chemokine ligand 11 (CXCL11) axes make them promising therapeutic targets (Nibbs and Graham, 2013; Sánchez-Martín et al., 2013; Benhadjeba et al., 2018; Quinn et al., 2018; Wang et al., 2018; Koenen et al., 2019). Modulation of CXCL12-scavenging activity of ACKR3 regulates CXCR4 function (Abe et al., 2014). Both receptors form heterodimers (Levoye et al., 2009; Fumagalli et al., 2019) and play an important role in CXCL12 biology (Hattermann and Mentlein, 2013; Krikun, 2018; Murphy and Heusinkveld, 2018). The CXCR4/ACKR3/CXCL12 system remains a contemporary target for therapeutic application, and the development of novel, potent, and selective modulators is of great interest to both academia and the pharmaceutical industry.
Pharmacological modulation of CXCR4 and ACKR3 has been of great interest, and considerable effort has been devoted to the discovery and development of a range of ligands, including small-molecule modulators, pepducins, synthetic peptides, and imaging tools such as fluorescent probes. This review aims to give a concise overview of such modulators.
Structural Determinants of Chemokine Receptor Ligand Binding
Chemokine receptor structures consist of seven α-helical TM (7TM) domains, linked through three extracellular (ECL) and three intracellular (ICL) loops, with an N- and a C-terminus (Fig. 1A). Features that define chemokine receptor structures include a particular helix positioning where the top of TM2 is tilted owing to the presence of the T2.56xP2.58 motif, a feature that is shared with other protein and peptide receptors (e.g., opioid receptors); a highly conserved disulfide bridge between the N-terminus and ECL3; the open, solvent-accessible 7TM domain; and a diversity of druggable pockets, in line with the chemical diversity among chemokine receptor ligands (Arimont et al., 2017).
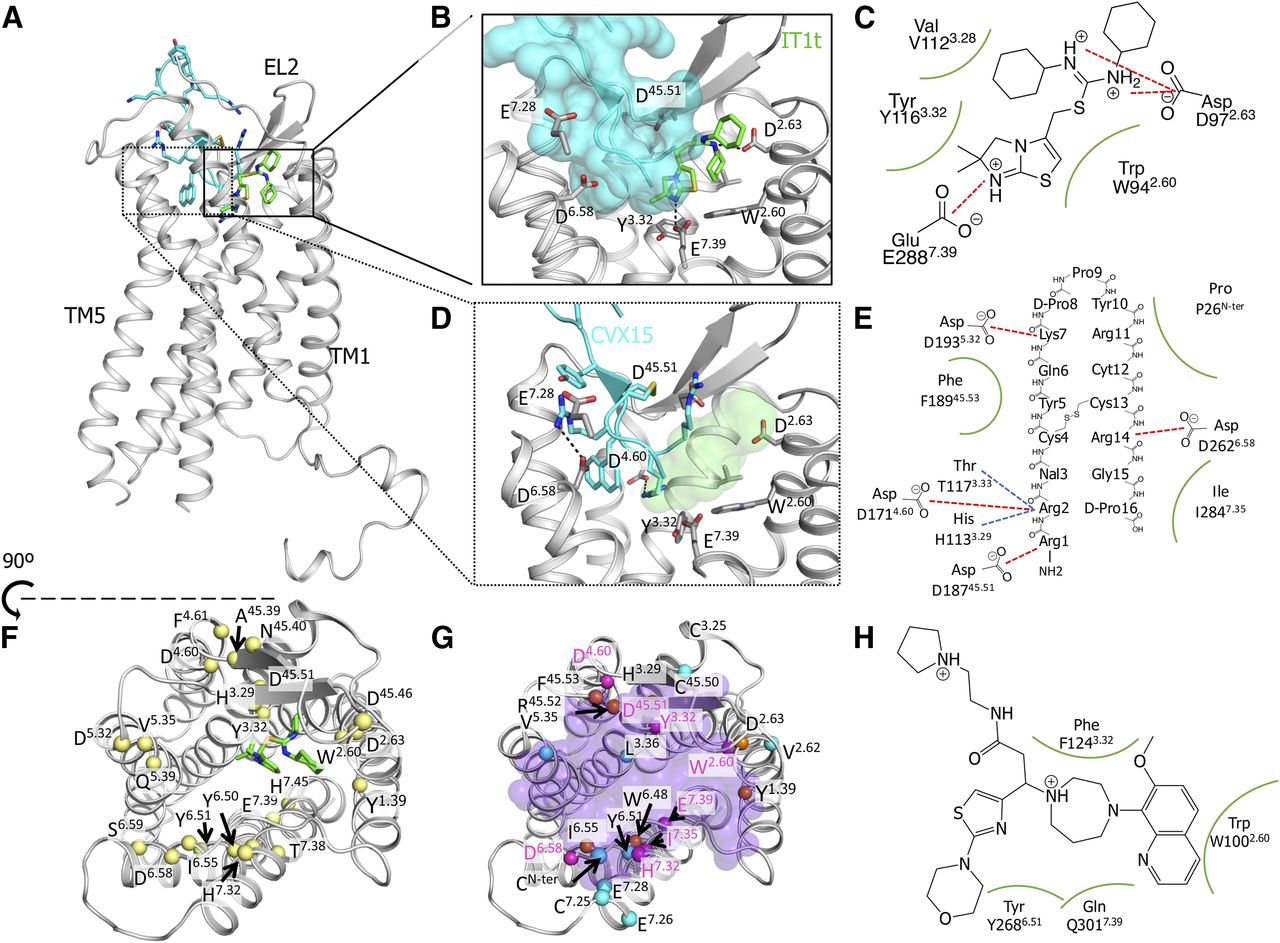
(A) Overview of the CXCR4 crystal structure (gray cartoon) bound to the small-molecule antagonist IT1t (green sticks, PDB ID: 3ODU) (Wu et al., 2010) and the synthetic peptide antagonist CVX15 (cyan cartoon and sticks, PDB ID: 3OE0) (Wu et al., 2010). (B) Binding details of IT1t to CXCR4. Side chains of relevant interacting residues are shown in gray sticks, and the CVX15 surface is shown transparent for reference. (C) IT1t-bound CXCR4 two-dimensional interaction map. Ionic interactions are represented as red dashed lines; hydrophobic contacts are represented as green lines. (D) Binding details of CVX15 to CXCR4. Side chains of relevant interacting residues are shown in gray sticks and IT1t surface is shown transparent for reference. (E) CVX15-bound CXCR4 two-dimensional interaction scheme. Ionic interactions are represented as red dashed lines; hydrogen bonds are represented as blue dashed lines; hydrophobic contacts are represented as green lines. (F) CXCR4 map of relevant residues (C-alpha shown as yellow spheres) for small-molecule binding and/or function as proven by site-directed mutagenesis (SDM) experiments. IT1t is shown as green sticks as a reference. (G) CXCR4 map of relevant residues for signaling of CXCL12 (C-alpha shown as blue spheres), peptidomimetics (orange spheres), or both (magenta spheres), as proven by SDM. Viral macrophage inflammatory protein-II (vMIP-II) is shown as transparent purple spheres as a reference. (H) CCX777 two-dimensional predicted-interaction map. Hydrophobic contacts predicted through mutant experiments are represented as green lines.
The endogenous ligands of chemokine receptors, chemokines (Fig. 2, A and B), are thought to bind in a two-step process: 1) The globular core of the chemokine binds to the N-terminus and extracellular loops of the receptor, allowing 2) the N-terminus of the chemokine to bind in the 7TM domain (Kufareva et al., 2014). The orthosteric pocket of chemokine receptors can be divided into a minor or small pocket, formed by residues in TM1–TM3, and TM7, and a major pocket, composed of residues in TM3–TM7. This binding site is wider and more solvent-accessible than that in aminergic GPCRs, and it contains a high number of negatively charged residue side chains that are often involved in ligand binding (Arimont et al., 2017). The negatively charged pocket of chemokine receptors is complementary to the positively charged nature of most of their ligands. Finally, X-ray and site-directed mutagenesis studies have proven the existence of an intracellular binding site that can be targeted by small molecules (de Kruijf et al., 2011; Scholten et al., 2014; Oswald et al., 2016; Zheng et al., 2016) and potentially by nanobodies, as exemplified by the stabilizing nanobody Nb7 (Burg et al., 2015).

Structure and structure-activity relationship of chemokine derivatives and peptidomimetics targeting CXCR4 and ACKR3. (A) Sequence alignment of chemokines CXCL11, CXCL12, and vMIP-II. The different domains and motifs are indicated on top. SAR is indicated in gray boxes. (B) Conserved tertiary structure of chemokines with CXCL12 crystal structure as a basis (PDB ID: 3GV3) (Murphy et al., 2010). The main structural motifs are conserved among different chemokines, including a free, flexible N-terminus (green) linked through a 310 loop to the globular core of the chemokine (gray), consisting of three antiparallel β-strands and a C-terminus α-helix (Scholten et al., 2014). This tertiary structure is maintained through one or more disulfide bridges (yellow), the number and spacing of which allow for classification of chemokines in the C-, CC-, CXC-, and CX3C- subfamilies. (C) Sequence and SAR of CXCR4 representative peptidomimetic modulators T22, T134, and FC131. SAR is indicated in gray boxes.
CXCR4 crystal structures reveal how chemokine receptors are regulated by different types of modulators, which bind in different (sometimes overlapping) binding pockets. The small-molecule isothiourea IT1t (Wu et al., 2010) binds exclusively in the minor pocket (Fig. 1B), where it makes ionic interactions with D972.63x63 and E2887.39x38 (Fig. 1C). The synthetic peptide CVX15 (Wu et al., 2010) binds into the major pocket (Fig. 1D) and makes interactions with D1714.60x61 and D2626x58x58 (Fig. 1E). Site-directed mutagenesis studies prove that binding of small-molecule ligands is not limited to the minor pocket. Rather, ligands can bind either the minor or major subpocket, or both (Fig. 1F). Binding of peptides and peptidomimetics often overlaps with the chemokine binding site (Fig. 1G) and comprises residues in the extracellular surface of the receptor, as well as more buried residues within the TM bundle, where it overlaps with small molecules. However, neither IT1t nor CVX15 fully overlap with the binding pocket of CXCL12 or viral macrophage inflammatory protein-II (vMIP-II) (Fig. 1, F and G). The partial overlap between the sites of chemokines, peptides, and small molecules explains the allosteric nature of a number of chemokine receptor ligands (Wu et al., 2010).
No ACKR3 crystal structures are available to date, and information on ACKR3 modulators is still scarce. The identification of the binding mode of ACKR3 modulators is therefore very challenging. Only one study on ACKR3 modulators reported potential interactions with the small molecule CCX777 (Gustavsson et al., 2017), but no key ionic interactions were identified (Fig. 1H). The lack of structural information and the challenging nature of chemokine receptor binding sites reflect the low success rate of drug discovery campaigns on noncrystallized receptors like ACKR3 (Oishi et al., 2015).
Chemokine and Peptide Binding to CXCR4 and ACKR3
CXCR4 and ACKR3 both bind the natural chemokine CXCL12 (Hesselgesser et al., 1998; Balabanian et al., 2005), the viral chemokine vMIP-II (Kledal et al., 1997; Szpakowska et al., 2016), and other endogenous proteins such as β-defensins 2 and 3 (Quiñones-Mateu et al., 2003; Feng et al., 2006). ACKR3 also binds CXCL11 (Burns et al., 2006) and adrenomedullin (Klein et al., 2014), and CXCR4 is reported to bind ubiquitin (Saini et al., 2010). For more than two decades, multiple studies focused on obtaining new or improved ligands using modifications of natural protein ligands as a basis to mimic their function. These modifications include point mutations, truncations, fragmentation, and the generation of mimetics and chimeras. An overview of chemokines, peptide, peptidomimetics, and modifications thereof that bind CXCR4 and/or ACKR3 is contained in Supplemental Table 1.
A number of studies focusing on mutagenesis of the endogenous agonists CXCL11 and CXCL12 allow the evaluation of the structural requirements for their activity (Fig. 2A). Importantly, the first two residues of CXCL12 are key for CXCR4 activity, as their mutation transforms it into an antagonist with lower affinity (Crump et al., 1997; Loetscher et al., 1998). Moreover, peptidomimetics of the N-terminus of the chemokines (Fig. 2B) are sufficient to promote CXCR4 and ACKR3 activity, which will allow the use of smaller and bioavailable peptide ligands as potential candidates for further drug discovery (Crump et al., 1997; Loetscher et al., 1998; Zhou et al., 2002; Ehrlich et al., 2013; Xu et al., 2013; Mo and Sun, 2015; Szpakowska et al., 2018).
Most CXCR4 peptide and peptide-like modulators are derivatives of polyphemusin II, a peptide isolated from immune cells of the horseshoe crab Limulus polyphemus (Murakami et al., 1997). The first reported CXCR4 antagonist analog of polyphemusin II, the 18-amino acid peptide T22 (Fig. 2C), inhibits the ability of HIV-1 to infect cells (Murakami et al., 1997). Since then, multiple studies have focused on the improvement of this peptide in terms of affinity; potency; biostability; and absorption, distribution, metabolism, excretion (ADME) properties, often through the incorporation of unnatural amino acids and always keeping a cyclic structure. Structure activity relationship (SAR) studies of T22 indicate that the presence of the disulfide loops and the amount of positive charges are key for anti-HIV activity (Tamamura et al., 1998a). Reducing the length and amount of disulfide bridges of T22 leads to peptides with higher potency without a significant change in secondary structure, such as in the peptide TW70 (Tamamura et al., 1998b). To stabilize the β-turn conformation in TW70, the unnatural amino acid d-Lys was introduced as a d-Lys-Pro motif into TW70. T22 and TW70 are highly basic peptides (eight positively charged residues), and as a consequence they show high collateral cytotoxicity. Analogs with reduced positive charges, such as T134, have higher selectivity indexes compared with T22 and TW70, which has been attributed to a decrease in cytotoxicity (Tamamura et al., 1998c) (Fig. 2C). Modification of a tryptophan in the sequence of T134 by a naphthyl-alanine results in a 5-fold higher anti-HIV activity than the lead peptide, as in the peptide T140 (Tamamura et al., 1998d). Amidation of the T140 C-terminus (TZ14004) increases the stability in serum but adds a positive charge, increasing cytotoxicity (Tamamura et al., 2001). Modification of positively charged residues by citrulline residues (x1 in TN14003, x2 in TC14012), in addition to the amidation, yields highly potent, less cytotoxic, and stable peptides (Tamamura et al., 2001). TC14012 has also proven to be an agonist of ACKR3 (Gravel et al., 2010). To improve biostability and keep selectivity and cytotoxicity in balance, a series of substitutions of Arg and/or Lys by Glu were designed, with an additional N-alkyl-amidation at the N-terminus. This yielded the (Ac-) TE14011 series (Tamamura et al., 2003a,b). Other N-terminal substitutions of TE14011 were also designed to prevent biodegradation, including guanylation (TF14002), Nα-tetramethylguanylation (TF14003), and Nα-benzoylation (TF14013 or 4F-benzoyl-TE14011) (Tamamura et al., 2003b). 4F-benzoyl-TN14003 and 4F-benzoyl-TE14011 show strong anti-HIV activity in vitro, antimetastatic activity against breast cancer and melanoma, and anti–rheumatoid arthritis activity in experimental models (Tamamura et al., 2003c, 2004; Takenaga et al., 2004). Using the β-hairpin structure of the natural product polyphemusin II and its analogs as a template, another series of macrocyclic template-bound β-hairpin mimetics was developed in iterative cycles, yielding compounds such as POL3026, also known as CVX15 (DeMarco et al., 2006) (Fig. 1). SAR studies on T140 identified four indispensable amino acids (Arg2, Nal3, Tyr5, and Arg14) responsible for its bioactivity (Fujii et al., 2003). These residues were therefore used to design cyclic pentapeptides, enabling the development of low-molecular-weight CXCR4 antagonists with bioactivities comparable to T140, including FC131 (Fujii et al., 2003). Multiple series of FC131 were developed aiming to reduce its peptide character and increase drug-likeness (Tamamura et al., 2005a-b), to generate conformationally constrained analogs (Tamamura et al., 2005a, b), and to develop bivalent ligands to study CXCR4 dimers (Tanaka et al., 2010). FC131 and derivatives are also agonists of ACKR3 (Oishi et al., 2015).
Other peptidomimetics not using polyphemusin II as a basis include peptides that mimic HIV proteins and mimetics of CXCR4 domains and of its ligand CXCL12. ALX40-4C was designed to mimic the basic domain of Tat, the HIV-1 transactivation protein. It is a nine-Arg polypeptide that includes d-amino acids and terminal protection for stabilization, and it has been proven to inhibit the use of CXCR4 by HIV-1 (Doranz et al., 1997). Multiple peptides were designed on the basis of the extracellular domain of CXCR4 (N-terminus and extracellular loops). Interactions between the highly conserved, positively charged residues of glycoprotein 120 (gp120) from the HIV envelope and the negatively charged extracellular domain of CXCR4 are key for the binding of gp120 to CXCR4 and ulterior entrance of the virus into the cell. These peptides show different ranges of anti-HIV activity (Hashimoto et al., 2013; Chevigné et al., 2014). In one particular study peptides were designed to mimic the N-terminus and extracellular loops 1 and 2, both in a linear and cyclic structure. All peptides show anti-HIV activity, but the cyclic peptides are more potent and less cytotoxic (Hashimoto et al., 2013). A CXCR4 ECL2-based peptide also inhibits ACKR3 internalization (Chevigné et al., 2014). A ligand-based approach used the motif Aro-Aro-Arg, where Aro is an aromatic residue, for peptidomimetics design. This motif is present in CXCL12 and also found in vMIP-II in reverse orientation, i.e., Arg-Aro-Aro. Four peptides, called R, S, T, and I, inhibit CXCL12-dependent cell migration, and decrease cancer growth and metastases in vitro. One particular CXCR4-binding cyclopeptide, POL6326 (Balixafortide), has progressed into clinical studies. The results for phase I studies have been published (Karpova et al., 2017; Pernas et al., 2018), and a phase III clinical trial was launched in early 2019 to evaluate the antagonist in combination with eribulin for the treatment of patients with metastatic breast cancer. Balixafortide is the only CXCR4 antagonist in development for breast cancer (ClinicalTrials.gov Identifier: NCT03786094).
Some studies report the use of chimeras consisting of different CXCR4 modulators. Examples include a chimera consisting of CXCL12 and the peptide T140 that acts as a partial CXCR4 agonist (Lefrançois et al., 2011), and one consisting of CXCL12 and the small-molecule antagonist IT1t, reported to be an antagonist with low affinity (Mona et al., 2016).
A chemokine-like chimera, named CXCL11_12, has been reported as a high-affinity ACKR3-selective ligand. The structure of the engineered chimera consists of the N-terminus of CXCL12 and the main body and C-terminus of CXCL11, and the study showed the internalization of CXCL11_12 by mouse ACKR3, making the chimera a useful tool to study ACKR3 (Puddinu et al., 2017; Ameti et al., 2018).
Pepducins
A specific subset of lipid-modified peptide-based modulators called pepducins (Covic et al., 2002; (O’Callaghan et al., 2012a,b) has been described for CXCR4 (Table 1). The peptide sequence of a pepducin is derived from one of the intracellular loops of a target GPCR and is typically connected to palmitic acid or other fatty acids via an amide bond. Pepducins can act as positive or negative allosteric GPCR modulators, and experimental evidence (Covic et al., 2002; Wielders et al., 2007; Janz et al., 2011) and modeling (Planesas et al., 2015) suggest that they bind to an allosteric, intracellular site on the target receptor. Their lipid tail is crucial for activity as it probably anchors the pepducin in the cell membrane (Covic et al., 2002).
Pepducins described for CXCR4a
All C-terminal ends are modified to an amide; N-terminal end Pal is Palmitic acid; all listed pepducins for CXCR4 are derived from ICL1 except PZ-210. R1 = R2 = L-Photo-Leucine.

ATI-2341 (Tchernychev et al., 2010) (Fig. 3), which is derived from the first intracellular loop of CXCR4, is probably the best described pepducin for CXCR4 and acts as a biased agonist (Quoyer et al., 2013), activating multiple G protein-activated signaling pathways downstream of CXCR4. ATI-2341 has been shown to variously cause a Gi-mediated inhibition of cAMP production, stimulate calcium mobilization, modify polymorphonuclear neutrophil mobilization, and elicit chemotaxis (Tchernychev et al., 2010). CXCR4 internalization has also been observed following addition of ATI-2341 in imaging techniques (Tchernychev et al., 2010; Dimond et al., 2011). However, it has also been shown using flow cytometry or bioluminescence resonance energy transfer (BRET)-based approaches that ATI-2341 does not or only partially recruits β-arrestins to cell surface CXCR4 and this appears to be accompanied by a weak receptor internalization compared with CXCL12. The authors suggest that these results might be attributable to different CXCR4 phosphorylation profiles promoted by ATI-2341 versus CXCL12 (Quoyer et al., 2013). The ability of ATI-2341 to recruit G proteins (Gα,i, G13) has also been investigated with BRET using a C-terminal renilla-luciferin 2-monooxygenase-tagged CXCR4 and a yellow fluorescent protein-tagged G protein. These studies concluded ATI2341 activates Gα,i in preference to G13, and this recruitment of G proteins is sensitive to pertussis toxin (Quoyer et al., 2013). Consistent with its low efficacy for β-arrestin recruitment, ATI-2341 also recruits lower levels of G protein-coupled receptor kinases (GRK) 2 and 3 (Quoyer et al., 2013). Furthermore, ATI-2341 only promotes protein kinase C–mediated phosphorylation of serine residues at the C-terminus of CXCR4, but not by GRK6 (Quoyer et al., 2013).

Schematic representation of ATI-2341 (peptide on the left) and CXCR4 (green) showing the amino acids of intracellular loop 1; representing the proposed localization of the pepducin in relation to the GPCR with the lipid of the pepducin serving as an anchor in the cell membrane and an intracellular interaction site.
Publications concerning ATI-2341 also describe pepducins with lower potencies and a fluorescent pepducin, ATI-2766, which can crosslink to CXCR4 and N-terminal-truncated variants (Janz et al., 2011). Mice and nonhuman primates release hematopoietic stem/progenitor cells from bone marrow after intravenous administration of ATI-2341, suggesting the potential of pepducins to induce stem cell mobilization (Tchernychev et al., 2010; O’Callaghan et al., 2012a).
PZ-218, another CXCR4 first internal loop pepducin, and PZ-210, derived from the third intracellular loop, are also CXCR4 antagonists (Kaneider et al., 2005; O’Callaghan et al., 2012b). They both antagonize CXCL12-mediated calcium mobilization in human neutrophils (Kaneider et al., 2005; O’Callaghan et al., 2012b), inhibit extracellular signal–regulated kinase activation in Jurkat cells (O’Callaghan et al., 2012b) and inhibit CXCL12-mediated chemotaxis in a range of cell lines (Kaneider et al., 2005; O’Callaghan et al., 2012). Combined with rituximab, these antagonist pepducins increase the cytotoxicity in leukemia cells from patients with chronic lymphocytic leukemia (Kaneider et al., 2005; O’Callaghan et al., 2012b) and in a mouse model of disseminated lymphoma—the combination with rituximab leads to an increase in survival (O’Callaghan et al., 2012b).
Small-Molecule Modulators of CXCR4 and ACKR3
The field of small-molecule CXCR4 antagonists has been reviewed before in detail (Zhan et al., 2007; Peng et al., 2018). Therefore, we focus on key examples. In a manner similar to the peptidomimetics that were first identified, the first small-molecule modulators of CXCR4 were initially identified as inhibitors of HIV entry. The most prominent CXCR4 antagonist, AMD3100, was discovered in 1994 as a highly potent and selective inhibitor of HIV entry (De Clercq et al., 1994). The actual target of AMD3100 turned out to be the CXC-chemokine receptor CXCR4, which is used by T-lymphotropic HIV strains to enter immune cells (Donzella et al., 1998). A phase I clinical trial with AMD3100 as an anti-HIV drug showed an increase in the white blood cell counts as an unexpected side effect. Further studies showed that AMD3100 specifically increases cluster of differentiation 34 (CD34+) hematopoietic stem cell (HSC) counts by antagonism of the CXCL12 interaction with CXCR4. De Clercq (2009) wrote a comprehensive overview of the discovery path of AMD3100 and recently reviewed the potential application of AMD3100 in other diseases, like the warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome (De Clercq, 2019).
As of 2008, AMD3100 (Fig. 4) [international nonproprietary name: plerixafor] is the first CXCR4 modulator approved for clinical use (Mozobil by Genzyme) in coadministration with granulocyte colony-stimulating factor for the mobilization of CD34+ cells in non-Hodgkin lymphoma (NHL) and multiple myeloma (MM). AMD3100 is still being examined in clinical trials for expanded use (ClinicalTrials.gov Identifiers: NCT03277209, NCT02231879, NCT01318317), but all phase II and phase III clinical trials for AMD3100 involve mobilization of HSCs in NHL, MM, and other hematologic diseases. Trials with AMD3100 for the treatment of HIV show a limited safety profile with heart- and gastrointestinally related adverse effects with the dose/time that would give a small therapeutic effect. Moreover, since AMD3100 is also not orally bioavailable, further development for the treatment of HIV-1 infection was discontinued (Hendrix et al., 2000, 2004). A series of ongoing trials are focused on the use of AMD3100 in combination with chemotherapy. The effect of metal chelation by AMD3100 has been assessed. For example, a complex with Zn2+ ions shows slightly higher anti-HIV activity, whereas complexes of Cu2+ and Pd2+ result in a 4-fold and a 7500-fold less active molecule compared with AMD3100, respectively (Bridger et al., 1995).
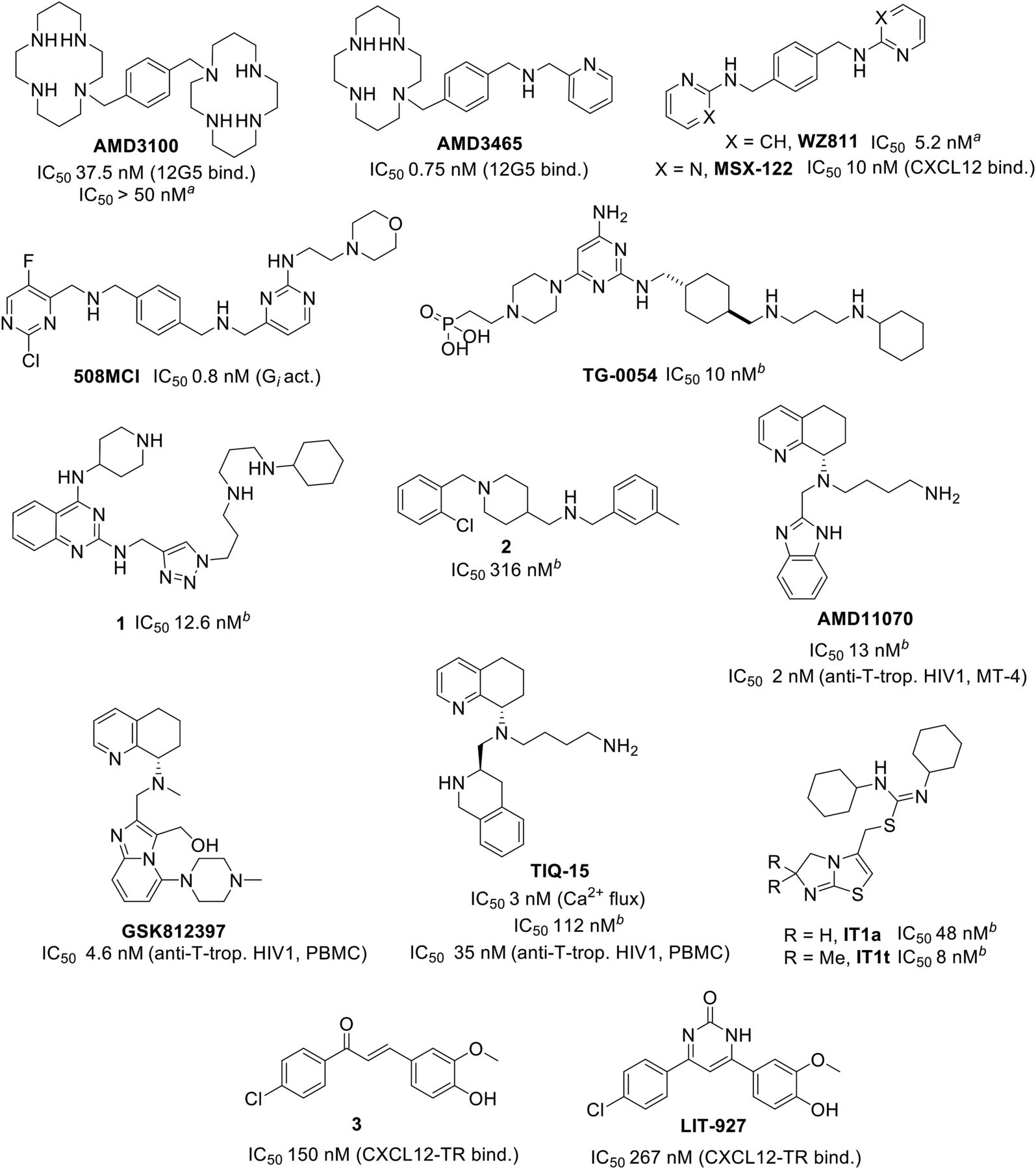
Selected small-molecule CXCR4 antagonists. aInhibition of CXCL12-mediated Matrigel invasion; bCXCR4 125I-CXCL12 inhibition binding assay.
An initial attempt to improve the bioavailability of bicyclams such as AMD3100 resulted in the discovery of AMD3465 with a more drug-like structure, and an improved potency. However the oral bioavailability was not improved (Hatse et al., 2005).
One of the first nonmacrocyclic small modulators that has been discovered is WZ811 (Zhan et al., 2007). However, this 1,4-phenylenedimethanamine has a poor pharmacokinetic profile owing to rapid metabolism. Optimization of WZ811 led to the discovery of MSX-122, a partial CXCR4 antagonist with superior Matrigel invasion inhibition compared with AMD3100. Additionally, MSX-122 is a selective inhibitor of the Gα,i pathway (cAMP inhibition), leaving the alternative Gα,q (calcium flux) pathway untouched. As a result, MSX-122 has a distinct functional profile of inhibition of CXCR4-mediated inflammation, metastasis, and chemotaxis without mobilizing stem cells (Liang et al., 2012). Interestingly, the ligand cannot block 125I-CXCL12 binding, whereas CXCL12 can block fluorine-18-labeled MSX-122 from binding to CXCR4, supporting the hypothesis that the small-molecule ligand blocks certain receptor functions via interference with CXCR4/CXCL12-mediated signaling (IC50 = 10 nM). Moreover, the therapeutic potential of MSX-122 was evaluated in three different murine models (Liang et al., 2012) and it was the first analog of AMD3100 to enter a clinical trial phase I in 2007 in patients with refractory metastatic or locally advanced solid tumors (ClinicalTrials.gov identifier: NCT00591682). However, the trial was suspended in 2008 and no further details on the clinical effectiveness of MSX-122 have been published.
In an attempt to address metabolic instability, another series of inhibitors were synthesized bearing scaffolds with a reduced electron density by introduction of pyrimidines, halogens, and other substituents. This led to the discovery of 508MCl, which displays subnanomolar potency in a CXCL12-mediated inhibition of cAMP production (IC50 = 0.8 nM); it is almost 1000-fold more potent than AMD3100 (IC50 = 695 nM). 508MCl has also been evaluated in vivo for inflammation, lung fibrosis, and tumor metastasis, and it was effective in all tested model systems. Although its biologic activity in vitro and in vivo is favorable, it has a very short half-life of about 15 minutes and is not orally bioavailable (Zhu et al., 2010).
Another CXCR4 inhibitor developed by TaiGen Biotechnology, compound TG-0054 (also known as burixafor) (Fig. 4) was applied in injections of poly(l-lactide) microparticle formulation for the treatment of choroid neovascularization (Shelke et al., 2011). In addition, its potential for treating the aftermath of a myocardial infarction was demonstrated in a pig model (Hsu et al., 2015), and the molecule has entered a clinical phase I trial to assess its use as a sensitizer in metastatic prostate cancer chemotherapy that would mobilize the cancer cells into the blood (NCT02478125) and a phase II trial for the mobilization of HSCs in patients with non-Hodgkin lymphoma or multiple myeloma (NCT01458288).
A high-throughput screening campaign to search for new CXCR4 antagonists revealed quinoline and quinazoline derivatives as candidates, where the hit ligand shows a 5-fold higher affinity in inhibition of 125I-CXCL12 binding to CXCR4 (IC50 = 4.7 nM) compared with AMD3100 (IC50 = 213 nM) (Wu et al., 2012). Another potent quinazoline-based selective CXCR4 antagonist (compound 1, Fig. 4) was characterized (IC50 = 13 nM) as a potential tool to study diseases mediated by the CXCR4/CXCL12 axis, was tested in vivo, and showed a therapeutic potential in an acute kidney injury disease model (Wu et al., 2015a).
Recently, virtual fragment screening, synthesis, and design have led to a series of N-substituted piperidin-4-yl-methanamine derivatives (for example, compound 2 in Fig. 4). Adlere et al. (2019) showed that small structural ligand changes lead to distinct interactions with CXCR4 and CXCL12, supporting the study with three-dimensional quantitative SAR (3D-QSAR) and proposed binding models.
Other purine-based small molecules display subnanomolar EC50 values for CXCR4-mediated HIV inhibition (up to 0.51 nM), show a better functional profile in chemotaxis than AMD3100, and mobilize CXCR4+ cells in mice (Wu et al., 2015b).
The 1,4-phenylenedimethanamine core, as in AMD3100 and AMD3465 (Fig. 4), is a prominent feature among CXCR4 small-molecule antagonists and serves as a starting point for ligand-based discovery of new analogs for improved potency against CXCR4. Likewise, several amide-sulfonamide derivatives have CXCR4 binding properties and outperform AMD3100 in Matrigel invasion inhibition (Bai et al., 2017a, 2017b).
Despite the potency of AMD3100 and other cyclam derivatives at CXCR4, oral bioavailability remains an issue for these lipophilic compounds. A stepwise discovery of nonmacrocyclam derivatives through structural modifications eventually led to the discovery of the first orally available, potent, and selective small molecule targeting CXCR4—AMD11070 (which was initially developed for the treatment of HIV-1 infection; Bridger et al., 2010; Skerlj et al., 2010). AMD11070 has successfully completed phase I and phase II clinical trials for the treatment of HIV (NCT00089466) and is also in a phase II/III trial (Dec 2016, NCT03005327) as a potential treatment of WHIM syndrome. Phase I trials of a combination therapy with pembrolizumab for the treatment of melanoma started in June 2016 (NCT02823405).
With AMD11070 as a basis, a variety of structural modifications have been made to improve metabolic stability, anti-HIV activity, and pharmacokinetic (PK) properties (Gudmundsson et al., 2009a,b; Catalano et al., 2010), including a highly potent hybrid-piperazine derivative, GSK812397 (Fig. 4), a nontoxic, selective, noncompetitive antagonist of CXCR4 with low nanomolar antiviral activity against a broad range of X4-utilizing strains of HIV-1 and with good pharmacokinetics (Jenkinson et al., 2010).
Among other AMD11070 derivatives targeting CXCR4 (Miller et al., 2010; Skerlj et al., 2011; Zhao et al., 2015), tetrahydroisoquinoline-based TIQ-15 (Fig. 4) was identified as a highly active scaffold for CXCR4 inhibition (Truax et al., 2013). More recently, new TIQ-15-derived CXCR4 antagonists were reported. Structural alterations of the molecule have led to improved properties, including enhanced metabolic stability and a reduced off-target liability, such as toward cytochrome P450 2D6 (Jecs et al., 2017; Wilson et al., 2017), and improvement of intestinal absorption (Miller et al., 2018). Overall, evidence suggests that TIQ-15 might have potential therapeutic application in anticancer therapy.
A high-throughput screening campaign at Novartis led to the discovery of a series of orally bioavailable isothiourea-derived CXCR4 antagonists (Thoma et al., 2008). Optimization of one of the hits, isothiourea IT1a (NIBR-1816), yielded the highly potent and selective ligand IT1t (Fig. 4), and its therapeutic potential as anti-AIDS drug was evaluated. The X-ray crystal structure of IT1t cocrystallized with CXCR4 is available (Wu et al., 2010) and is advancing structure-based ligand discovery studies for small molecules targeting CXCR4 (Mysinger et al., 2012; Cox et al., 2015; Das et al., 2015). Noteworthy among the small molecules, including the ones found via virtual screening approaches, some potent ligands, although retaining a functional effect, did not displace 125I-CXCL12, indicating the allosteric interactions or multiple binding sites for small molecules.
Another approach to preventing CXCR4 signaling with small molecules is the targeting of CXCL12; thus, small molecules would act as chemokine scavengers. A set of small molecules with a micromolar range of affinities was discovered through a virtual screen for compounds that bind to CXCL12 occupied by CXCR4 residues D20 and sY21 and using the nuclear magnetic resonance structure of the receptor-peptide complex (Veldkamp et al., 2010; Ziarek et al., 2012). Another series of ligands that inhibit CXCL12-CXCR4 interaction via binding of the chemokine, called “chemokine neutraligands,” was identified after screening of a small (3200 molecules) chemical library in a fluorescent binding assay on living cells, where a single submicromolar CXCL12 binder that inhibits CXCL12-evoked CXCR4 internalization (Fig. 4, compound 3, IC50 = 150 nM) was identified (Hachet-Haas et al., 2008). Additionally, chalcone 3 was also found to be an inhibitor of CXCL12-mediated ACKR3 activation, further establishing its mechanism of action via the scavenging of the chemokine. A prodrug analog of a neutraligand 2 locally inhibits airway inflammation in a mouse model (IC50 = 10 nmol/kg) (Gasparik et al., 2011). Recently, modifications of the CXCL12 neutraligand 2 led to a discovery of a novel non-Michael acceptor with anti-inflammatory effect, LIT-927 (Fig. 4), with better solubility (Regenass et al., 2018).
To summarize the current status in the field of small-molecule modulators for CXCR4, it is fair to state that AMD3100, AMD11070, and IT1t still remain as reference ligands in many studies targeting CXCR4/CXCL12-mediated processes. Yet, there are several small molecules showing similar or better properties regarding potency, affinity, PK, and ADME properties. Owing to the early stage of the development or incomplete characterization, they are arguably not yet considered as reference molecules.
In contrast to reports on CXCR4, there are only a small number of studies describing small molecules that target ACKR3. In 2009, the classic CXCR4 antagonist AMD3100 was shown to be an allosteric ACKR3 agonist at high concentration (>10 μM), enhancing CXCL12 binding to ACKR3, and, alone, recruiting β-arrestin to ACKR3 in a BRET assay with a small (13%) increase in BRET (Kalatskaya et al., 2009). Interestingly, preincubation with isothiourea IT1a (NIBR-1816) significantly affects the uptake of chimeric ACKR3-selective ligand CXCL11_12, indicating that IT1a targets both receptors, although possibly by different mechanisms (Puddinu et al., 2017).
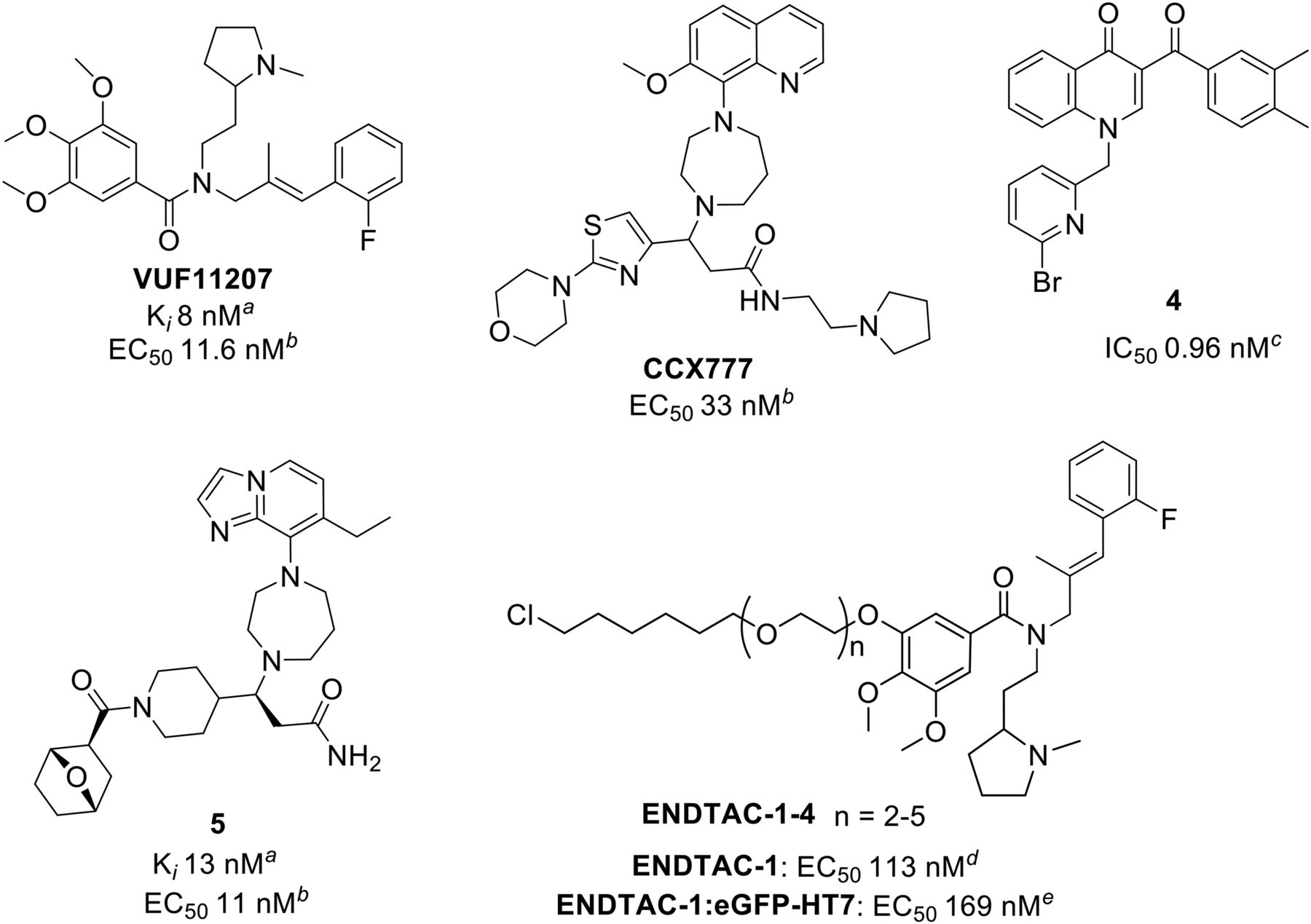
Among a series of substituted styrene amides patented by Chemocentryx, VUF11207 (Fig. 5) was found to be a high-affinity ACKR3 agonist able to recruit β-arrestin2 and reduce ACKR3 surface expression (Wijtmans et al., 2012). Indeed, Chemocentryx has developed a number of ACKR3 ligands. A selective ACKR3 agonist CCX771 (IC50 4.1 nM) (Zabel et al., 2009, 2011; Yamada et al., 2015) is patented by the company (structure not revealed) and is often used in the characterization of ACKR3. CCX771 inhibits tumor growth, lung metastasis, and tumor angiogenesis in vivo, and further evidence suggests that the CXCL12-ACKR3 autocrine loop affects proangiogenic properties of tumor endothelial cells (Yamada et al., 2015). However, the signaling pathway and the mode of action of this small molecule are still not clear. VUF11207 and CCX771 block X7-HIV-1-strain entry with IC50 approx. 50 nM. VUF11207 is selective toward the HIV-1 HE #10 viral strain and both compounds do not block the HIV-2 EHO strain. CCX771 was further evaluated in HIV-1 replication using quantitative real-time polymerase chain reaction showing HIV entry-blocking properties (D’Huys et al., 2018).

Structures of selected small-molecule ACKR3 agonists. aACKR3 125I-CXCL12 inhibition binding assay. bβ-arrestin recruitment BRET assay. cInhibition of CXCL12-induced tube formation in human umbilical vein endothelial cells, the exact EC50 for β-arrestin recruitment BRET assay not given. dPulse chase Tango assay (compound alone). ePulse chase Tango assay for precomplexed compound.
Another Chemocentryx ligand, CCX777 (Fig. 5), can be used as a tool probe for mapping the binding pocket of ACKR3. The compound acts as a partial agonist of ACKR3 with an efficacy of 52% relative to the endogenous ligand and potency of 33 nM (8.5 nM for CXCL12) in β-arrestin2-recruitment assay (Gustavsson et al., 2017). Using radiolytic footprinting, two different complexes of ACKR3 with CCX777 and CXCL12 were compared (Gustavsson et al., 2017). Another ACKR3 ligand, compound 4 (Fig. 5), inhibits CXCL12-induced tube formation in human umbilical vein endothelial cells (0.96 nM), demonstrating selective ACKR3 agonist potential as angiogenesis inhibitors. Moreover, it recruits β-arrestin2 in a BRET assay. An antagonistic antibody for ACKR3 did not affect tube formation under the same conditions (Uto-Konomi et al., 2013).
A novel 1,4-diazepine-ACKR3 ligand was discovered using the chemical scaffold of one of the Chemocentryx ligands as a basis. Exhaustive SAR studies led to a hit, compound 5 (Fig. 5), with equal high-affinity agonism on human (13 nM) and mouse (51 nM) ACKR3. In vivo tests showed reduction of cardiac fibrosis in a mouse model (Menhaji-Klotz et al., 2018). A range of pharmacological data in this study can facilitate the use of this ligand to study ACKR3.
Very recently, Nalawansha et al. (2019) showed a proof-of-concept study of heterodimeric molecules, termed “ENDosome TArgeting Chimeras” (ENDTACs), and their internalization and degradation of an intracellular recombinant enhanced green fluorescent protein (eGFP) and Halotag7 (HT7) fusion by capturing ACKR3. ENDTACs-1–4 (Fig. 5) were obtained using a warhead of VUF11207, a potent ACKR3 agonist (vide supra), that incorporated an HT7-recruiting chloroalkane at a position of one of the methoxy groups of VUF11207.
ENDTACs-1–4 display only slightly reduced potency (EC50 111–158 nm) compared with the parent VUF11207 (EC50 68 nM, same assay.) Additionally, prereacted ENDTACs1–4:eGFP-F complex showed similar potency for ACKR3 activation (EC50 169–287 nm) (Nalawansha et al., 2019).
CXCR4 and ACKR3 Radioligands
For the characterization of ligand binding to GPCRs in systems in vitro and in vivo, tagged molecules are often required to measure or visualize ligand-GPCR binding. Radioligands are very useful tools for such a purpose.
For in vitro studies, a common method is to use radiolabeled peptides whose tyrosine residues are labeled with iodine-125 (125I). This technique has been used for labeling CXCL12 since the initial discovery of CXCR4 as a chemokine receptor for CXCL12 (Bleul et al., 1996; Oberlin et al., 1996; Hesselgesser et al., 1997, 1998; Di Salvo et al., 2000). The use of 125I-CXCL12 remains a robust method for the measurement of the binding of ligands to CXCR4, but as CXCL12 also binds to ACKR3 some interference can be caused if both receptors are present in the same cell population. As ACKR3 has a 10-fold higher affinity for CXCL12, 125I-CXCL12 is also used for ACKR3 binding studies. It is worth noting that the endogenous peptide CXCL12 may not compete with the small molecules binding to the same receptor. A small-molecule CXCR4 allosteric modulator, MSX-122, has been 18F-radiolabeled and used for characterization of its nonlabeled analog in vitro (Liang et al., 2012). Radioiodination has been applied to a number of selective endogenous CXCR4 ligands, such as the highly specific anti-CXCR4 monoclonal antibody (mAb) 12G5 (Endres et al., 1996), which is less affected by mutations of CXCR4 than CXCL12 (Thiele et al., 2014). Others include the BKT-140 (4-F-benzoyl-TN14003) (Fig. 6) analog Ac-TZ14011 for in vivo and in vitro studies (Han et al., 2010) and the 125I-labeled pentapeptide FC131 (Koglin et al., 2006; Demmer et al., 2011a).

Selected CXCR4-specific PET tracers.
For in vivo imaging of receptor expression, the use of radiotracers is practically indispensable. Depending on the radioisotope that is used, the basis of in vivo imaging is gamma ray detection (single-photon emission computed tomography or SPECT) or detection of photon pairs resulting from positron annihilation (positron-emission tomography or PET). Nowadays, PET is a detection method used both in clinics and research and PET tracers are useful tools in theranostics (Yordanova et al., 2017).
89Zr-labeled ACKR3-mAb (clone 11G8) was evaluated in vitro and in vivo by PET, which showed enhanced labeled-antibody uptake in high-ACKR3-expressing tumors, and serves as a viable diagnostic marker (Behnam Azad et al., 2016).
Radiofluorinated ligands are commonly used for PET tracer development. For CXCR4-expressing tumor imaging, the peptide BKT-140 isotopologue 4-[18F]Bn-TN14003 (Jacobson et al., 2010) and [18F]fluoropropionate-labeled Ac-TC14012 peptide (Zhang et al., 2013) show a high tumor uptake and low nonspecific binding. 18F has also been incorporated in the FC131 analog CCIC15, in which the arginine residue was changed to a (2-fluoroethyl)-1H-1,2,3-triazol-4-ylalanine (George et al., 2013); however, no specific tumor uptake was detected. An 18F-labeled noncyclam AMD3465 analog was also evaluated as a CXCR4 PET tracer, which revealed that reduced receptor binding and fast excretion are the limiting factors for CXCR4 imaging (Demoin et al., 2016). An 11C-labeled derivative of AMD3465 (Fig. 6), however, was used for the determination of the receptor occupancy by AMD3100 in a C6 glioma tumor model (Hartimath et al., 2014, 2017).
Chelation of radioactive metal ions is another well known technique for labeling PET tracers. Small-molecule tracers such as 64Cu-AMD3100 (Fig. 6) (Jacobson et al., 2009; De Silva et al., 2011; Weiss et al., 2012), 64Cu-labeled AMD3465 and its bridged analog (De Silva et al., 2011; Woodard et al., 2014) and, recently, 99mTc-AMD3465 (Zhang et al., 2018) have been used as tools for successful identification of CXCR4+ tumors.
BKT-140 has also been used as a template for metal-ion chelates. This was enabled by conjugation of either the d-lysine or both lysine residues of the peptide with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelator moieties (68Ga-DOTA-4-FBn-TN14003, Fig. 6) (Hennrich et al., 2012; Jacobson et al., 2012). Moreover, specificity of BKT-140 was improved and this peptide has been labeled with 68Ga3+s, resulting in 68Ga-NOTA-NFB using 1,4,7-triazacyclononane-N,N′,N′′-triacetic acid as a chelator (Fig. 6), which was successfully applied in glioma patients (Wang et al., 2015).
The N-methylated derivative of the CXCR4-selective peptide FC131 was also developed as a PET tracer. Substitution of the N-methylated d-arginine for a N-methylated d-ornithine and subsequent conjugation with a benzoic acid-linked DOTA chelating with 68Ga3+ resulted in 68Ga-pentixafor (Fig. 6) (Demmer et al., 2011b). The tracer was used to localize multiple myeloma and other tumor types, visualization of CXCR4-expression after acute myocardial infarction, in atherosclerotic plaques, and for the identification of leukocytes after transplantation (Herrmann et al., 2015; Thackeray et al., 2015; Derlin et al., 2016, 2017; Lapa et al., 2016a,b; Rischpler et al., 2016; Vag et al., 2016; Hyafil et al., 2017; Watts et al., 2017). A tyrosine monoiodinated analog of FC131, pentixather, labeled with 177Lu provides a tracer with higher liver uptake than 68Ga-pentixafor but also high tumor uptake, making it promising for clinical application in combination with 68Ga-pentixafor (Schottelius et al., 2017). Ulocuplumab, a CXCR4-selective human monoclonal antibody labeled with 89Zr, shows a specific accumulation in CXCR4 expressing tumors (Azad et al., 2016).
Fluorescent Probes
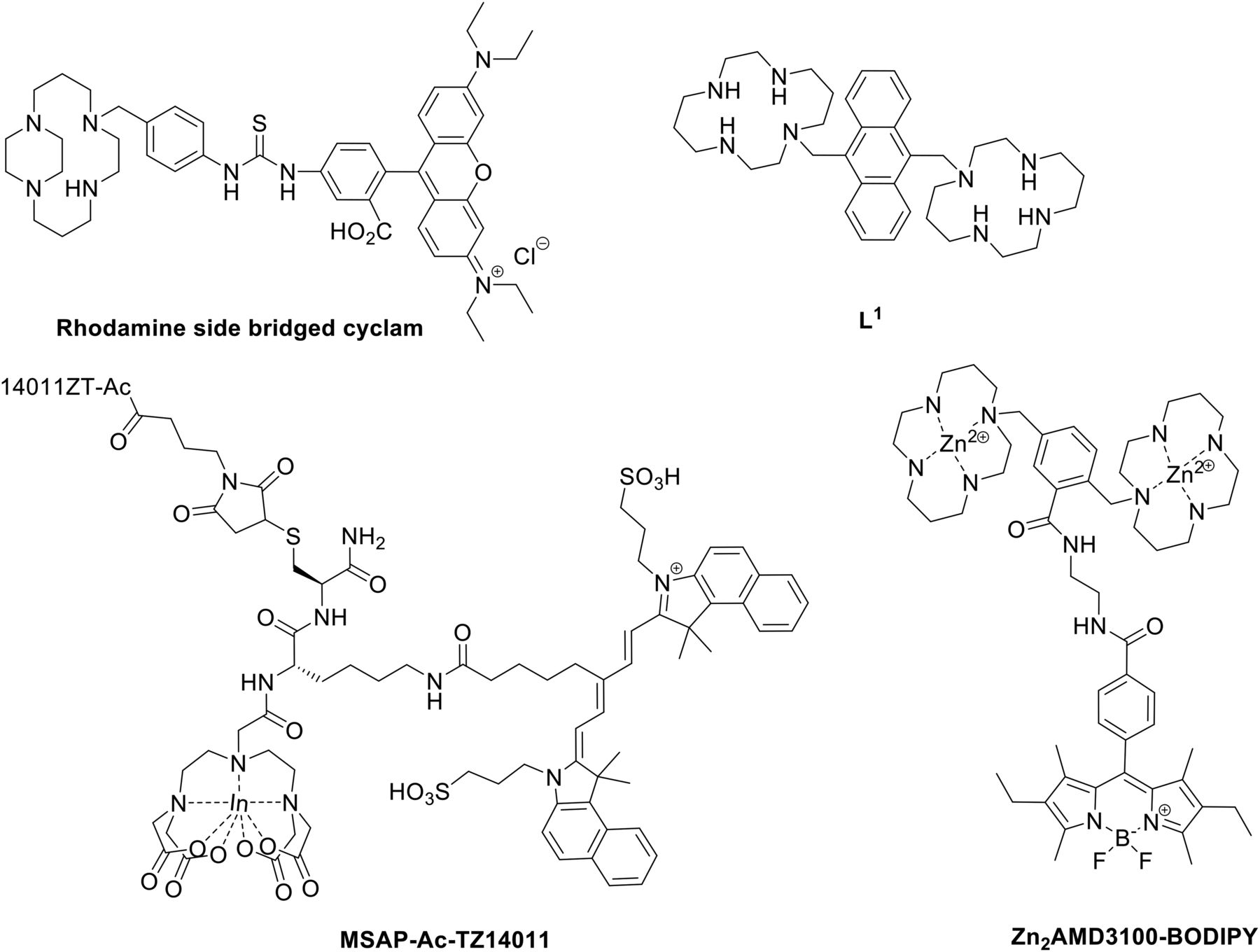
Fluorescent labeling of appropriate chemokine receptor ligands provides a unique way to study the complex biologic interactions of CXCR4 and ACKR3. Early efforts to develop a CXCR4-targeting fluorescent probe used an AMD3100 derivative incorporating a rhodamine fluorophore for visualization (Khan et al., 2007) (Fig. 7). Metalation of AMD3100 using Zn2+, Ni2+, and Cu2+ ions yields highly stable metal complexes and even increases affinity toward CXCR4 (Gerlach et al., 2003). Binding data of the fluorescent probe disclosed by Khan et al. (2007) were not reported. However, flow cytometry studies showed that the copper-complexed probe actively competes with CXCR4-specific antibodies. The uncomplexed fluorophore displays no CXCR4-specific binding, indicating that metal ions are necessary for receptor binding by conformationally restricting the cyclam.

Selected fluorescent probes targeting CXCR4.
Another fluorescent derivative of AMD3100 has also been reported by (Knight et al. (2011) and used an anthracene moiety to link two cyclam rings (Fig. 7). The fluorescent ligand (L1) inhibits binding of anti-CXCR4 antibody 12G5, albeit with a significantly reduced affinity for CXCR4 compared with AMD3100 (IC50 = 0.25–0.9 μM for L1, IC50 = 10 nM for AMD3100). Further exploration of the AMD3100 scaffold led to the discovery of a boron-dipyrromethene-labeled (BODIPY) CXCR4 antagonist (Fig. 7) (Poty et al., 2015). The use of a BODIPY as a fluorophore is well documented and has several advantages (Wiederschain, 2011). The BODIPY-conjugated probe displays binding properties inferior to the parent ligand. Whereas AMD3100 has an EC50 of 11 nM in an anti-HIV assay, the most potent AMD3100-BODIPY conjugate gives an EC50 of only 0.8 μM. Furthermore, this fluorescent conjugate was not found to compete directly with CXCL12 in a calcium flux competition assay. Overall these imaging probes with a small-molecule basis display poor pharmacological properties for CXCR4, showing attenuation in affinity compared with parent AMD3100, thus limiting their applicability.
A cyclic peptide-based fluorescent probe Ac-TZ14011 displays an IC50 of 1.2 nM and 2.6 nM in an CXCL12 binding assay and a Ca2+ mobilization assay, respectively (Hanaoka et al., 2006). Utilizing the d-lysine of this peptide, it was possible to introduce carboxytetramethylrhodamine-based and fluorescein dyes (Nomura et al., 2008) with the resulting fluorescent probes displaying IC50 values of 14 and 11 nM, respectively, in a competitive binding assay. Use of Ac-TZ14011 in the development of CXCR4-targeting probes was further exploited by labeling the peptide with a multifunctional single attachment point reagent containing a DTPA (diethylenetriaminepentaacetic acid) chelate and a fluorescent dye (Fig. 7) (Kuil et al., 2011). Although a decrease in binding affinity was observed (KD = 177.1 ± 37.2 nM for the conjugate, KD = 8.61 ± 1.42 nM for the unlabeled peptide), the probe was successfully used to image CXCR4-expressing cells both in vitro and in vivo. A different fluorescent peptide was also developed consisting of a fluorescein isothiocyanate conjugated to DV1 (Yang et al., 2014). DV1 is an all d-amino acid analog of V1, a synthetic peptide derived from the N-terminus of the viral macrophage protein vMIP-II (Zhou et al., 2002). This fluorescent conjugate shows a binding affinity for CXCR4 in the micromolar range and was competitively displaced by small molecules AMD3100 and IT1t, as well as by parent peptide DV1.
Small-molecule- or peptide-based fluorescent probes that specifically target ACKR3 have not been reported to date. Fluorescence-based (imaging) techniques so far have instead relied on the use of fluorescently tagged CXCL12- or ACKR3-specific antibodies (Meincke et al., 2011; Humpert et al., 2012; Masuda et al., 2012; Melo et al., 2018).
Concluding Remarks
In this review, we describe the relationships between ligand binding and structural motifs of the two related chemokine receptors CXCR4 and ACKR3 and provide an overview of the known modulators of both GPCRs, with a specific emphasis on validated research tools.
Structurally, CXCR4 shows the same common features as other GPCRs of the chemokine receptor subclass. The N-terminal end of the receptor is of particular interest for binding of the endogenous ligand CXCL12, which binds in the orthosteric binding site, which can be divided into a minor and major pocket (Arimont et al., 2017). This orthosteric binding site overlaps with the binding site of several small molecules and peptide ligands as seen in crystal structures and deduced from mutational studies (Wu et al., 2010; Arimont et al., 2017). CXCR4 and ACKR3 both bind CXCL12 (Hesselgesser et al., 1998; Balabanian et al., 2005), the viral chemokine vMIP-II (Kledal et al., 1997; Szpakowska et al., 2016), and β-defensins 2 and 3 (Quiñones-Mateu et al., 2003; Feng et al., 2006), but they also have endogenous ligands binding only one of the two. CXCR4 binds ubiquitin (Saini et al., 2010) and ACKR3 binds to CXCL11 (Burns et al., 2006) and adrenomedullin (Klein et al., 2014).
Most CXCR4 peptide ligands are derived from polyphemusin II and have been shown to inhibit the ability of HIV-1 to infect cells (Murakami et al., 1997). There has been an array of peptide variants with this original structure as a basis showing different affinities, potencies, stabilities, and selectivities and some of them also interact with ACKR3 (Oishi et al., 2011). Other peptidomimetics have as a basis peptides that mimic HIV proteins, CXCR4 domains, or parts of CXCL12 and show that they also inhibit the ability of HIV-1 to infect cells (Hashimoto et al., 2013; Chevigné et al., 2014). The last group of peptide ligands consists of chimeric ligands fusing two ligands (Lefrançois et al., 2011, Mona et al., 2016).
Pepducins have been described as agonists and antagonists for CXCR4, but not for ACKR3, (Tchernychev et al., 2010) and although they are a new interesting class of receptor modulators, their exact mode of action is unclear and designing new pepducins is difficult. However, they act as an interesting class of ligands with their postulated binding site being an allosteric intracellular one (Covic et al., 2002; Wielders et al., 2007; Janz et al., 2011).
AMD3100, AMD11070, and IT1t are currently the most widely used CXCR4 small-molecule reference ligands, even though there is a range of compounds showing similar or better potency, affinity, and PK and ADME properties. The first small molecules targeting CXCR4 were originally identified as HIV-entry inhibitors, with the most prominent compound approved for clinical use being AMD3100. This drug is now used for the mobilization of CD34+ cells in NHL and MM rather than for HIV treatment (De Clercq et al., 1994; De Clercq, 2009).
Apart from AMD3100-like compounds, nonmacrocyclic modulators were discovered in various screens in efforts to solve the problem of bioavailability. TG-0054 (burixafor) is in clinical trials for assessing its use as a sensitizer in metastatic prostate cancer (Shelke et al., 2011) and AMD11070 for the treatment of HIV and WHIM syndrome (Bridger et al., 2010; Skerlj et al., 2010).
Only a small number of compounds have been described as modulators of ACKR3. Some of the CXCR4 ligands, such as AMD3100 (Kalatskaya et al., 2009) and isothiourea IT1a (NIBR-1816) (Puddinu et al., 2017), interact with ACKR3 at high concentrations. VUF11207 (Wijtmans et al., 2012) and CCX771 (Zabel et al., 2009, structure unpublished) have been described as ACKR3 agonists, and CCX777 (Gustavsson et al., 2017) acts as a partial agonist.
Radiolabeled and fluorescent ligands have been developed as useful research tools. CXCL12 has been labeled using an iodine-125 label on tyrosine residues and also with Alexa Fluor 647 (Bleul et al., 1996; Oberlin et al., 1996; Hesselgesser et al., 1997, 1998; Di Salvo et al., 2000). Radioiodination has also been introduced to CXCR4 and ACKR3 antibodies and peptides (Endres et al., 1996; Koglin et al., 2006; Han et al., 2010; Demmer et al., 2011a; Behnam Azad et al., 2016). Furthermore, small molecules have been labeled using an array of radiolabels with different tracers (18F, 11C, 64Cu, 68Ga, 89Zr, or 177Lu) either as substitutes of unlabeled atoms or in a chelate complex. These small molecules can also be labeled using fluorescent tags (Khan et al., 2007; Wiederschain, 2011). However, the tag often influences the binding properties of the compounds owing to their size and they have to be studied as a new compound. For ACKR3, only fluorescently labeled antibodies and CXCL12 have been described as tools to label the receptor.
Efforts to find suitable ligands for CXCR4 and ACKR3 are a continuous process to provide the research community with better research tools and to seize the therapeutic opportunities in the area of HIV infection and cancer. Continuing structural biology efforts are of fundamental importance for the understanding of drug-receptor interactions at both GPCR proteins and for the ongoing drug discovery programs targeting CXCR4 and ACKR3.
Acknowledgments
The authors dedicate this article to the memory of Kirsten Visser, who was an outstanding student, talented young researcher, and beloved colleague at the Division of Medicinal Chemistry (Vrije Universiteit Amsterdam).
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Adlere, Caspar, Arimont, Dekkers, Visser, Stuijt, de Graaf, Stocks, Kellam, Briddon, Wijtmans, de Esch, Hill, Leurs.
Footnotes
- Received July 3, 2019.
- Accepted September 14, 2019.
↵1 I.A., B.C., and M.A. contributed equally to this work.
This work was supported by European Union’s Horizon2020 MSCA Program under grant agreement 641833 [ONCORNET]. This minireview is part of the series “From Insight to Modulation of CXCR4 and ACKR3 (CXCR7) Function.”
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- 7TM
- seven transmembrane
- ACKR3
- atypical chemokine receptor 3
- ADME
- absorption, distribution, metabolism, excretion
- BRET
- bioluminescence resonance energy transfer
- CD34+
- cluster of differentiation 34
- CXCL11
- C-X-C chemokine ligand 11
- CXCL12
- C-X-C chemokine ligand 12
- CXCR4
- C-X-C chemokine receptor type 4
- CXCR7
- C-X-C chemokine receptor type 7
- DOTA
- tetraazacyclododecane-1,4,7,10-tetraacetic acid
- DTPA
- diethylenetriaminepentaacetic acid
- ECL
- extracellular loop
- gp120
- glycoprotein 120
- GPCR
- G protein-coupled receptor
- HIV
- human immunodeficiency virus
- HSC
- hematopoietic stem cell
- HT7
- Halotag7 protein
- MM
- multiple myeloma
- NHL
- non-Hodgkin lymphoma
- PET
- positron-emission tomography
- PK
- pharmacokinetic
- SAR
- structure-activity relationship
- TM
- transmembrane
- vMIP-II
- viral macrophage inflammatory protein-II
- WHIM
- warts, hypogammaglobulinemia, infections and myelokathexis
- Copyright © 2019 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Structural Determinants of Chemokine Receptor Ligand Binding
- Chemokine and Peptide Binding to CXCR4 and ACKR3
- Pepducins
- Small-Molecule Modulators of CXCR4 and ACKR3
- CXCR4 and ACKR3 Radioligands
- Fluorescent Probes
- Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI