


Abstract
The sigma-1 receptor (S1R) is a 223 amino acid two transmembrane (TM) pass protein. It is a non-ATP-binding nonglycosylated ligand-regulated molecular chaperone of unknown three-dimensional structure. The S1R is resident to eukaryotic mitochondrial-associated endoplasmic reticulum and plasma membranes with broad functions that regulate cellular calcium homeostasis and reduce oxidative stress. Several multitasking functions of the S1R are underwritten by chaperone-mediated direct (and indirect) interactions with ion channels, G-protein coupled receptors and cell-signaling molecules involved in the regulation of cell growth. The S1R is a promising drug target for the treatment of several neurodegenerative diseases related to cellular stress. In vitro and in vivo functional and molecular characteristics of the S1R and its interactions with endogenous and synthetic small molecules have been discovered by the use of pharmacologic, biochemical, biophysical, and molecular biology approaches. The S1R exists in monomer, dimer, tetramer, hexamer/octamer, and higher oligomeric forms that may be important determinants in defining the pharmacology and mechanism(s) of action of the S1R. A canonical GXXXG in putative TM2 is important for S1R oligomerization. The ligand-binding regions of S1R have been identified and include portions of TM2 and the TM proximal regions of the C terminus. Some client protein chaperone functions and interactions with the cochaperone 78-kDa glucose-regulated protein (binding immunoglobulin protein) involve the C terminus. Based on its biochemical features and mechanisms of chaperone action the possibility that the S1R is a member of the small heat shock protein family is discussed.
Introduction
This minireview summarizes current aspects of the molecular pharmacology and biochemistry of the sigma-1 receptor (S1R). The remarkable history of the S1R extends from the late 1970s and early 1980s (Martin et al., 1976; Su, 1981, 1988; Tam, 1983; Vaupel, 1983; Tam and Cook, 1984; Largent et al., 1986, 1987; Sircar et al., 1986a; Weber et al., 1986; Itzhak, 1987; McLean and Weber, 1988; Sharkey et al., 1988; Bowen et al., 1989; de Costa et al., 1989; Hellewell and Bowen, 1990) to the present day. The reader is directed to the earlier publications and reviews that outline the original pharmacologic identification of this important receptor (Martin, 1983; Su and Hayashi, 2003; Glennon, 2005).
The guinea pig S1R was originally purified and cloned by Hanner et al. (1996), who found that the S1R is a unique 223 amino acid protein with 90% sequence conservation in many mammalian species. The S1R is located in eukaryotic mitochondrial-associated endoplasmic reticulum membranes (Hayashi and Su, 2007) and in some plasma membrane compartments (Mavlyutov and Ruoho, 2007; Mavlyutov et al., 2010, 2012). Notable molecular features of the S1R include 1) two putative transmembrane sequences, which predicts that both the N and C termini are located on the same side of the membrane, 2) the presence of several splice variants (Ganapathy et al., 1999), and 3) endogenous and synthetic ligand regulation of its chaperone functions (Hayashi and Su, 2007).
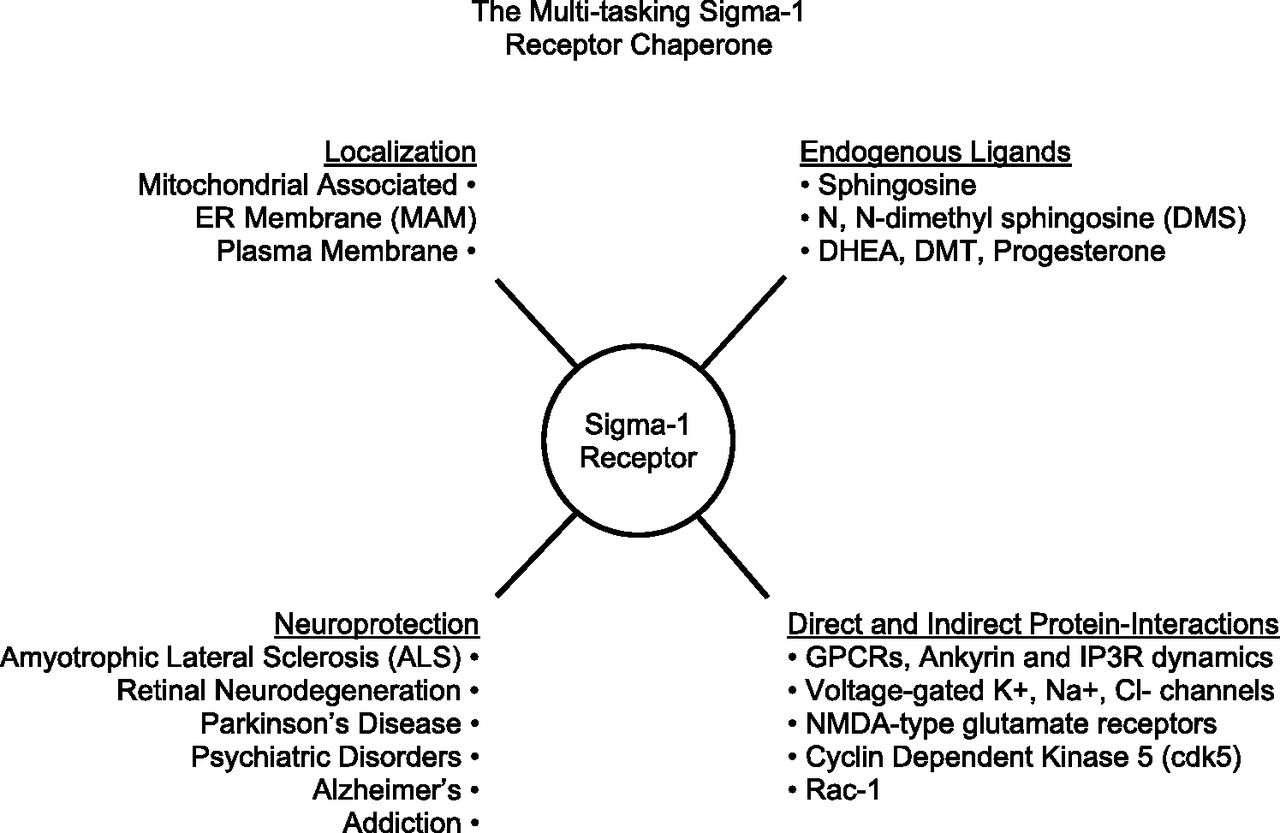
The gene that encodes for the human S1R is located on chromosome 9 band p13 and contains four exons and three introns. The S1R shares 60% amino acid identity to a yeast sterol isomerase, ERG2, however, the mammalian S1R does not possess sterol isomerase activity, nor can it rescue the growth of sterol isomerase-deficient yeast (Moebius et al., 1996, 1997). The S1R is a ligand-regulated non–ATP-binding membrane bound chaperone protein (Hayashi and Su, 2007). In its multitasking functions, the S1R has been demonstrated to physically interact with client proteins in a functional manner including ion channels such as inositol triphosphate (IP3) receptors (Hayashi and Su, 2001; Wu and Bowen, 2008), voltage-gated potassium (Aydar et al., 2002), sodium (Balasuriya et al., 2012), and calcium channels (Tchedre et al., 2008), calcium-activated chloride channels (Zhang and Cuevas, 2002), and N-methyl-d-aspartate receptors (Sircar et al., 1986b; Monnet et al., 1990; Balasuriya et al., 2013; Pabba et al., 2014). Specific interactions of the S1R with several types of G-protein coupled receptors (GPCRs) (Kim et al., 2010). Navarro et al. (2010) have also been reported. The S1R directly and indirectly modulates neuronal mitochondrial Rac-1 GTPase (Tsai et al., 2009; Natsvlishvili et al., 2015), which is involved in dendritic spine formation. S1R reduces tau phosphorylation (Tsai et al., 2015) via indirect regulation of cyclin-dependent kinase 5 (cdk5) and thus protects neuronal axon elongation. (Fig. 1).

The multitasking sigma-1 receptor chaperone. Overview of some sigma-1 receptor molecular interactions and potential therapeutic applications.
The S1R has been established as a therapeutic target for many neurodegenerative conditions in humans that involve various forms of cellular metabolic stress, including amyotrophic lateral sclerosis (Al-Saif et al., 2011; Mancuso et al., 2012; Mavlyutov et al., 2013, 2015; Prause et al., 2013; Gromek et al., 2014; Fukunaga et al., 2015; Mishra et al., 2015), frontotemporal lobar dementia (Luty et al., 2010), Alzheimer disease (Feher et al., 2012; Yin et al., 2015), Parkinson disease (Mishina et al., 2005; Mori et al., 2012), retinal neurodegeneration (Smith et al., 2008; Mavlyutov et al., 2011; Shimazawa et al., 2015), addiction to drugs of abuse (Navarro et al., 2010; Nguyen et al., 2015), and psychiatric disorders (Tsai et al., 2014) (Fig. 1).
Commonly Used Agonists/Antagonists for the S1R
Glennon (2005) proposed that the pharmacophore for small molecule binding to the S1R is defined by two hydrophobic constituents attached to a central nitrogen atom containing unshared electrons. An added feature of the general pharmacophore is that the primary hydrophobic group would optimally be an N-aralkyl substituent. After analysis of many structure-activity studies compiled from at least 2 to 3 decades of intense activity involving small molecule S1R selective syntheses, it has been generally agreed that this primary hydrophobic component is optimally situated at approximately five carbons (6–10 Å) from the nitrogen. A secondary hydrophobic group can be located closer to the obligate nitrogen (2.5–4 Å) than the primary hydrophobic group. Additionally, Ruoho et al. (2012) have shown that para-nitro or para-azido electron-withdrawing groups on the phenyl ring of molecules such as cocaine (Chen et al., 2007) greatly enhance the specific S1R-binding affinity (discussed later). Similarly the binding of a series of N-3-(4-nitrophenyl) propyl-Nʹ-alkylamines to the pure S1R in which the Nʹ-alkyl chains varied from propyl to octadecyl showed a similar increase in affinity compared with their parent N-phenylalkyl amines (Ruoho et al., 2012).
A minimal pharmacophore for S1R binding based on a phenylalkylamine platform (Maeda et al., 2002; Fontanilla et al., 2009) has led to the identification of a plethora of compounds with reasonable (and in some cases exceptional) S1R-binding affinity as either agonists or antagonists (selected compounds are shown in Table 1). Upon examination of the benzomorphans, such as pentazocine and SKF10047 ([2S-(2α,6α,11R*]-1,2,3,4,5,6-hexahydro-6,11-dimethyl-3-(2-propenyl)-2,6-methano-3-benzazocin-8-ol), an important feature involving the stereospecificity of binding to the S1R is emphasized; that is, the dextrorotatory forms bind to the S1R with significantly higher affinity than the levorotatory forms. Given this generally promiscuous pharmacophore, several small molecules that are able to bind to other drug targets also interact with the S1R, such as fenpropimorph (antifungal), haloperidol (antipsychotic), chlorpheniramine (antihistamine), N,Nʹ dimethyl tryptamine (DMT) and methamphetamine (psychostimulants), fluvoxamine (antidepressant), and verapamil (calcium channel blocker). Endogenous compounds such as sphingosine and sphingosine derivatives (discussed later), progesterone, dehydroepiandrosterone (DHEA), and cholesterol also bind to the S1R.
Representative sigma-1 receptor agonists and antagonists
Structural Features of the S1R
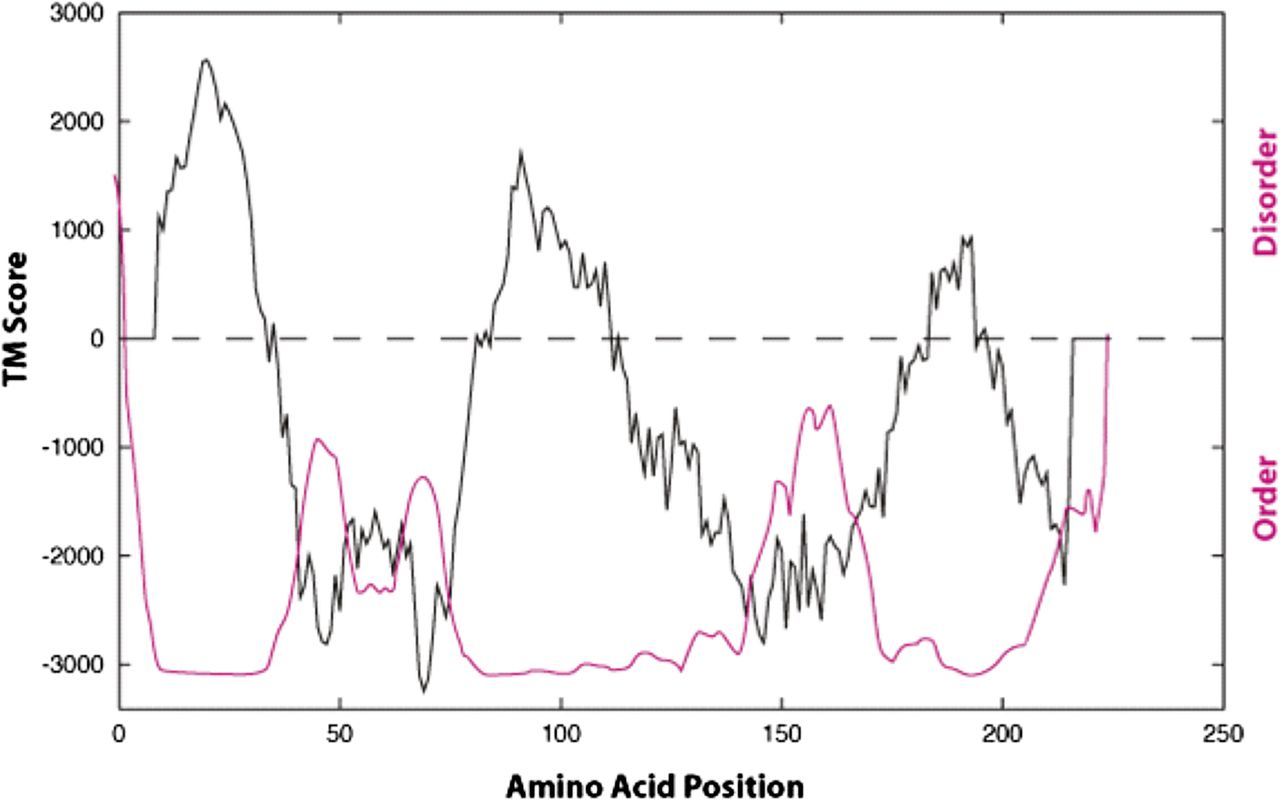
The three-dimensional structure of the S1R is currently unknown. Originally, sequence-based algorithms that predict amino acid hydrophobicity [TM BASE (Hofmann, 1993)] and structural flexibility assessment plots [PONDR (Garner et al., 1999; Li et al., 1999)] have indicated two major hydrophobic helical transmembrane sequences (Aydar et al., 2002) (Fig. 2). Depending on the algorithm used to predict the transmembrane (TM) segments, TM1 is predicted to span between amino acids 8 and 34, and TM2 region spans between amino acids 83 and 111 (Ortega-Roldan et al., 2015). This disposition of the receptor in mammalian endoplasmic reticulum (ER) membranes indicates that the N and C termini are both located in the ER lumen (Hayashi and Su, 2007). Further, this orientation predicts that if S1R enters the plasma membrane (as is the case in some mammalian cells) the S1R N and C termini are located outside the cell.

Hydropathy and PONDR plots of the guinea pig sigma-1 receptor. The hydropathy TM plot in black and PONDR plot in red (PONDR.com). Adapted from Aydar et al. (2002) with permission from Elsevier.
This orientation has been confirmed in vivo in intact mouse nucleus accumbens neurons using C-terminal sequence–specific antibodies (Kourrich et al., 2013). Based on sequence identity to the yeast sterol isomerase ERG2, two regions of the S1R have been referred to as steroid-binding-like domains I and II (SBLD I and SBLD II) (Fig. 3). Proposed structural features of the pure S1R have been recently reported using in silico approaches based on the use of several reasonable model platforms reported in the Protein Data Bank (Brune et al., 2013; Brune et al., 2014) and by proton NMR assessments of regions of the S1R (Ortega-Roldan et al., 2013, 2015).

Model of the key structural motifs of the sigma-1 receptor. (A) A model of the sigma-1 receptor. The structure was determined using solution NMR studies of the S1R(∆35) construct where the first 35 amino acids of the S1R were deleted. Helices are represented as blue, gray, green, and red cylinders, and the length of each helix is scaled relative to each other. Flexible regions are not drawn to scale. The residues making up the transmembrane domain 1 (TM1), depicted as a gray cylinder and indicated by asterisks, is predicted based on several computer algorithms. Nomenclature of each helical region is adapted from Ortega-Roldan et al. (2015); the SBDL I and SBDL II regions (shown in red) are modeled after Pal et al. (2007), and the cholesterol recognition motif (CRM) I and II were identified by Palmer et al. (2007). Based on photolabeling data (Fig. 4) and site-directed mutagenesis data (Table 2), the shaded area broadly defines the ligand-binding site and perhaps a dimerization-dependent agonist binding interface between monomers. An ER retention sequence was determined by Hayashi and Su (2003), and the oligomerization sequence (GGMWG) of the sigma-1 receptor was based on studies published by Gromek et al. (2014). Note: this model of the sigma-1 receptor is drawn from the perspective of the ER membrane; it is worth mentioning that the sigma-1 receptor will assume a reverse orientation on the plasma membrane where the N and C termini are extracellular. (B) Structures of photoprobes that have been used to study the S1R that are referenced in the text. Photoprobes i–iii were reported for S1R covalent labeling. Photoprobes iv–ix have been used to identify individual residues in the S1R binding sites and/or regions of the receptor as indicated. Adapted from Ortega-Roldan et al. (2015).
Specific Structural Features of the S1R N-Terminus, Proposed TM1 and TM2 and TM1/TM2 Loop.
The S1R N terminus sequence before entrance into the membrane bilayer involves approximately 9 amino acids, including an ER retention sequence of double arginines at positions 7 and 8 in the guinea pig (Hayashi and Su, 2003). TM1 is predicted to span between amino acids 9 and 34 while TM2 is predicted to contain amino acids 83–111. While the precise amino acids that comprise the transmembrane sequences have not been established with certainty, recent NMR data on a truncated form of the S1R (S1R∆35) in which the N-terminal 35 amino acids were removed have indicated unusual flexibility of serine 87 beyond that expected for a membrane-imbedded residue (Ortega-Roldan et al., 2015). The conclusion made was that TM2 enters at residue 87 (instead of 83) from the N terminus and extends to residue 108 (Fig. 3). This truncated form of S1R, however, would likely enter the Escherichia coli membrane translocon during expression in a manner that could reverse the TM2 sequence in the membrane with respect to the constitutive full-length S1R TM1 and remain in this orientation in the dodecylphosphocholine detergent micelles (Ortega-Roldan et al., 2015).
Significant additional structural features of TM2 include a canonical membrane-imbedded GxxxG dimerization sequence (Overton et al., 2003) at positions 87–91, which is involved in oligomerization of the S1R (Gromek et al., 2014) (Fig. 3), and a single cysteine residue that occurs at position 94 in TM2. A specific functional role has not been ascribed to this cysteine. Because the S1R has been associated with various in vivo mechanisms to suppress cellular oxidative stress (Bucolo et al., 2006; Tuerxun et al., 2010; Pal et al., 2012), perhaps cysteine 94 participates in redox reactions to maintain an intracellular reducing environment. An alanine residue when substituted for cysteine 94 does not significantly affect the binding of the S1R agonist [3H]-(+)-pentazocine (unpublished results).
The 50 amino acid loop joining the two TM sequences has been examined by NMR and found to contain three short helical segments, labeled as cH1, cH2, and cH3 (Ortega-Roldan et al., 2015) (Fig. 3). The cH2 and cH3 helices appear to be constrained in some manner either by constitutive strong helical interactions or by interactions with the detergent used in the NMR experiments. A compact three-dimensional structure for this region is consistent with previous observations that lysine 60 is relatively insensitive to Endolys C proteolytic cleavage even in the presence of sodium dodecylsulfate (SDS) (Pal et al., 2008).
Specific Structural and Functional Features of the S1R C Terminus.
The in vitro structural and functional studies of the S1R have been achieved using the C-terminal approximately 123 amino acids of the S1R. Structurally, this region of the S1R C terminus is predicted to contain varying levels of order and disorder (Aydar et al., 2002) (Fig. 2). High-resolution NMR analysis of S1R C-terminal residues 112–223 when expressed in E. coli, purified as a polyhistidine construct and reconstituted into dodecylphosphocholine micelles, revealed five helical segments, H1 (∼121 to 137), H2 (∼167 to 175), H3 (∼180 to 189), H4 (∼193 to 210), H5 (∼212 to 219), and two short extended regions (Ortega-Roldan et al., 2013) (Fig. 3). The mobility of the H1 helix, which is most proximal to TM2, appears to depend in part on its connection to TM2 (Ortega-Roldan et al., 2015).
The ER luminal S1R C-terminal amino acids contain at least a portion of the structural and functional information needed for the S1R to interact with protein partners; for example, the C-terminal construct was found to interact with the nucleotide-binding domain of the ATP requiring cochaperone binding immunoglobulin protein (BiP) (78-kDa glucose-regulated protein, GRP78), and not the substrate domain of BiP (Ortega-Roldan et al., 2013). Residues 116–223, when expressed as a fusion with glutathione S-transferase, were shown in vitro to mitigate heat-mediated aggregation of the mitochondrial enzyme citrate synthase and to interact with GRP78 (BiP) in a calcium-dependent fashion (Hayashi and Su, 2007). In vivo overexpression of the S1R C-terminal 102–223 in MCF-7 cells enhanced bradykinin-stimulated calcium release by promoting removal of ankyrin 220 from the IP3-type 3 receptor in ER membranes. This latter action occurred via a chaperone-mediated function by binding directly to ankyrin 220 with resultant dissociation from the IP3-type 3 receptor (Hayashi and Su, 2001; Wu and Bowen, 2008). It is important to point out that the C-terminal amino acids (encompassing amino acids 100–223 approximately) do not bind ligands but can chaperone partner proteins. This indicates that although the chaperone function is ligand regulated in the intact protein, ligand-binding functions and chaperone functions are separable.
Structural Features of the S1R Ligand-Binding Region
Two main approaches have been informative for identification of the amino acid residues that affect S1R ligand binding: 1) site-directed mutagenesis and 2) use of radiolabeled photoaffinity probes.
Site-Directed Mutagenesis.
Carbodiimide-based chemical modifications of anionic amino acids (or combinations of amino acids) in the C terminus of the wild-type receptor or in the recombinantly expressed S1R were reported to reduce [3H]-haloperidol and [3H]-(+)-pentazocine binding. Reduction of [3H]-haloperidol binding was further supported by glycine substitutions that identified Asp126 and Glu172 as important residues (Seth et al., 2001) (Fig. 3A and Table 2). Yamamoto et al. (1999) showed that S99A, Y103F, and di-L105, 106 di-A which are in, or in close proximity to, TM2 reduced [3H]-(+)-pentazocine binding (Fig. 3A and Table 2). Recently a study of residues in the human S1R that were transiently expressed in a quail fibroblast line QT-6, null for constitutive S1Rs, identified amino acid residues important for specific [3H]-(+)-pentazocine binding (Brune et al., 2014). When percentiled against the human wild-type S1R the investigators found the following residues to reduce binding by greater than 70%: R119A, I128A, Y173A, S101A, Y103A, F107A, R175A, and E172D (see Table 2).
Amino acid residues of the sigma-1 receptor affected by mutation
Essentially, complete loss of binding, when compared with wild type, was seen with I128A, Y173A, and E172D. Deletion of 15 and 23 amino acids from the C terminus appeared to result in a partially unfolded receptor. Deletion of the C-terminal 7 residue YLFGQDP sequence reduced [3H]-(+)-pentazocine–specific binding by approximately 60%, presumably by a “Hasp-like” effect that altered or reduced the strength of interaction of R119, D126, and E172 with (+)-pentazocine. Additionally, 30%–70% reductions in (3H)-(+)-pentazocine binding were noted for Y120A, S127A, T127A, and F146A.
Based on current knowledge that the S1R exists both in vitro and in vivo in oligomeric forms (discussed later), it would be of great interest to establish whether only certain functional forms of the S1R are binding to agonists and antagonists and further to determine the stoichiometries of agonist/antagonist/S1R subunit binding. Multisubunit receptors such as the GABA A (Sigel and Steinmann, 2012) and the nicotinic acetylcholine receptors (Albuquerque et al., 2009) bind regulatory ligands at the interface between subunits. Is this also the case for ligands that interact with the homo-oligomeric S1R?
Use of Photoaffinity Probes.
Specific covalent derivatization of the binding site of the S1R in membrane preparations has been reported using several types of S1R ligand derivatives, including [3H]-azido-1,3-di-o-tolylguanidine (Kavanaugh et al., 1989), [3H]-azidoemopamil (Moebius et al., 1993), and [3H]-haloperidol (Largent et al., 1984; Tam and Cook, 1984) (Fig. 3B, i–iii). Several radioiodinated photoprobes have been developed in the Ruoho laboratory (Kahoun and Ruoho, 1992; Chen et al., 2007; Pal et al., 2007, 2008; Fontanilla et al., 2008) (Fig. 3B, iv–ix).
With the advent of effective methods to express the S1R in E. coli and purification of the S1R in native forms (Ramachandran et al., 2007; Gromek et al., 2013, 2014), identification of the general location of certain ligand binding regions has been possible using various small molecule radiolabeled photoprobes. Selective cleavage of the specifically covalently photolabeled pure S1R has been achieved by EndoLys C at the lysine at position 142, followed in some cases, by additional cleavage with cyanogen bromide at methionines 90 and 170 followed by SDS-PAGE analyses. The data from these experiments are summarized in Fig. 3B, iv–ix.
[125I]-Iodoazidococaine ([125I]-IACoc) specifically derivatized aspartate 188 in the SBLD II region (Chen et al., 2007). Electron-withdrawing groups on the para position of the phenyl ring such as azido and/or nitro enhanced the affinity of the cocaine derivatives for the S1R by at least 100-fold compared with para position electron-donating or nonderivatized phenyl ring tropane derivatives. These data presage the notion (Chen et al., 2007) that T-stacking pi/pi interactions of the cocaine phenyl ring with nearby aromatic amino acid containing side chains occur in the binding site. When the pi electron density of the cocaine phenyl ring was reduced by withdrawing groups, the pi electronic density of surrounding aromatic amino acid side chains strongly compensated to result in the enhancement of IACoc affinity. These aromatic amino acid residues in the S1R-binding site are likely to be Tyr120 and/or Trp121, as shown for the interaction of the heteraromatic condensed ring of (+)-pentazocine (Brune et al., 2014) with the S1R.
Binding and photoincorporation of N-3-(4-nitrophenylpropyl) dodecan-1-amine (4-NPPC12) (Fig. 3B, ix) (Chu et al., 2013) into His154 was similarly dependent on the electron-withdrawing properties of 4-NPPC12. Further, [125I]-iodoazidofenpropimorph ([125I]-IAF) labeled both the SBLD I and the SBLD II regions (Pal et al., 2007). When the radioiodinated phenyl azide was repositioned to provide greater probe flexibility and to identify additional binding regions such as with [125I]-N-IACoc and N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl) propylamine ([125I]-IAC44), the covalent derivatization expanded to include TM1 (Fontanilla et al., 2008).
The involvement of SBLD II in the ligand binding site as indicated by covalent derivatizations of the purified guinea pig S1R with the cocaine and fenpropimorph derived photoprobes is of interest. The juxtaposition of SBLD I and SBLD II as predicted by use of the reversible photocrosslinker [125I]-IABM (S-[[4-(4-amino-3-[125I] iodobenzoyl phenyl] methyl] ester) (Pal et al., 2008) is of further interest because SBLD II residues were not implicated in the specific binding of [3H]-(+)-pentazocine as assessed by mutagenesis in S1Rs overexpressed in quail cells (Brune et al., 2014). Because the S1R has been shown both in vitro and in vivo to exist in various oligomeric forms (Gromek et al., 2014; Mishra et al., 2015) it is not clear, at present, which forms are binding to (+)-pentazocine and which forms are binding to the photoprobes. Subtle structural changes in the ligand-binding regions in the various forms could underwrite the differences observed between the [3H]-(+)-pentazocine binding (Brune et al., 2014) and the results from [125I]-IABM (Pal et al., 2008) photolabeling.
Interestingly, when the final 15 residues of the C terminus were removed, specific photolabeling of the S1R by the S1R ligand [125I]-IACoc could not be achieved (Chen et al., 2007). These data have been further supported, as previously mentioned, by demonstration of a complete loss of specific [3H]-(+)-pentazocine binding after removal of 7 (YLFGQDP), 15, or 23 residues from the C terminus (Brune et al., 2014). These data, collectively, indicate an important role for the C terminus of the S1R for support of ligand binding.
S1R Interactions with Lipids
Although the S1R interacts with an impressive array of xenobiotic compounds, there are very few endogenous small molecules that show high-affinity binding to the receptor. It was noted that synthetic long-chain N-alkyl amines interact with the S1R. Based on this observation it was discovered that endogenous long-chain sphingoid bases such as d-erythro sphingosine, which conform to the S1R pharmacophore motif of a long N-alkyl chain with a nitrogen atom containing unshared electrons, interact with the S1R (Ramachandran et al., 2009; Chu et al., 2011, 2013). The endogenous long-chain sphingoid bases d-erythro-sphingosine, l-threo-sphingosine, dihydrosphingosine (sphinganine), and N,Nʹ-dimethyl sphingosine inhibited [3H]-(+)-pentazocine binding to the purified S1R with Kd values in the 100 nM range. The Kd values for inhibition were lower by an order of magnitude for the pure S1R as compared with guinea pig liver membranes (Ramachandran et al., 2009). The major long-chain base d-erythro-sphingosine (and to a lesser extent d-erythro-sphingosine-1-phosphate) was detected in S1R immunoprecipitates from receptor-overexpressing human embryonic kidney 293 (HEK293) cells, further confirming that such an interaction between sphingosine and the S1R occurs in vivo (Ruoho et al., 2012).
d-Erythro sphingosine and its derivatives are important endogenous lipid mediators variously involved in multiple signal transduction pathways (Hannun and Obeid, 2008). Sphingosine can be phosphorylated to sphingosine-1 phosphate by the enzyme sphingosine kinase (Spiegel and Milstien, 2007). However, compared with sphingosine and N,N-dimethylsphingosine, sphingosine-1 phosphate was not as efficient in inhibiting [3H]-(+)-pentazocine binding to the S1R (Ramachandran et al., 2009). Ceramide and ceramide-1 phosphate, the acylated derivatives of sphingosine, also failed to inhibit [3H]-(+)-pentazocine binding to the S1R. High-affinity sphingosine binding to the S1R raises the possibility that sphingosine and N-methylated derivatives of sphingosine may be endogenous regulators of the S1R. Tsai et al. (2015) have also implicated myristic acid as a S1R agonist that, when bound to the receptor, mitigates aberrant tau phosphorylation via cdk5 to allow for proper axon extension in mouse brain.
Sigma receptors have been previously reported to be associated with lipid-containing microdomains. The S1R was found to be present in cholesterol-enriched, detergent-insoluble lipid rafts of the ER in NG108 neuroblastoma cells where they were shown to be important for the compartmentalization of ER synthesized lipids (Hayashi and Su, 2003). In ER lipid droplets, the S1R colocalized with caveolin-2, a cholesterol-binding protein. Treatment with the S1R ligand (+)-pentazocine resulted in translocation of S1Rs from the ER lipid microdomains to cytoplasmic domains (Hayashi and Su, 2003). Furthermore, treatment of Chinese hamster ovary K1 cells with sigma ligands enhanced the appearance of the S1R in focal adhesions (Mavlyutov and Ruoho, 2007). In rat primary hippocampal cultures, S1Rs were shown to form galactoceramide-enriched lipid rafts and to promote differentiation of oligodendrocytes (Hayashi and Su, 2004).
It has been suggested that the S1R plays an important role in modulating the stability of the glycosphingolipid biosynthetic enzyme ceramide galactosyl synthase. This enzyme is expressed predominantly at the endoplasmic reticulum of oligodendrocytes and is responsible for synthesizing galactosylceramides that play important roles in regulation of axon conductance. Knockdown of the S1R increased the lifetime and enzymatic activity of ceramide galactosylsynthase in a D6P2T Schwann cell line (Hayashi et al., 2012). In light of these findings in cell culture studies, it would be of interest to determine whether the S1R knockout mice show aberrant myelination of axons and exhibit differences in neuronal conduction.
When the purified S1R was exchanged from Triton X-100 into lauryl dimethylamine N-oxide (LDAO), a detergent often preferred for the crystallization of membrane proteins, a complete loss of specific S1R [3H]-(+)-pentazocine binding was observed. It was subsequently noted that LDAO is a specific inhibitor of [3H]-(+)-pentazocine binding to the S1R in guinea pig liver membranes as well as to the pure S1R protein with Ki values of 5.4 nM and 72.1 nM, respectively (Ramachandran, 2008). The high-affinity binding of this detergent to the S1R may afford an opportunity for S1R crystallization trials using LDAO specifically bound to the S1R ligand-binding site at concentrations below its critical micelle concentration.
In addition to LDAO, molecules that share a common long alkyl chain that extend from a secondary or tertiary nitrogen atom and bind to the S1R include tridemorph and (2R-trans)-2-butyl-5-heptylpyrrolidine, a compound isolated from the culture broth of Streptomyces longispororuber (Kumagai et al., 2000). Finally, the S1R has been shown to bind cholesterol (Palmer et al., 2007). Using cholesteryl beads, Palmer et al. (2007) were able to demonstrate that purified myc-tagged S1R peptides containing residues 161–180 and 191–210 bound to cholesterol (Fig. 3A).
Functions of the S1R May Be Based on Its Oligomerization States
As initially demonstrated by use of the radioiodinated photoaffinity label [125I]-iodoazidofenpropimorph ([125I]-IAF), the S1R in rat liver membranes exists in oligomeric forms (Pal et al., 2007). This observation was further supported by in vitro experiments demonstrating the separation of the purified S1R and the maltose-binding protein–S1R fusion protein into monomers, tetramers, and hexamer/octamers using size exclusion chromatography (Ramachandran et al., 2007; Gromek et al., 2014). The tetramer and hexamer/octamer forms bound specifically to the S1R agonist [3H]-(+)-pentazocine but not to the monomer form. By calculation, the tetramer form of the maltose binding protein–S1R fusion bound the S1R antagonist BD-1047 (Nʹ-[2-(3,4-dichlorophenyl)ethyl]-N,N,Nʹ-trimethylethane-1,2-diamine), with a stoichiometry of 1:2 (1 BD-1047:2 S1R monomers).
The presence of a S1R ligand-binding dimer form, shown both in vitro and in vivo, has been further supported by use of the photoprobe N-3-(4-nitrophenylpropyl) dodecan-1-amine (4 NPPC12) (Fig. 3B, ix) (Chu et al., 2013) and by in situ oxidative crosslinking of a C94A, M170C guinea pig S1R construct. Further, when the S1R was coexpressed in COS-7 cells as C-terminal fluorescent protein (GFP2 and CYP) fusion constructs and analyzed by Forster resonance energy transfer (spectral FRET) (Mishra et al., 2015), the S1R was shown to exist constitutively in monomeric and oligomeric forms. Compared with non-drug-treated controls, dimer and monomer forms were favored in the presence of the agonist (+)-pentazocine, whereas higher order oligomers were favored in the presence of the S1R antagonist haloperidol.
The presence of oligomeric forms of the S1R in vivo that are modified in their ratios by S1R ligands offers the likelihood that constitutively active forms of the S1R exist in vivo and that the populations of these forms are regulated by agonists and antagonist (or inverse agonists). Because the C-terminal tail by itself (amino acids approximately 110–223) has been clearly shown to possess chaperone functions (Hayashi and Su, 2007; Wu and Bowen, 2008) and interactions with the cochaperone BiP (Ortega-Roldan et al., 2013), the role of S1R agonists may be to dissociate or “expose” C-terminal regions of the S1R in the dimer and/or monomer forms. In the tetramer and higher oligomeric forms, the C terminus may be stabilized by “cryptic” intramolecular interactions by antagonists (or as inverse agonists, as discussed later) and thus be functionally unavailable.
Do Antagonists of the S1R Function as Inverse Agonists?
A consideration of inverse agonists for receptors has many components (Milligan, 2003), but its basic tenants are 1) that a receptor can exist in functionally active constitutive forms in vivo, 2) that defined alternative structural forms of a receptor occur that can be stabilized by compounds that reduce the constitutive activity of the receptor, and 3) that endogenous agonists do not account for the observed “constitutive” receptor activity. Several examples of constitutive activity of the S1R have been reported. When washed neuronal membranes prepared from mouse brain were treated with the putative S1R antagonists BD-1047 or haloperidol, GPCRs such as the µ-opioid receptor and the muscarinic acetylcholine receptor showed enhanced potency for their respective agonists as measured by increased GTPγ35S binding (Kim et al., 2010). The investigators concluded that the S1R in the neuronal membrane preparations was constitutively active and that inhibition of the S1R “released” the suppressed signaling efficiency of the GPCR agonists. Overexpression of the S1R in MCF7 cells was shown to result in enhanced bradykinin-induced calcium release in the absence of exogenously added agonist (Wu and Bowen, 2008). Because this constitutive response was inhibited by the S1R ligand, BD-1063 [N-[2-(3,4-dichlorophenyl) ethyl]-4-methylpiperazine dihydrochloride], Wu and Bowen (2008) concluded that the S1R was constitutively active in these experiments and that BD-1063 behaved as an S1R inverse agonist.
Aydar et al. (2002) have demonstrated that expression of the S1R in Xenopus oocytes in the absence of agonist resulted in regulation of voltage-gated 1.4 and 1.5 potassium channels, indicating constitutive activity of the S1R. It is therefore reasonable to consider the following scenario: monomer, dimer, tetramer, hexamer/octamer states of the S1R may have important consequences regarding the chaperone functions of the S1R. As previously described when COS cells containing the S1R were incubated with the S1R agonist (+)-pentazocine, the relative ratio of dimer and monomers increased (Mishra et al., 2015) whereas treatment of the cells with the S1R inhibitor haloperidol increased higher oligomeric forms. It is tempting to speculate that higher oligomeric forms of the S1R are functionally inactive. In this way, S1R functional inhibitors such as haloperidol, BD-1047, and BD-1063 may be more accurately considered to be inverse agonists rather than neutral antagonists (see Fig. 4 for a proposed model of the chaperone mechanism (s) of S1R).

Proposed mechanistic model of the sigma-1 receptor chaperone function in vivo. In vivo (and in vitro) evidence suggests that the sigma-1 receptor exists as dimers, tetramers, and hexamers/octamers (and perhaps even higher order oligomers). In vivo, the sigma-1 receptor is proposed to bind agonists as a dimer and/or monomer (or the agonists create functionally active dimers and/or monomers from higher order oligomers) while putative antagonists (or inverse agonists) shift the sigma-1 receptor to the tetramer/oligomer forms. As such, dimer and monomer forms may represent the functional chaperone states while higher oligomers of the sigma-1 receptor may serve as a reservoir for the active forms. The monomer form has been shown to bind to protein partners at the plasma membrane as a functional unit, which may suggest a secondary function independent of the chaperone activity of the sigma-1 receptor.
This situation is not unlike that of the β2-adrenergic receptor (and other GPCRs) in which an Ro state, which is structurally differentiated from the R basal or constitutive states (Rʹ, Rʹʹ, Rʹʹ, R*), is functionally associated with inverse agonists (Gether and Kobilka, 1998; Manglik and Kobilka, 2014). Because many states (or functional forms) of the S1R may occur in situ, the function of inhibitors such as haloperidol, BD-1047, and BD-1063 may be more accurately considered as inverse S1R agonists rather than neutral antagonists.
Proposed Dependence of S1R Oligomeric Equilibria for Chaperone Functions: Is the S1R a Member of the Small Heat Shock Protein Family?
The multitasking chaperone nature of the S1R has been well established (Su et al., 2010). As previously described in this review, in vitro and in vivo the S1R exists in oligomeric forms that appear to be modulated by (+)-pentazocine and by haloperidol (Gromek et al., 2014; Mishra et al., 2015). Based on current knowledge in the field, the S1R does not bind ATP, in contrast to many large mammalian chaperone-like heat shock proteins such as GRP78/BiP Hsp 70, and Hsp 90 (Hayashi and Su, 2007). Families of small heat shock proteins (sHSPs) consisting of monomer sizes ranging from 12 to 42 kDa that do not bind ATP have been identified in prokaryotes, plants, and mammals (reviewed in Carver et al., 2003; Poulain et al., 2010; Basha et al., 2012). Humans have 10 paralogous sHSPs, HspB1 to HspB10 (Basha et al., 2012). These sHSPs are both soluble proteins and lipid-mediated membrane-bound proteins that localize to lipid rafts especially under cellular stress conditions (Horvath et al., 2008). The function of these sHSPs is to temporarily stabilize proteins that are partially disordered under cellular stress to prevent complete aggregation and denaturation. The sHSPs-protected proteins are then handed off to larger HSPs (such as HSP70) where the energy of ATP is used to complete the folding process. The sHSPs therefore provide initial “paramedic” life support for a failing protein in preparation for a final ATP-dependent “physician” rescue. In this regard it is noteworthy that Mori et al. (2013) have demonstrated that temporary stabilization of the cellular stress protein IRE1-α by S1R is necessary to allow appropriate phosphorylation of IRE1-α and thus escape from ubiqitination and proteosomal destruction.
As previously discussed, a direct interaction of the S1R with the ATP chaperone GRP78 (BiP) has been well documented (Hayashi and Su, 2007) and further elaborated by the Schnell laboratory (Ortega-Roldan et al., 2013) to involve the S1R C terminus interaction with the nucleotide-binding domain (not the “substrate” domain of BiP). This observation is in total agreement with the interaction regions of sHSPs with their ATP-dependent chaperone partners such as Hsp70 (Poulain et al., 2010). Additionally, although the S1R functions mainly as an intracellular chaperone protein, an extracellular presence of the S1R secreted from NG108 neuroblastoma glioma hybrid cells has been reported (Hayashi and Su, 2003) although a chaperoning target has not been unequivocally identified. A similar extracellular presence has been observed for Clusterin (Poon et al., 2000; Wilson and Easterbrook-Smith, 2000; Hatters et al., 2002; Carver et al., 2003), a glycosylated sHSP that presumably chaperones extracellular client proteins that require stabilization. Finally, the subunit-subunit interfaces of sHSPs appear to vary in some oligomeric complexes (Van Montfort et al., 2001; McHaourab et al., 2009; Basha et al., 2012) providing for potential pleiotropic molecular interactions. Can these properties also extend to the S1R to account partially for the wide range of small molecules that bind to and regulate S1R functions?
The sHSPs are further defined by a C-terminal domain of approximately 90–100 amino acids known as the alpha crystallins domain that contains seven or eight antiparallel beta strands (for a review, see Basha et al., 2012) and a L/V/I X L/V/I sequence near the C terminus that is important for stabilizing dimer formation (Poulain et al., 2010). A striking feature of many of the sHSPs (especially those in mammals) that function as dimers is that the interaction of two specific antiparallel beta strand sequences that are stabilized by the L/V/I X L/V/I sequence (Poulain et al., 2010) further promote the subsequent formation of oligomers from the preformed dimers (Basha et al., 2012).
It has been suggested that the higher oligomeric forms of some sHSPs may subsequently serve as reservoirs for the active dimer forms. The mammalian S1R shares many of the features described here for the sHSPs, including the presence of C-terminal beta strands (Ortega-Roldan et al., 2015) and a C-terminal LELQL sequence that may be involved in stabilizing dimerization of the S1R. One additional feature regarding attainment of maximum steady-state–specific binding of (3H)-(+)-pentazocine to S1R in membrane preparations or in cells is that incubation is usually required for at least 90 minutes at 32–37°C. This may be time needed to “trap” an active (+)-pentazocine binding form of the S1R, perhaps a dimer, which has been generated from higher oligomeric forms (Gromek et al., 2014; Mishra et al., 2015). The data are therefore consistent with the occurrence of a dynamic equilibrium between octamer/hexamer/tetramer/dimer and monomer states of the S1R that may presage the functional mechanisms of the S1R. Similar to sHSPs, the hexamer/octamer/tetramer forms are likely to serve as storage forms of the S1R that equilibrate and are stabilized in active states by agonists such as (+)-pentazocine to eventually generate the dimer and monomer forms that constitute the chaperoning structures.
Inhibitory tetramers and higher order forms may be stabilized by inverse agonists such as haloperidol and BD-1047 (see Fig. 4). In agreement with these general ideas, using atomic force microscopy, Balasuriya et al. (2012) have shown that the monomer form of the S1R (or possibly a monomer form generated from the dimer) has been identified as directly bound to the voltage-gated Nav1.5 sodium channel with 4-fold symmetry, to the human ether-à-go-go (hERG) voltage-gated potassium channel (Balasuriya et al., 2014) with 4-fold symmetry, to the acid-sensing ion channel-1a with 3-fold symmetry (Carnally et al., 2010), and with selectivity to the GLuN1 subunit of the GluN1/GluN2a N-methyl-d-apartate receptor (Balasuriya et al., 2013) (see Table 3 for further examples of client proteins that have been shown to interact with the S1R).
Client proteins that bind to the sigma-1 receptor
List of client proteins that have been shown to directly bind to the sigma-1 receptor as demonstrated by coimmunoprecipitation or by Forster resonance energy transfer (FRET; readers are directed to the references listed to review the data).
Beyond the chaperone functions of the S1R that involve its first-responder–like sHSP functions to client proteins, the S1R is speculated to provide permanent regulatory support for their signal transduction roles—for example, interactions with the aforementioned ion channels at the plasma membrane. However, because the S1R has been demonstrated to bind to the nonglycosylated form of the hERG channel at the plasma membrane (Balasuriya et al., 2014), questions remain as to whether the S1R binds to the semiunfolded state or the native form of these protein substrates as a functional subunit.
In sum, despite these apparent similarities with the sHSPs, the S1R have evolved in mammalian cells such that the various “states” of the S1R (including perhaps also the 18–21 kDa sigma-2 receptor) (Bowen et al., 1989; Hellewell and Bowen, 1990) require regulation by small molecules (both endogenous and synthetic). However, important and unique differences differentiate the S1R from the sHSPs: 1) the sHSPs are only variously tethered to the membrane but do not possess transmembrane regions as is the case for the S1R, and 2) the sHSPs have not been reported to regulate ion channels or other receptors (such as GPCRs) on plasma membranes.
Conclusions and Summary
The S1R is a unique multitasking lipid microdomain ER and plasma membrane localized chaperone protein that is likely to be an important therapeutic target for treatment of many forms of neurodegenerative conditions in humans. As a molecular chaperone the S1R responds to various cellular stresses to stabilize and/or support the stability of proteins involved in signal transduction pathways that involve calcium homeostasis of the cytoplasm, ER, and mitochondria. Cell-signaling protein partners such as voltage-gated ion channels and G-protein coupled receptors appear to be constitutively regulated by direct interaction with the S1R. Direct and indirect actions of S1Rs that involve alterations in enzyme activities that can regulate signaling pathways—for example, Rac-1 GTPase and cdk5 control maturation of neuronal dendrites and axon elongation.
S1Rs reduce stress-related protein aggregations associated with neurodegenerative diseases. The S1R exists in ligand-regulated monomeric and oligomeric forms in vitro and in vivo that likely underwrite its multitasking functions. In vitro, S1R dimers and tetramers may bind to regulatory small molecules at monomer/monomer interfaces (2:1 ligand/monomer ratio). Higher oligomeric forms of the S1R are proposed to be stabilized by inhibitors in a manner consistent with inverse agonist actions. The molecular features of the membrane-bound S1R that show pharmacologic regulation via endogenous and synthetic small molecules place it in a unique niche of the superfamily of small heat shock protein chaperones.
Acknowledgments
The authors thank Brian Torres for assistance in formatting of the text, figures, and tables.
Authorship Contributions
Wrote or contributed to the writing of the entire manuscript: Chu, Ruoho.
Footnotes
- Received July 31, 2015.
- Accepted November 6, 2015.
Supported by a University of Wisconsin School of Medicine and Public Health grant (to A.E.R.).
Abbreviations
- BD-1047
- Nʹ-[2-(3,4-dichlorophenyl)ethyl]-N,N,Nʹ-trimethylethane-1,2-diamine
- BD-1063
- N-[2-(3,4-dichlorophenyl) ethyl]-4-methylpiperazine dihydrochloride
- BiP
- binding immunoglobulin protein
- cdk5
- cyclin-dependent kinase 5
- ER
- endoplasmic reticulum
- GPCR
- G-protein coupled receptor
- GRP78
- 78-kDa glucose-regulated protein
- hERG
- human ether-à-go-go
- HSP
- heat shock protein
- [125I]-IACoc
- [125I]-iodoazidococaine
- [125I]-IABM
- S-[[4-(4-amino-3-[125I] iodobenzoyl phenyl] methyl] ester
- [125I]-IAC44
- N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl) propylamine
- (125I)-IAF
- (125I)-iodoazidofenpropimorph
- IP3
- inositol triphosphate
- LDAO
- lauryl dimethylamine N-oxide
- NE-100
- N,N-dipropyl-2-[4-methoxy-3-(2-phenylethoxy)phenyl]-ethylamine monohydrochloride
- 4-NPPC12
- N-3-(4-nitrophenylpropyl) dodecan-1-amine
- PRE-084
- 2-(4-morpholinethyl) 1-phenylcyclohexane-1-carboxylate hydrochloride
- SKF10047
- [2S-(2α,6α,11R*]-1,2,3,4,5,6-hexahydro-6,11-dimethyl-3-(2-propenyl)-2,6-methano-3-benzazocin-8-ol
- S1R
- sigma-1 receptor
- SBLD
- steroid-binding-like domain
- sHSP
- small heat shock protein
- TM
- transmembrane
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Commonly Used Agonists/Antagonists for the S1R
- Structural Features of the S1R
- Structural Features of the S1R Ligand-Binding Region
- S1R Interactions with Lipids
- Functions of the S1R May Be Based on Its Oligomerization States
- Do Antagonists of the S1R Function as Inverse Agonists?
- Proposed Dependence of S1R Oligomeric Equilibria for Chaperone Functions: Is the S1R a Member of the Small Heat Shock Protein Family?
- Conclusions and Summary
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters