


Abstract
Westernization of dietary habits increases lipid intake and is responsible for increased numbers of patients with atherosclerotic diseases. Niemann-Pick C1-Like 1 (NPC1L1)—a cholesterol importer—plays a crucial role in dietary cholesterol absorption in the intestine and is closely associated with several lipid-related diseases, including atherosclerosis. NPC1L1 is highly expressed in the liver and intestine in humans, whereas NPC1L1 expression is low in the rodent liver. Due to species differences in the tissue distribution of NPC1L1, there are limited studies on the pathophysiological role of hepatic NPC1L1, a cholesterol reabsorber from bile. In the present study, to explore whether hepatic NPC1L1 is involved in the development/progression of atherosclerosis, we compared four kinds of atherosclerosis mouse models with different expression levels of NPC1L1 in the intestinal and liver tissues in a genetic background of dysfunctional low-density lipoprotein receptor mutation. Western diet (WD)–induced hyperlipidemia and atherosclerotic plaque formation were more severe in mice expressing NPC1L1 in both the liver and intestine (plasma cholesterol, 839.5 mg/dl; plaque area, 29.5% of total aorta), compared with mice expressing NPC1L1 only in the intestine (plasma cholesterol, 573.1 mg/dl; plaque area, 13.3% of total aorta). Such hepatic NPC1L1-mediated promotion of hyperlipidemia and atherosclerosis was not observed in mice not expressing intestinal NPC1L1 and mice treated with ezetimibe, an NPC1L1 inhibitor used clinically for dyslipidemia. These results suggested that hepatic NPC1L1 promotes WD-induced dyslipidemia and atherosclerosis in concert with intestinal NPC1L1. Our findings provide novel insights into the pathophysiological importance of hepatic NPC1L1 in development/progression of atherosclerosis.
Significance Statement Niemann-Pick C1-Like 1 (NPC1L1) protein, a cholesterol importer and a molecular target of ezetimibe clinically used for dyslipidemia, is highly expressed not only in the intestine, but also in the liver in humans, although the pathophysiological importance of hepatic NPC1L1 in atherosclerotic diseases remained unclear. By using novel mouse models to separately analyze the effects of hepatic and intestinal NPC1L1 on the development/progression of atherosclerosis, we first demonstrated that hepatic NPC1L1 accelerates Western diet–induced atherosclerotic plaque formation in an intestinal NPC1L1-dependent and an ezetimibe-sensitive manner.
Introduction
Because of the global changes in dietary habits suiting Western styles—such as high consumption of fats, including cholesterol—the number of patients with atherosclerosis—a chronic and progressive disease involving the arterial walls and resulting in lethal myocardial infarction and stroke—is increasing worldwide. It is well known that a high plasma cholesterol concentration is one of the major risk factors for the development of atherosclerosis. Several processes such as de novo cholesterol synthesis, catabolism to bile acids, intestinal cholesterol absorption, and biliary cholesterol excretion regulate plasma cholesterol levels. Among these processes, intestinal cholesterol absorption and biliary cholesterol excretion are controlled by Niemann-Pick C1-Like 1 (NPC1L1) protein (Altmann et al., 2004; Temel et al., 2007; Yamanashi et al., 2007, 2009, 2017, 2018).
NPC1L1 is a cholesterol importer, which plays a key role in intestinal cholesterol absorption and is a molecular target of ezetimibe, a cholesterol absorption inhibitor clinically used for treatment of dyslipidemia (Altmann et al., 2004; Garcia-Calvo et al., 2005; Yamanashi et al., 2007). It has been reported that there are species differences in the tissue distribution of NPC1L1 (Altmann et al., 2004). Indeed, NPC1L1 is highly expressed in both the liver and intestine in humans, whereas NPC1L1 is predominantly expressed in the intestine but its expression is low in the liver of rodents. To date, the physiologic functions of hepatic NPC1L1 have been studied extensively (Temel et al., 2007; Tang et al., 2011; Yamanashi et al., 2012; Xie et al., 2013; Wang et al., 2018), and several studies with liver-specific human NPC1L1 transgenic (1L1Tg) mice have demonstrated that hepatic NPC1L1 is expressed in the bile canalicular membrane and is involved in the reabsorption of biliary cholesterol by hepatocytes in an ezetimibe-sensitive manner (Temel et al., 2007; Tang et al., 2011; Xie et al., 2013; Wang et al., 2018). These findings suggest that hepatic NPC1L1 negatively regulates biliary cholesterol excretion in humans.
To date, a number of in vivo studies with atherosclerosis mouse models, such as LDL receptor (LDLR)–deficient (or mutant) mice and apolipoprotein E–deficient mice, have been conducted to reveal the molecular mechanisms of atherosclerosis development/progression and to identify novel therapeutic targets for atherosclerotic diseases (Knowles and Maeda, 2000; Davis et al., 2007; Zadelaar et al., 2007; Lee et al., 2017). Some of these studies indicated that NPC1L1-mediated intestinal cholesterol absorption is involved in atherosclerotic plaque formation and progression (Davis et al., 2007). However, due to differences in the tissue distribution of NPC1L1 between humans and rodents, a feasible pathophysiological involvement of hepatic NPC1L1 in atherosclerosis has been overlooked in many studies using mouse models. Indeed, although the importance of hepatic NPC1L1 in the physiology and pathophysiology of bile formation and liver diseases has been intensively studied so far (Temel et al., 2007; Toyoda et al., 2019), there is little information about its effect on atherosclerosis progression. Therefore, the purpose of our study was to explore the effect of hepatic NPC1L1 on atherosclerotic plaque formation in mouse models.
For this purpose, we generated novel atherosclerosis mouse models by crossing LDLR mutant (Ldlrmt) mice with liver-specific 1L1Tg mice and/or NPC1L1 knockout (1l1KO) mice, allowing us to independently analyze the effect of hepatic NPC1L1 and that of intestinal NPC1L1 on Western diet (WD)–induced atherosclerosis development/progression. By comparing the generation of four kinds of distinct mouse models with or without the expression of intestinal or hepatic NPC1L1 (Ldlrmt mice, Ldlrmt/1L1Tg mice, Ldlrmt/1l1KO mice, and Ldlrmt/1L1Tg/1l1KO mice), we revealed that hepatic NPC1L1 contributes to increased plasma concentrations of cholesterol and triglycerides and exacerbates atherosclerotic plaque formation when intestinal NPC1L1 is functional. These observations indicated the pathophysiological importance of NPC1L1 not only in the intestine, but also in the liver in diet-induced hyperlipidemia and atherosclerosis.
Materials and Methods
Animals.
Ldlrmt mice with a C57BL/6 background (RBRC-GSC0247) were provided by RIKEN BRC through the National BioResource Project of MEXT (Yamamoto et al., 2017). 1l1KO mice with a C57BL/6 background were previously generated (Takada et al., 2015). Liver-specific 1L1Tg mice were purchased from The Jackson Laboratory and backcrossed with C57BL/6 mice. Ldlrmt and 1l1KO mice were crossed to generate Ldlr-mutated 1l1KO mice (Ldlrmt/1l1KO). In addition, Ldlr-mutated 1L1Tg mice (Ldlrmt/1L1Tg) were generated by crossing Ldlrmt mice with 1L1Tg mice. Furthermore, Ldlrmt/1l1KO mice and Ldlrmt/1L1Tg mice were crossed to generate Ldlrmt mice expressing NPC1L1 in the liver, but not in the intestine (Ldlrmt/1L1Tg/1l1KO). In the present study, the following four kinds of genetically distinct mice with or without NPC1L1 expression in the intestine or liver were tested: Ldlrmt mice, Ldlrmt/1l1KO mice, Ldlrmt/1L1Tg mice, and Ldlrmt/1L1Tg/1l1KO mice. All mice were housed in temperature-controlled and humidity-controlled cages with a 12-hour light/dark cycle and with free access to water and a normal control diet (FR-1; Funabashi Farm Co., Ltd., Chiba, Japan) or WD [D12079B (41 kcal% fat and 0.21% cholesterol); Research Diets Inc., New Brunswick, NJ] in the absence or presence of ezetimibe (20 μg/g diet). Hepatic concentrations of ezetimibe (approximately 0.10 μg/g liver) and ezetimibe-glucuronide (approximately 1.0 μg/g liver), an active metabolite of ezetimibe, were not significantly different among mice of each genotype fed with the ezetimibe-containing WD. All experiments with mice were conducted in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals, and with protocols approved by the Animal Studies Committee of the University of Tokyo. All mice studied were euthanized by cervical dislocation.
Western Blot Analysis.
For preparing crude membranes from intestinal epithelial cells and hepatocytes, isolated mouse small intestine and liver were homogenized with buffer A [50 mM Tris-HCl (pH 7.4) containing 2 mM EDTA, 2 mM EGTA, 2 mM PMSF (phenylmethylsulfonyl fluoride), leupeptin (5 mg/ml), pepstatin (1 mg/ml), and oprotinin (5 mg/ml)] and were centrifuged at 1500g for 15 minutes. After centrifugation, the supernatant was recentrifuged at 20,000g for 60 minutes. The precipitated crude membrane preparation was resuspended in buffer A and stored at −80°C before use in Western blot analysis. The protein concentration of each specimen was measured by BCA protein assay using bovine serum albumin as a standard. Total extracted crude membrane proteins were diluted with 2× SDS loading buffer and subjected to Western blot analysis, as described previously (Yamanashi et al., 2007). An SDS-polyacrylamide gel (7%) was used to separate proteins in each specimen. The molecular weights were determined using a prestained protein marker (New England Biolabs Inc., Ipswich, MA). The primary antibodies used for experiments were 500-fold diluted rabbit anti-NPC1L1 antibody (NB400-128; Novus Biologicals, Centennial, CO) for NPC1L1, or 200-fold diluted rabbit anti-Na+/K+-ATPase (Na+- and K+-dependent ATPase) α antibody (H-300, catalog number sc-28800; Santa Cruz Biotechnology Inc., Dallas, TX) for endogenous Na+/K+-ATPase α. For detection, the membrane was incubated in 5000-fold diluted horseradish peroxidase–labeled anti-rabbit IgG antibody (catalog number NA934V; GE Healthcare UK Ltd., Buckinghamshire, UK) in Tris-buffered saline containing 0.05% Tween 20 with 0.1% bovine serum albumin for 1 hour at room temperature. Enzyme activity was assessed using an ECL Prime Western Blotting Detection Reagent (GE Healthcare UK Ltd.) with a luminescent image analyzer (Bio-Rad Laboratories, Hercules, CA).
Emulsion Preparation.
Emulsions were prepared as described previously (Yamamoto et al., 2007; Narushima et al., 2008; Takada et al., 2015) with minor modifications. Briefly, stock lipid solutions were mixed to give a final concentration of 13.3 mM triolein, 2.6 mM cholesterol, 3 mM l-α-phosphatidylcholine, and 3 μCi/ml [3H]cholesterol (American Radiolabeled Chemical Inc., St. Louis, MO). The solvent was evaporated, and 19 mM sodium taurocholate (dissolved in PBS) was added to give the required lipid concentrations. The mixture was then sonicated three times for 3 minutes using an ultrasonic homogenizer (catalog number UP 200H; Hielscher Ultrasonics, Teltow, Germany).
In Vivo Acute Cholesterol Absorption Study.
In vivo acute cholesterol absorption studies were conducted as described previously (Yamamoto et al., 2007; Narushima et al., 2008; Takada et al., 2015) with minor modifications. The 10- to 12-week-old male mice were fasted for 18 hours and anesthetized with urethane. Next, a [3H]cholesterol-containing emulsion (5 ml/kg) was delivered directly into the small intestine via the duodenal cannula. Two hours after the emulsion loading, mice were sacrificed, and plasma and liver were isolated to quantify the level of [3H]cholesterol. The level of [3H]cholesterol in each specimen was measured using the liquid scintillation counter.
Quantification of Biliary Lipids.
The 10- to 12-week-old male mice were anesthetized with urethane. Then, the cystic duct was ligated and a common bile duct fistula was created using a Teflon catheter (catalog number UT-03; Unique Medical Co., Ltd., Tokyo, Japan) to collect hepatic bile specimens, as described previously (Yamanashi et al., 2011). Concentrations of biliary cholesterol, total bile acids, and phospholipids were quantified using Total Cholesterol E-test Wako (Wako Pure Chemical Industries, Ltd., Osaka, Japan), Total Bile Acids-test Wako (Wako Pure Chemical Industries, Ltd.), and Phospholipids C-test Wako (Wako Pure Chemical Industries, Ltd.), respectively.
Lipoprotein Fractionation.
Lipoprotein fractionation was performed as described previously (Yamamoto et al., 2017). Briefly, plasma specimens collected from mice were fractionated by size-exclusion chromatography using a Pharmacia Smart System fast protein liquid chromatography (FPLC) system equipped with a Superose 6 column (GE Healthcare UK Ltd.). Elution was performed in PBS containing 1 mM EDTA and 3 mM sodium azide as a running buffer. After loading 300 μl of serum, the system was run with a constant flow of 200 μl/min, with 500-μl fractions collected. Cholesterol and triglyceride concentrations in each fraction were quantified with the Total Cholesterol E-test Wako and Triglyceride E-test Wako (Wako Pure Chemical Industries, Ltd.), respectively.
Quantification of Hepatic Lipids.
Lipid extraction from liver samples was performed according to the well-known method of Bligh and Dyer (1959). In brief, 200 μl of homogenized liver solution (50 mg liver/ml PBS) was mixed with 250 μl of chloroform and 500 μl of methanol, then was well vortexed. After 10 minutes, 250 μl of chloroform was added to the mixture, and after blending, the homogenates mixture was centrifuged at 1800g for 10 minutes. After complete removal of the alcoholic (top) layer, 250 μl of the resulting chloroform (bottom) layer was transferred to a new glass tube, then evaporated to dryness under a stream of nitrogen, dissolved in isopropanol containing 10% (w/w) Triton X-100, and subjected to measurements of hepatic cholesterol and triglyceride. For quantitative calibration curves, standard samples containing known concentrations of cholesterol and triglyceride were prepared in a similar manner. The concentrations of cholesterol and triglyceride in each sample were measured using the Total Cholesterol E-test Wako and the Triglyceride E-test Wako, respectively.
RNA Extraction and Quantitative Polymerase Chain Reaction.
Total RNA was extracted from mouse liver using the RNA isoPlus Reagent (Takara Bio, Inc., Shiga, Japan), according to the manufacturer protocol. The extracted total RNA was reverse-transcribed with ReverTraAce (Toyobo, Osaka, Japan). Quantitative polymerase chain reaction (PCR) was performed using SYBR GreenER qPCR SuperMix Universal (Life Technologies, Tokyo Japan) and the Eco Real-Time PCR System (Illumina Inc., San Diego, CA) at 95°C for 10 minutes followed by 40 cycles at 95°C for 15 seconds and at 60°C for 1 minute. Primers for mouse 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase (sense and antisense primers were 5′-gtggcaatgctgatcgg-3′ and 5′-ggatcttcttgcggtaggct-3′, respectively), mouse HMG-CoA reductase (sense and antisense primers were 5′-tgggcatgaacatgatctcta-3′ and 5′-ggcttcacaaaccacagtc-3′, respectively), and mouse glyceraldehyde-3-phosphate dehydrogenase (sense and antisense primers were 5′-atgtgtccgtcgtggatctg-3′ and 5′-tgaagtcgcaggagacaacc-3′, respectively) were used.
Atherosclerotic Lesion Analysis.
The 6- to 8-week-old male mice were fed with a control diet or a WD for 20 weeks. After the 20 weeks of feeding, mice were sacrificed and plasma was collected, and the entire aorta was isolated. The collected aorta was opened longitudinally and stained with Oil Red O (Merck KGaA, Darmstadt, Germany). Oil Red O–stained plaque lesions were quantified using Adobe Photoshop, and the extent of atherosclerosis was expressed as the percentage of the lesion area extending from the ascending aorta to the abdominal bifurcation. The quantification was performed by an investigator who was blinded to group allocation.
Sample Sizes.
Each experiment was designed to use the minimum number of mice to obtain informative results. Although no statistical methods were used to predetermine sample sizes, based on preliminary results or an empirical approach we determined sufficient sample size. In assays for Figs. 3 and 5 requiring a long-term WD feeding, we used all mice born for the assays. Because the birth rates of mice of each genotype were different, sample sizes of these assays differed among genotypes.
Data Analysis and Statistics.
Individual data points are shown as dot plots, and graph bars represent the mean ± S.D. All statistical analyses were performed by using GraphPad Prism 8 (GraphPad Software, San Diego, CA). Different statistical tests were used for different experiments, as described in the figure legends. Briefly, we predetermined to statistically compare Ldlrmt/1l1KO mice, Ldlrmt/1L1Tg mice, and Ldlrmt/1L1Tg/1l1KO mice to Ldlrmt mice under conditions without ezetimibe administration by Dunnett’s test. In addition, we predetermined statistical comparison of ezetimibe-administered mice and nonadministered mice in each genotype by Student’s t test. Statistical significance was defined in terms of P values less than 0.05.
Results
Generated Mouse Models Showed Expected NPC1L1 Protein Expression Patterns in the Intestine and Liver.
First, we examined NPC1L1 protein expression in the liver and intestine in our generated mouse models (Fig. 1). Consistent with previous observations in wild-type (WT) mice (Altmann et al., 2004), Ldlrmt mice exhibited distinct expression of NPC1L1 in the intestine, whereas hepatic NPC1L1 was hardly detected. Meanwhile, both intestinal and hepatic expression of NPC1L1 protein was observed in Ldlrmt/1L1Tg mice, whereas neither intestinal nor hepatic NPC1L1 was detected in Ldlrmt/1l1KO mice. In addition, we confirmed the expression of NPC1L1 in the liver, but little in the intestine, in Ldlrmt/1L1Tg/1l1KO mice. Based on the tissue distribution of NPC1L1, we have described Ldlrmt, Ldlrmt/1L1Tg, Ldlrmt/1l1KO, and Ldlrmt/1L1Tg/1l1KO mice as I+L−, I+L+, I−L−, and I−L+ mice (where “I” is for intestine and “L” is for liver), respectively, in the following text.

NPC1L1 protein expression in NPC1L1 gene-modified Ldlrmt mice. Protein expression of NPC1L1 in the intestine and liver from 10- to 12-week-old male mice was analyzed by Western blot analysis. Na+/K+-ATPase α was used as loading control.
Generated Mouse Models Have Appropriate Activities of Intestinal Cholesterol Absorption and Biliary Cholesterol Excretion.
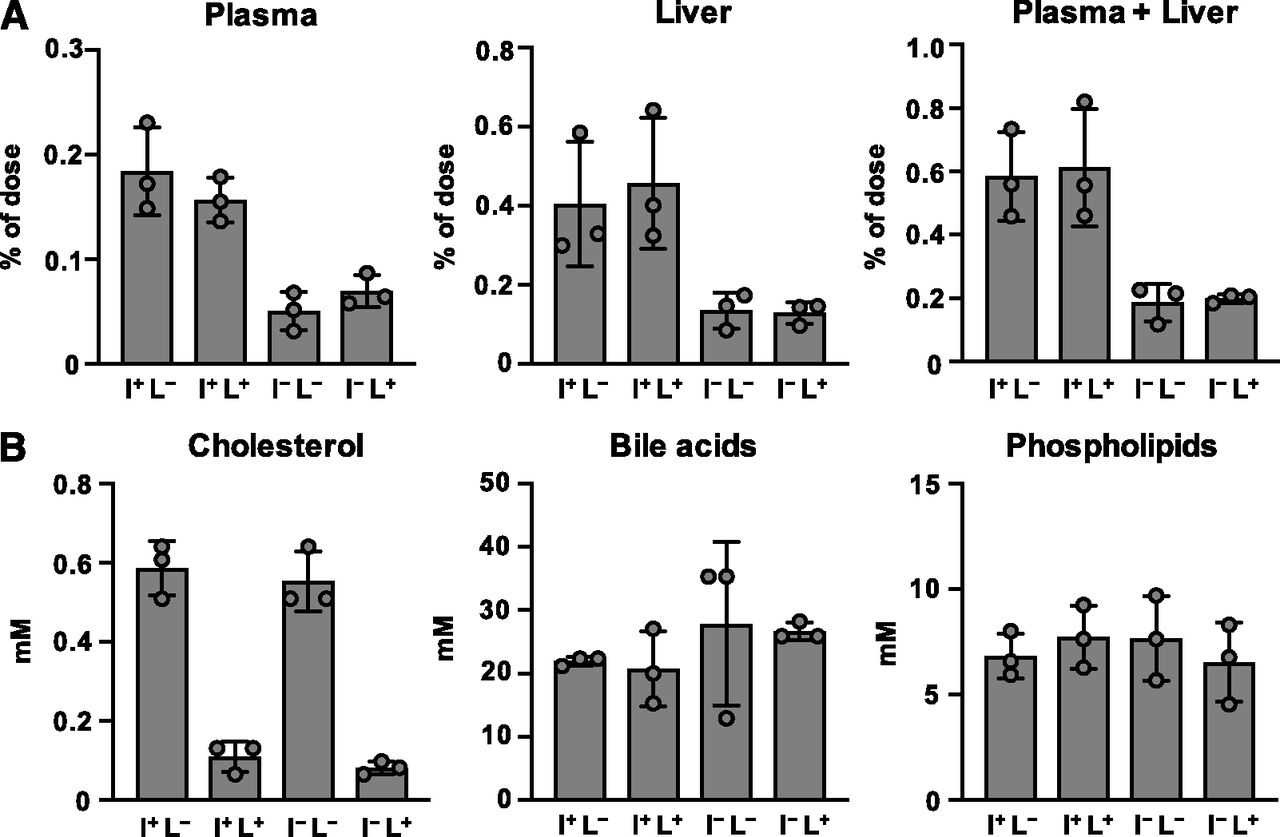
We next examined intestinal cholesterol absorption in mice of each genotype (Fig. 2A). Consistent with the expression level of NPC1L1 protein in the intestine, cholesterol absorption into the plasma and the liver after oral administration was lower in I−L− and I−L+ mice, compared with that in I+L− mice [WT mice for tissue distribution of NPC1L1 (WT1L1)]. Meanwhile, little difference was observed between I+L− (WT1L1) mice and I+L+ mice.

Characterization of NPC1L1 gene-modified Ldlrmt mice. (A) Cholesterol absorption was examined in 10- to 12-week-old male I+L−, I+L+, I−L−, and I−L+ mice. [3H]cholesterol concentrations in plasma, liver, and plasma plus liver were examined 2 hours after the administration of [3H]cholesterol-containing emulsion. (B) Biliary concentrations of cholesterol, bile acids, and phospholipids were examined in 10- to 12-week-old male I+L−, I+L+, I−L−, and I−L+ mice. Bar graphs represent the mean ± S.D. The dots on bar graphs represent each individual data point (n = 3: no statistical analysis was performed in Fig. 2 because the sample size was too small to justify statistical testing for multiple comparisons, although the observed differences among genotypes were sufficiently clear to validate the constructed mouse models).
We also elucidated biliary cholesterol excretion in the mice of each genotype (Fig. 2B). Although the biliary cholesterol concentration in I−L− mice was equivalent to that in I+L− (WT1L1) mice, I−L+ and I+L+ mice exhibited a lower concentration of biliary cholesterol than I+L− (WT1L1) mice. Next, we confirmed that the concentration of other biliary components, such as bile acids and phospholipids, was similar among all groups. These observations indicated that, consistent with previous reports (Temel et al., 2007), hepatic NPC1L1 selectively reabsorbs biliary cholesterol in the liver.
All of these data indicated that we successfully generated mouse models with different expression patterns of NPC1L1, and varied cholesterol uptake and reuptake activity in the intestine and liver.
Hepatic NPC1L1 Exacerbated WD-Induced Dyslipidemia.
Next, we analyzed plasma lipid levels and lipoprotein profiles in the mice of each genotype after WD feeding for 20 weeks, with or without ezetimibe. We observed that the body weights of all mice increased appropriately during the assay period, indicating that mice of each genotype grew normally under our experimental conditions (Supplemental Table 1). In addition, before WD feeding, we did not observe a significant difference in the plasma levels of cholesterol (Supplemental Fig. 1A) and triglycerides (Supplemental Fig. 1B) among the four genotypes. Then, we analyzed the effect of WD feeding on plasma lipid levels and found that, in I+L− (WT1L1) mice and I+L+ mice, plasma concentrations of cholesterol and triglyceride were dramatically increased and that these increases were effectively inhibited upon ezetimibe treatment (Fig. 3, A and B). Meanwhile, in intestinal NPC1L1-deficient mice (I−L− and I−L+ mice), an ezetimibe-sensitive increase in plasma lipids was hardly observed, and consequently, plasma lipid levels in these mice were significantly lower than those in mice expressing NPC1L1 in the intestine [I+L− (WT1L1) and I+L+ mice]. These results indicated that intestinal NPC1L1 plays a critical role in the diet-induced increase in plasma lipid levels. Moreover, we observed that I+L+ mice exhibited significantly higher levels of plasma lipids than I+L− (WT1L1) mice, whereas little difference in plasma lipid levels was observed between I−L− and I−L+ mice (Fig. 3, A and B). These results suggested that hepatic NPC1L1 also contributes to the increase in plasma lipids when intestinal NPC1L1 is functional.

Plasma lipid profiles in NPC1L1 gene–modified Ldlrmt mice after WD feeding. Cholesterol (A) and triglyceride (B) concentrations in plasma from male mice (initially 6–8 weeks old) fed with a WD for 20 weeks in the absence (−) or presence (+) of ezetimibe (Eze). Bar graphs represent the mean ± S.D. The dots on bar graphs represent each individual data point [n = 15, I+L− Eze (−) mice; n = 8, I+L− Eze (+) mice; n = 9, I+L+ Eze (−) mice; n = 4, I+L+ Eze (+) mice; n = 12, I−L− Eze (−) mice; n = 4, I−L− Eze (+) mice; n = 7, I−L+ Eze (−) mice; n = 4, I−L+ Eze (+) mice]. *P < 0.05; **P < 0.01, significantly different by Dunnett’s test comparing I+L+ Eze (−) mice, I−L− Eze (−) mice, and I−L+ Eze (−) mice with I+L− Eze (−) mice. †P < 0.05; ††P < 0.01, significantly different by Student’s t test comparing two groups [Eze (−) and Eze (+)] in each genotype. N.S., not significantly different between the groups. Cholesterol (C) and triglyceride (D) concentrations in lipoprotein fractions collected from male mice (initially, 6–8 weeks old) fed with a WD for 20 weeks in the absence (left panel) or presence (right panel) of ezetimibe. Representative lipoprotein profiles from at least two independent experiments are shown.
We also analyzed lipoprotein profiles in the mice of each genotype after WD feeding and found that the ezetimibe-sensitive increases in plasma cholesterol and triglyceride were primarily accounted for by ezetimibe-sensitive increases in VLDL and/or LDL (VLDL/LDL) cholesterol (Fig. 3C) and VLDL/LDL triglyceride (Fig. 3D), respectively. These findings indicated that both hepatic and intestinal NPC1L1 are somehow associated with increased levels of VLDL/LDL lipids in particular.
Since the liver is a major source of plasma lipids, we analyzed hepatic concentrations of cholesterol and triglyceride in the mice of each genotype after WD feeding for 20 weeks. An ezetimibe-sensitive WD-induced hepatic cholesterol accumulation was significantly increased in I+L+ mice compared with that in I+L− (WT1L1) mice, and such an accumulation was not observed in intestinal NPC1L1-deficient mice (I−L− and I−L+ mice) (Fig. 4A). A similar tendency was observed in triglyceride levels in the liver, although the difference between I+L− (WT1L1) and I+L+ mice was not statistically significant (Fig. 4B). We also analyzed hepatic mRNA expressions of genes involved in de novo cholesterol synthesis (HMG-CoA synthase and HMG-CoA reductase) and found that no significant difference was observed between I+L− (WT1L1) and I+L+ mice, whereas the expression levels of HMG-CoA reductase in I−L− and I−L+ mice were significantly higher than that in I+L− (WT1L1) mice (Supplemental Fig. 2). These results, together with the fact that hepatic NPC1L1 is involved in biliary cholesterol reabsorption (Fig. 2B), suggested that higher levels of plasma and hepatic cholesterol in I+L+ mice compared with I+L− (WT1L1) mice (Figs. 3A and 4A) were probably not due to an enhancement of de novo cholesterol synthesis in the liver, but rather to the reduced biliary cholesterol excretion.

Hepatic lipid concentrations in NPC1L1 gene–modified Ldlrmt mice after WD feeding. Cholesterol (A) and triglyceride (B) concentrations in the liver from male mice (initially, 6–8 weeks old) fed with a WD for 20 weeks in the absence (−) or presence (+) of ezetimibe (Eze). Bar graphs represent the mean ± S.D. The dots on bar graphs represent each individual data point (n = 4). **P < 0.01, significantly different by Dunnett’s test comparing I+L+ Eze (−) mice, I−L− Eze (−) mice, and I−L+ Eze (−) mice with I+L− Eze (−) mice. ††P < 0.01, significantly different by Student’s t test comparing two groups [Eze (−) and Eze (+)] in each genotype. N.S., not significantly different between the groups.
Hepatic NPC1L1 Exacerbated WD-Induced Atherosclerotic Plaque Formation.
Next, we analyzed progression of WD-induced atherosclerosis in the mice of each genotype (Fig. 5A). As a result, atherosclerotic plaques, which were stained by Oil Red O, were observed in I+L− (WT1L1) mice after WD feeding for 20 weeks. Notably, I+L+ mice developed atherosclerotic plaques more severely than I+L− (WT1L1) mice. In addition, we observed that mice that did not express intestinal NPC1L1 (I−L− mice and I−L+ mice) hardly developed atherosclerotic plaques after WD feeding. These results were in accordance with plasma lipid levels. Indeed, we observed significant positive correlations of atherosclerotic plaque area with plasma cholesterol levels (Fig. 5B) and triglyceride levels (Fig. 5C). In particular, plasma cholesterol levels exhibited a strong correlation with the plaque area. These observations indicated that, although intestinal NPC1L1 clearly contributes to atherosclerotic plaque formation as well as hyperlipidemia progression, hepatic NPC1L1 can also exacerbate diet-induced hyperlipidemia and atherosclerotic plaque formation/progression in concert with intestinal NPC1L1 (Fig. 6). Furthermore, we also found that ezetimibe administration inhibited plaque formation in not only I+L− (WT1L1) mice, but also in I+L+ mice (Fig. 5). These results, together with previous findings suggesting that ezetimibe can inhibit both intestinal and hepatic NPC1L1 in vivo (Temel et al., 2007), provide a strong rationale for using ezetimibe to prevent atherosclerosis in humans, who express NPC1L1 in both the intestine and liver.

Atherosclerotic lesions of the aorta in NPC1L1 gene–modified Ldlrmt mice after WD feeding. The excised aorta from the mice analyzed in Fig. 3 was opened longitudinally, followed by Oil Red O staining. (A) Representative pictures (top panels) and percentage of areas of intimal atherosclerotic plaque lesions of the aortic wall (bottom graph) are shown. Bar graphs represent the mean ± S.D. The dots on bar graphs represent each individual data point [n = 15, I+L− Eze (−) mice; n = 8, I+L− Eze (+) mice; n = 9, I+L+ Eze (−) mice; n = 4, I+L+ Eze (+) mice; n = 12, I−L− Eze (−) mice; n = 4, I−L− Eze (+) mice; n = 7, I−L+ Eze (−) mice; n = 4, I−L+ Eze (+) mice]. **P < 0.01, significantly different by Dunnett’s test comparing I+L+ Eze (−) mice, I−L− Eze (−) mice, and I−L+ Eze (−) mice with I+L− Eze (−) mice. ††P < 0.01, significantly different by Student’s t test comparing two groups [Eze (−) and Eze (+)] in each genotype. N.D., not detected. (B and C) Correlation of the percentage of areas of intimal atherosclerotic plaque lesions with plasma concentrations of cholesterol (B) and triglyceride (C). Correlation analysis was performed using Pearson’s method. R value represents Pearson’s correlation coefficient.

Schematic illustration of involvement of intestinal and hepatic NPC1L1 in the development/progression of dyslipidemia and atherosclerosis in Ldlrmt mice. Intestinal NPC1L1 is fundamentally involved in the development/progression of WD-induced dyslipidemia and atherosclerosis via intestinal absorption of dietary lipids, including cholesterol. In addition to intestinal NPC1L1, hepatic NPC1L1 can exacerbate dyslipidemia and atherosclerosis, probably via the reabsorption of biliary lipids when intestinal NPC1L1 is functional.
Discussion
In this study, we generated mouse models to analyze the effects of NPC1L1 in the intestine and liver on atherosclerosis development/progression (Figs. 1 and 2), and we demonstrated that, under a WD-fed condition, mice expressing NPC1L1 in both the intestine and liver (I+L+ mice) showed greater increases in serum VLDL/LDL lipids (Fig. 3) and hepatic lipids (Fig. 4) and more severe atherosclerotic plaque formation (Fig. 5) than mice expressing NPC1L1 predominantly in the intestine and to a lesser extent in the liver [I+L− (WT1L1) mice]. In addition, consistent with a previous study using apolipoprotein E–deficient mouse models (Davis et al., 2007), 1l1KO mice (I−L− mice) hardly exhibited WD-induced increases in serum lipids (Fig. 3) and atherosclerotic plaques (Fig. 5). Taken together, our observations suggest that hepatic NPC1L1 can exacerbate diet-induced dyslipidemia and atherosclerosis, in concert with intestinal NPC1L1 (Fig. 6). Considering that NPC1L1 is highly expressed in the liver, in addition to the intestine in humans (Altmann et al., 2004), I+L+ mice will be useful as an animal model for deeper understanding of the pathophysiological function (involvement) of not only intestinal, but also hepatic, NPC1L1 in atherosclerotic diseases.
WD-fed mice showed an increase in VLDL/LDL lipids in an intestinal NPC1L1-dependent manner, whereas high-density lipoprotein lipids were hardly affected by NPC1L1 expression (Fig. 3, C and D). This is consistent with a previous report demonstrating that intestinal NPC1L1 is a major determinant of plasma VLDL/LDL cholesterol levels in LDLR-deficient mice (Xie et al., 2014). In addition, we revealed that, besides intestinal NPC1L1, hepatic NPC1L1 also contributes to increased VLDL/LDL lipid levels (Fig. 3, C and D), indicating that NPC1L1-mediated reabsorption of biliary cholesterol is involved in the regulation of VLDL/LDL lipid levels. In accordance with all of these observations, a clinical study demonstrated that plasma LDL cholesterol levels were significantly lower in carriers of NPC1L1-inactivating mutations than those in noncarriers of such mutations, whereas high-density lipoprotein cholesterol levels were not significantly different between carriers and noncarriers (Stitziel et al., 2014). The detailed mechanisms involving how NPC1L1 controls the concentrations of VLDL/LDL lipids remains unknown. However, considering our observations that hepatic NPC1L1 increased the hepatic cholesterol level when intestinal NPC1L1 is functional (Fig. 4A), and a previous finding that cholesterol (ester) in the liver stimulates hepatic production and/or secretion of VLDL into the blood (Fungwe et al., 1992), in addition to the fact that VLDL is converted to LDL in the bloodstream, it is highly possible that NPC1L1 indirectly regulates blood VLDL/LDL levels by controlling hepatic cholesterol levels through dietary cholesterol absorption and biliary cholesterol reabsorption. Previous findings that hepatic overexpression of a heterodimer of ATP-binding cassette transporters G5 and G8, cholesterol exporters from hepatocytes to the bile (Berge et al., 2000; Yu et al., 2002), reduced blood VLDL/LDL levels and inhibited atherosclerosis progression in LDLR-deficient mice (Basso et al., 2007) also support the potential importance of hepatic cholesterol homeostasis regulated by biliary cholesterol excretion via the cholesterol transporter–mediated pathway.
We observed that ezetimibe treatment could improve dyslipidemia and effectively inhibited diet-induced atherosclerotic plaque formation in I+L+ mice and I+L− (WT1L1) mice (Figs. 3 and 5). Consistent with our results using mouse models, a number of clinical research studies demonstrated that ezetimibe therapy was effective in improving dyslipidemia in humans (Bays, 2002). However, it seems that the extent of the therapeutic (or preventive) effects of ezetimibe on atherosclerotic diseases are not as high in humans as those observed in I+L+ mice in this study. Indeed, several large-scale clinical trials demonstrated that the positive effects of ezetimibe on atherosclerotic diseases in humans were not observed (intima-media thickness was not improved by ezetimibe administration) (Kastelein et al., 2008; Taylor et al., 2009) or were very limited (only 2% reduction in the rate of cardiovascular events with ezetimibe therapy) (Cannon et al., 2015). The reason why such a difference exits has not yet been clarified, but the timing of starting ezetimibe treatment may be a key factor. In our study as well as many other studies with mouse models, ezetimibe treatment was initiated at the same timing as starting WD feeding, and, therefore, the tested mice had not experienced severe dyslipidemia. Meanwhile, most of the clinical studies examining the effect of ezetimibe were conducted with patients who had already received a diagnosis of dyslipidemia. These facts, together with results of meta-analyses showing that LDL cholesterol has a cumulative effect on the risk of cardiovascular diseases (Horton et al., 2009), suggest that the difference in timing of starting ezetimibe therapy may account for the difference in therapeutic effects of ezetimibe on atherosclerotic diseases. In support of this, it has been revealed that humans with dysfunctional mutations of the NPC1L1 gene exhibited significant decreases in the risk of cardiovascular diseases, compared with noncarriers of such mutations (Stitziel et al., 2014), indicating that lifelong reduction in dietary and biliary cholesterol absorption and reabsorption can also prevent cardiovascular diseases in humans. From all of these results, starting ezetimibe therapy and/or improvement of dietary habits at the earliest possible stage of dyslipidemia could be important in preventing the development/progression of atherosclerotic diseases.
In conclusion, we revealed that hepatic NPC1L1 exacerbates diet-induced dyslipidemia and atherosclerosis when intestinal NPC1L1 is functional (Fig. 6). To our knowledge, this is the first report demonstrating that hepatic NPC1L1 affects diet-induced atherosclerosis development/progression. Unfortunately, the detailed mechanisms involved have not been clarified. We recently revealed that NPC1L1 has uptake activity not only for cholesterol, but also for vitamins E and K1 (Narushima et al., 2008; Takada and Suzuki, 2010; Takada et al., 2015; Yamanashi et al., 2017). As the chemical structures of cholesterol, vitamin E, and vitamin K1 are largely different (Yamanashi et al., 2017), it is possible that NPC1L1 might exhibit unexpectedly broad substrate specificity and might be involved in the absorption and reabsorption of various dietary and/or biliary lipids, including unknown harmful lipids causing and/or promoting atherosclerosis. Considering that vitamins E and K1 demonstrate positive effects on inhibiting atherosclerosis (vitamin E, antioxidative effect; vitamin K1, anticalcification effect), the balance of harmful lipids and beneficial lipids (e.g., vitamin E and vitamin K1) controlled by NPC1L1 should be taken into consideration for understanding atherosclerosis development/progression. From this point of view, comprehensive analyses of plasma, hepatic, and biliary lipids in our generated mouse models will be useful for deeper understanding of the NPC1L1-mediated exacerbation of atherosclerosis.
Acknowledgments
We thank Dr. Ayaori, Dr. Kondoh, and Dr. Ikewaki at National Defense Medical College (Division of Cardiovascular Medicine, Department of Internal Medicine, Saitama, Japan) for instructions on the technique of atherosclerotic lesion analysis.
Authorship Contributions
Participated in research design: Yamamoto, Yamanashi, Takada, Suzuki.
Conducted experiments: Yamamoto, Yamanashi, Mu, Tanaka, Komine.
Performed data analysis: Yamamoto, Yamanashi, Takada.
Wrote or contributed to the writing of the manuscript: Yamanashi, Takada, Suzuki.
Footnotes
- Received January 17, 2019.
- Accepted May 2, 2019.
↵1 H.Y., Y.Y., and T.T. contributed equally to this work.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grant Numbers [16H06219] (to Y.Y.) and [16K15155] (to T.T.) as well as a grant-in-aid for Scientific Research on Innovative Areas “HD-physiology” [22136015] (to H.S.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. Y.Y. has received research grants from Takeda Science Foundation and Mochida Memorial Foundation for Medical and Pharmaceutical Research. T.T. has received a Lotte Research Promotion grant from the Lotte Foundation. H.Y., Y.Y., T.T., and H.S. have a patent pending related to the work reported in this article.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- HMG
- 3-hydroxy-3-methylglutaryl
- LDL
- low-density lipoprotein
- LDLR
- low-density lipoprotein receptor
- NPC1L1
- Niemann-Pick C1-Like 1
- PCR
- polymerase chain reaction
- VLDL
- very low-density lipoprotein
- WD
- Western diet
- WT
- wild-type
- Copyright © 2019 The Author(s).
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}