


Abstract
Tamoxifen is used to prevent and treat estrogen receptor–positive (ER+) breast cancer (BC); however, its chronic use can increase uterine cancer risk and induce tamoxifen resistance. Novel melatonin-tamoxifen drug conjugates may be promising to treat BC and may help offset the adverse effects of tamoxifen usage alone due to the presence of melatonin. We synthesized and screened five drug conjugates (C2, C4, C5, C9, and C15 linked) for their effects on BC cell (MCF-7, tamoxifen-resistant MCF-7, mouse mammary carcinoma, MDA-MB-231, and BT-549) viability, migration, and binding affinity to melatonin receptor 1 (MT1R) and estrogen receptor 1 (ESR1). C4 and C5 demonstrated the most favorable pharmacological characteristics with respect to binding profiles (affinity for ESR1 and MT1R) and their potency/efficacy to inhibit BC cell viability and migration in four phenotypically diverse invasive ductal BC cell lines. C4 and C5 were further assessed for their actions against tamoxifen-resistant MCF-7 cells and a patient-derived xenograft triple-negative BC cell line (TU-BcX-4IC) and for their mechanisms of action using selective mitogen-activated protein kinase kinase MEK1/2, MEK5, and phosphoinositide 3-kinase (PI3K) inhibitors. C4 and C5 inhibited tamoxifen-resistant MCF-7 cells with equal potency (IC50 = 4–8 μM) and efficacy (∼90% inhibition of viability and migration) but demonstrated increased potency (IC50 = 80–211 μM) and efficacy (∼140% inhibition) to inhibit migration versus cell viability (IC50 = 181–304 mM; efficacy ∼80% inhibition) in TU-BcX-4IC cells. Unique pharmacokinetic profiles were observed, with C4 having greater bioavailability than C5. Further assessment of C4 and C5 demonstrates that they create novel pharmacophores within each BC cell that is context specific and involves MEK1/2/pERK1/2, MEK5/pERK5, PI3K, and nuclear factor κB. These melatonin-tamoxifen drug conjugates show promise as novel anticancer drugs and further preclinical and clinical evaluation is warranted.
Introduction
Tamoxifen, a first-generation selective estrogen receptor (ER) modulator, is one of the main treatment choices for ER-positive (ER+) breast cancer (BC) (Ring and Dowsett, 2004). The U.S. Food and Drug Administration approved tamoxifen for treatment of ER+ BC in pre- and postmenopausal women (Jaiyesimi et al., 1995; Chang, 2012). Several studies demonstrate the efficacy of tamoxifen in reducing the risk of invasive and noninvasive BC (Wapnir et al., 2011). Despite having clinical benefit for treating BC, long-term use of tamoxifen can result in relapse (Ring and Dowsett, 2004) and increase uterine cancer risk (van Leeuwen et al., 1994; Assikis and Jordan, 1997; Bernstein et al., 1999; Jones et al., 2012).
Melatonin, a chronobiotic molecule released from the pineal gland in response to darkness, demonstrates protection against BC where depletion of melatonin through shift work or light exposure at night increases BC risk and tumor growth (Davis et al., 2001; Schernhammer et al., 2001; Blask et al., 2005; Grant et al., 2009). Supplementation of endogenous nocturnal melatonin peaks with exogenous melatonin and/or high nocturnal levels of melatonin are associated with BC protection (Blask et al., 2005; Grant et al., 2009; Davis et al., 2011; Dauchy et al., 2014; Hill et al., 2015). In addition to being protective on its own, melatonin used as an adjuvant to existing BC therapies has been proposed to not only enhance the anticancer actions of BC therapies, including tamoxifen-resistant (TamR) BC, but also to lessen adverse effects and toxicities associated with these therapies (Lissoni et al., 1996; Witt-Enderby et al., 2006; Grant et al., 2009; Davis et al., 2011; Sabzichi et al., 2016). Support for this comes from the following: melatonin can increase the efficacy of tamoxifen in MCF-7 cells (Sabzichi et al., 2016) and in patients with metastatic BC unresponsive to tamoxifen alone (Lissoni et al., 1995); melatonin reverses tamoxifen resistance due to light exposure at night and depletion of nocturnal melatonin levels (Blask et al., 2005; Grant et al., 2009; Dauchy et al., 2014); and the addition of melatonin to other antiestrogenic therapies (e.g., tamoxifen and anastrozole) has provided clinical benefit in improving both survival and quality of life in patients with metastatic BC (Lissoni et al., 1995, 1996, 2009). The soporific effects of melatonin coupled with its positive effects on well-being and menopausal symptoms demonstrated in peri- and postmenopausal women (Kotlarczyk et al., 2012; Maria et al., 2018) may offset other negative effects of tamoxifen usage such as hot flashes, vaginal dryness, and sleep disturbances (Lorizio et al., 2012).
The mechanisms underlying melatonin-mediated inhibition of TamR BC may involve modification and/or upregulation of melatonin receptor 1 (MT1R) and ER binding sites enhancing the efficacy of melatonin and tamoxifen to stimulate or antagonize, respectively, MT1Rs and ERs. Support for this comes from findings demonstrating that increases in MT1R expression inhibit BC cell proliferation (Hill et al., 2009) and gain in ER expression enhances the inhibitory actions of tamoxifen at ESR2 (ER-β) (Hopp et al., 2004). Comodulation of MT1Rs and ERs after exposure to tamoxifen (i.e., 4-OH-tamoxifen) has been demonstrated in breast (i.e., MCF-7 cells) and ER+ ovarian cancer cells (Treeck et al., 2006). Melatonin may also prevent tamoxifen resistance through some unique interaction between ERs and MT1Rs, enhancing the anticancer actions of tamoxifen as shown (Dauchy et al., 2014; Sabzichi et al., 2016).
Melatonin-tamoxifen drug conjugates were recently reported as promising drugs for the prevention and treatment of BC without tamoxifen’s untoward side effects on the uterus (Witt-Enderby et al., 2014; Hasan et al., 2018). The drug conjugate, C5 (6-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]hexanamide), with a spacer of five CH2 groups was reported in the patent (Witt-Enderby et al., 2014). To assess the influence of spacer length and based on the commercial availability of the alkylating agents Br(CH2)nCO2Me, the following drug conjugates were additionally synthesized: C2 (3-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]propionamide; n = 2, shorter spacer), C4 (5-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]pentanamide; n = 4, spacer of a similar length), and C9 (10-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]decanamide) and C15 (16-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]hexadecanamide; n = 9 and 15, respectively; longer spacers). Drug conjugates are emerging as novel anticancer agents, as reviewed previously (Hasan et al., 2018). Benefits of using drug conjugates lie in their ability to modulate diverse targets (receptors and/or intracellular signaling proteins), which has been shown to increase their efficacy as anticancer agents and may also prevent drug resistance and toxicity (Hasan et al., 2018). In this study, we examined a series of melatonin and tamoxifen drug conjugates linked by carbon chains of varying lengths—C2, C4, C5, C9, and C15—against BC cells that express ERs (e.g., MCF-7) and TamR cells, in human epidermal growth factor receptor 2 (HER2)–expressing BC such as mouse mammary carcinoma (MMC) cells, and in triple-negative breast cancer (TNBC) cells (e.g., MDA-MB-231 and BT-549). The results from this study identified two novel BC drug candidates, C4 and C5, demonstrating unique pharmacokinetics, pharmacology, and potential drug targets that include pERK1/2, pERK5, pAKT, RUNX2, nuclear factor κB (NF-κB), and β1-INTEGRIN.
Utilizing patient-derived xenografts (PDXs) in drug discovery research incorporates a translational approach to evaluate drug effects in the laboratory setting (Whittle et al., 2015; Byrne et al., 2017). Although established cell line–based research provides important insight into drug effects on cancer cells and basic mechanisms of action, treating patient tumors ex vivo and primary cells derived from patient tumors facilitates translation of these findings to clinical observation. In our study, we validated our findings using PDX models that represent clinically aggressive TNBC tumors (TU-BcX-4IC and TU-BcX-4QAN).
Materials and Methods
Synthesis of Melatonin-Tamoxifen Drug Conjugates
The synthetic approach toward the drug conjugates C2, C4, C5, C9, and C15 is shown in Supplemental Fig. 1. Briefly, N-desmethyltamoxifen (Gryder et al., 2013) was N-alkylated with Br(CH2)nCO2Me (n = 2, 4, 5, 9, and 15). The products were subjected to ester hydrolysis, and the resulting acids were coupled with N-desacetylmelatonin (Chatterjie et al., 2001) to give the final drug conjugates. Detailed experimental procedures for the synthesis of C4, including full analytical characterization by 1H NMR, 13C NMR, and mass spectrometry (MS), and the NMR spectra of all five drug conjugates are shown in Supplemental Figs. 1–5.
Competition Binding for Melatonin and ERs
Binding affinities of each of the drug conjugates (C2, C4, C5, C9, and C15) or melatonin, tamoxifen, or 4-OH-tamoxifen alone were measured using 2-[125I]-iodomelatonin, [125I]-estradiol, and [3H]-estradiol, respectively, as described previously with modification (Witt-Enderby and Dubocovich, 1996; Kirker et al., 2013; Suofu et al., 2017). In brief, experiments were conducted on the mouse uterus (Witt-Enderby et al., 2014) and in whole cell lysates prepared from human MT1 Chinese hamster ovary cells or MCF-7 BC cells grown to confluence on 10-cm plates. Cell lysates were prepared by first washing cells with 5 ml PBS and then lifting them into the buffer (10 mM KPO4 and 1 mM EDTA, pH 7.4). Cells were then pelleted by centrifugation (277g for 5 minutes) and then resuspended in Tris-HCl buffer (50 mM, pH 7.4). Next, cells were added to tubes containing 115 pM 2-[125I]-iodomelatonin (2200 Ci/mmol; PerkinElmer, Waltham, MA) in the presence of melatonin (1 pM to 100 μM) or drug conjugate (1 pM to 100 μM) or the absence of melatonin or drug conjugate (total binding). For 4-OH-tamoxifen or drug conjugate binding in MCF-7 cells, 2 nM [3H]-estradiol was used in the presence of 4-OH-tamoxifen (1 pM to 100 μM) or drug conjugate (1 pM to 100 μM). The incubation volume was 260 µl. Next, cells were incubated for 1 hour at room temperature and then terminated by the addition of ice-cold 50 mM Tris-HCl solution and rapid filtration over glass-fiber filters (0.22 μm; Schleicher & Schuell, Keene, NH) presoaked in 0.5% polyethyleneimine solution (v/v) (Sigma-Aldrich, St. Louis, MO). Each filter was washed twice with 5 ml cold buffer. Radioactivity of [125I]-iodomelatonin and [3H]-estradiol was determined in a gamma counter and a scintillation counter, respectively.
Total Binding Assay
G protein–coupled estrogen receptor (GPR30) total binding to C4 and C5 in MCF-7 cells was assessed using [3H]-estradiol as described (Kirker et al., 2013 with modification) on MCF-7 cell lysates. Briefly, 200 μl cell lysate was added to tubes containing 3 nM [3H]-estradiol (PerkinElmer) in the absence (total binding) or presence of the following: 10 μM 4-OH-tamoxifen, 1 μM C4, 1 μM C5, and/or 1 μM G1 GPR30 agonist (Cayman Chemical Company, Ann Arbor, MI). The whole incubation volume was 260 µl. Binding data, initially expressed as moles per milligram protein, were repeated three times and subjected to one-way ANOVA.
Cell Culture
MCF-7, MMC, MDA-MB-231, and BT-549 cells were obtained from American Type Culture Collection (Manassas, VA) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. BT-549 cells were cultured in the same condition (10% FBS and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2) using RPMI-1640 medium.
Development and Characterization of TamR MCF-7 Cells
The characterization of TamR MCF-7 cells is described in Supplemental Fig. 6. In brief, MCF-7 cells, obtained from American Type Culture Collection, were cultured at 37°C in a humidified atmosphere with 5% CO2 for 6 months in DMEM (phenol red-free)/F-12 containing 10% charcoal-stripped FBS and 1% penicillin/streptomycin and 100 nM 4-OH-tamoxifen; 6 months was the amount of time required for MCF-7 cells to become resistant to tamoxifen. For growth assays, wild-type (WT) and TamR MCF-7 cells were counted using the trypan blue exclusion assay after exposure to 100 nM 4-OH-tamoxifen for 1–7 days. For immunocytochemistry images, MCF-7 and TamR cells were treated with either vehicle or 100 nM 4-hydroxytamoxifen for 24 hours. After treatment, cells were fixed in 4% paraformaldehyde for 15 minutes. Cells were then permeabilized with 0.3% Triton-X followed by the addition of rabbit Ki-67 (1:1000, catalog no. 9129S; Cell Signaling Technology, Danvers, MA) and mouse ∝-tubulin (1:1000, catalog no. 3873S; Cell Signaling Technology) primary antibodies. Goat anti-mouse Alexa Fluor 488 nm (1:1000, catalog no. A-11001; Life Technologies, Carlsbad, CA) and goat anti-rabbit Alexa Fluor 555 nm (1:1000, catalog no. A-21429; Life Technologies) were used as secondary antibodies. A Hoechst (Fisher) stain was used to visualize the nucleus. Images were obtained with the EVOS fluorescent inverted microscope (Life Technologies) under 20× objective (scale bar = 200 μm). Basal expression of pERK5, pERK1/2, ER-α, NF-κB, and pAKT in TamR MCF-7 and MCF-7 cells was measured by Western blot. Bands were quantified using Image Studio Lite software (LI-COR Biosciences, Lincoln, NE) and normalized against β-actin (see the “Western Blot” section for further details).
Ex Vivo Treatment of PDX Tumors
The TU-BcX-4QAN PDX tumor was established from a mastectomy specimen of an African American woman and represents a drug-resistant TNBC tumor. The PDX tumor was propagated and maintained in SCID/Beige immunodeficient mice (CB17.cg-PrkdcscidLystbg/Crl) obtained from Charles River (Wilmington, MA). After two serial transplantation passages in mice, the tumor was removed and small tumor pieces were dissected (∼3 × 3 mm2). The tumor pieces were kept intact and placed in individual wells of a 24-well plate. Drug solutions of DMSO/PBS vehicle control, melatonin (10 µM), tamoxifen (10 µM), melatonin plus tamoxifen (10 µM of each), C4 (10 µM), and C5 (10 µM) were made and added to the intact tumor pieces (1 ml/well) in triplicate. After 24 hours of incubation at 37°C in 5% CO2, tumor pieces were removed and placed in 1.5-ml centrifuge tubes and stored at −20°C.
In Vitro Treatment of PDX-Derived Cells for Western Blot
TU-BcX-4IC cells were derived from a mastectomy specimen of a Caucasian woman. The TU-BcX-4IC tumor was a metaplastic breast carcinoma that had a TNBC PAM50 subtype. The cells were maintained in DMEM supplemented with 10% FBS, insulin, nonessential amino acids, minimal essential amino acids, antibiotics/antimycotics, and sodium pyruvate at 37°C in 5% CO2. TU-BcX-4IC cells were plated in T-25 flasks and exposed to serum-free media for 24 hours before treatment. DMSO/PBS vehicle control, melatonin (10 µM), tamoxifen (10 µM), melatonin plus tamoxifen (10 µM of each), C4 hybrid (10 µM), and C5 hybrid (10 µM) were added to the cells in triplicate. After 36 hours of incubation at 37°C in 5% CO2, images were obtained using brightfield microscopy (40× magnification) and cells were harvested. Adherent cells were harvested from the DMSO and melatonin treatments. Detached cells were harvested in the media from the tamoxifen, melatonin plus tamoxifen, C4, and C5 treatments.
Cell Viability
The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay method was used to determine cellular viability after exposure to each of the drug conjugates (C2, C4, C5, C9, and C15) per the manufacturer’s instructions. Subconfluent monolayer cell lines grown on 10-cm culture plates were trypsinized and plated equally into each well of a 48-well plate. The next day, cells were exposed to concentrations (1 pM, 1 nM, and 1, 10, and 100 μM) of melatonin, each of the drug conjugates, or the controls tamoxifen (Millipore Sigma, Burlington, MA) or 4-OH-tamoxifen (Millipore Sigma) alone or in combination with the inhibitors for 24 hours. For the inhibitor studies assessing the involvement of mitogen-activated protein kinase kinase MEK1/2, MEK5, or PI3K, PD98059 (Santa Cruz Biotechnology, Dallas, TX), Bix02189 (Santa Cruz Biotechnology), and pictilisib (PI3K inhibitor; SellerChem, Houston, TX) were all added at a final concentration of 10 μM. After 24 hours, 25 μl MTT (dissolved in sterile water; 5 mg/ml; Sigma-Aldrich) was added to each well (0.5 mg/ml final concentration), and the plates were placed in the incubator for 3 hours (5% CO2 and 37°C). Next, the plates were centrifuged at 50g (37°C for 5 minutes). The entire medium was aspirated, and 250 µl DMSO (Fisher Scientific, Pittsburgh, PA) was added to each well to stop the reaction. The plate was wrapped in aluminum foil and incubated at room temperature for 15 minutes to dissolve the MTT-formazan crystals. Absorbance was measured at 570 nm (VICTOR3 1420 multilabel counter; PerkinElmer).
Cell Migration
Wound/scratch assays were performed in 24-well cell plates to determine the invasiveness of the cells after exposure to each of the drug conjugates (C2, C4, C5, C9, and C15). Twenty-four hours before the assay, cells were plated and allowed to settle. Next, a border was created using a 200-μl pipette tip that was dragged across the bottom of each well to make a visible and uniform space between the cells. The floating cells were aspirated, and the cells were refed fresh media containing the various drug treatments described for the MTT assays above. The border width was measured at baseline (time 0 hours) and then 24 hours after exposure to the treatments (time 24 hours) added alone or in combination with the inhibitors (where indicated) using an EVOS digital inverted fluorescence microscope. Any width changes were expressed as percentages. For the inhibitor studies assessing the involvement of MEK1/2, MEK5, or PI3K, PD98059, Bix02189, and pictilisib were all added at a final concentration of 10 μM. Border widths were calculated as (24 − 0 hours)/0 hours × 100, where a more positive number indicates inhibition of migration and a more negative number indicates stimulation of migration. To calculate the maximum inhibition of TamR MCF-7 cell migration, the percent change in border width was normalized against the minimum inhibition of migration, whereas the minimum inhibition of migration was considered as 0% and no border change was considered as 100%.
Western Blot
To identify potential intracellular targets, C4- or C5-mediated effects on pERK1/2, pERK5, p-AKT, NF-κB, RUNX2, and β1-INTEGRIN proteins were analyzed by Western blot analysis. BC cells were treated with each drug conjugate (1 nM and 10 μM) in the absence or presence of 10 μM PD98059, BIX02189, or pictilisib for 0 minutes (baseline) or 15 minutes, after which the cells were scraped into Laemmli sample buffer containing β-mercaptoethanol, heated for 5 minutes at 95°C, cooled, and then stored at −20°C until use. Western blot analysis was conducted using the Odyssey Western Blotting Kit IV RD (catalog no. 926-31084; LI-COR Biosciences). Equal amounts of each sample (20 μl) and 5 μl molecular weight marker (Precision Plus Protein, catalog no. 161-0373; Bio-Rad, Hercules, CA) were loaded onto 10% gels. Proteins, separated by SDS-PAGE, were transferred to nitrocellulose membranes. Membranes were incubated in blocking buffer (LI-COR Biosciences) for 30 minutes and then incubated with the following respective primary antibodies along with mouse anti–β-actin (catalog no. 926-42212; LI-COR Biosciences) overnight at 4°C: rabbit anti-pERK1/2 and anti-pERK5 (T218/Y220, 1:1000 dilution, catalog no. 3371; Cell Signaling Technology), rabbit anti–phospho-AKT (1:750 dilution, catalog no. 9271; Cell Signaling Technology), rabbit anti–NF-κB (p52, 1:1000 dilution, catalog no. sc-298; Santa Cruz Biotechnology), rabbit anti-Runx2 (1:1000 dilution, catalog no. sc-10758; Santa Cruz Biotechnology), rat ER-α (1:750 dilution, catalog no. sc-53493; Santa Cruz Biotechnology), and rabbit anti–β1-integrin (M-106, 1:1000 dilution, catalog no. sc-8978; Santa Cruz Biotechnology). Next, the blots were washed with PBS containing Tween 20 and then incubated with the following secondary antibodies for 30 minutes at room temperature: goat anti-rabbit (IRDye 800CW, 1:20,000 dilution, catalog no. 925-32211; LI-COR Biosciences) and goat anti-mouse (IRDye 680RD, 1:20,000 dilution, catalog no. 925-68070; LI-COR Biosciences). For the ER-α primary antibody, goat anti-rat (IRDye 800CW, 1:20,000 dilution, catalog no. 925-32219; LI-COR Biosciences) was used for 30 minutes at room temperature. Bands were quantified using Image Studio Lite software (LI-COR Biosciences) and normalized against β-actin to control for differences in protein loading between treatment groups.
Microsomal Incubation Method
Human liver microsomes (Ultrapool HLM 150, catalog no. 452117; Corning, Woburn, MA) and mouse liver microsomes (pooled female MLMs, catalog no. 452702; Corning) were used to assess susceptibility to hepatic metabolism of C4 and C5. Specifically, microsomes were incubated with 5 μM of either C4, C5, or tamoxifen in the presence of an NADPH regenerating system (0.5 mM NADPH, 1 U/ml glucose-6-phosphate dehydrogenase, and 10 mM glucose 6-phosphate) at 37°C for 0, 5, and 10 minutes in mouse microsomes and for 0, 10, and 20 minutes in human microsomes. The longer incubation time was required for human microsomes compared with mouse microsomes because of slow metabolism. Reactions were stopped and precipitated using 50% acetonitrile. Reaction vials were centrifuged at 9500g (4°C for 15 minutes). The rate of drug loss was measured by high-performance liquid chromatography (HPLC)-UV detection. After optimization of the HPLC conditions [column prewash 2 minutes with 70% solvent A (10% methanol in phosphate buffer) and 30% solvent B (100% methanol) for 2 minutes; sample runs = 25 minutes at a gradient flow rate of 0.4 ml/min, where solvent B was changed from 30% to 90%], retention times for C4 (20.46 minutes) and C5 (20.93 minutes) were similar to that of tamoxifen (20.46 minutes). Peak absorbance of each sample was normalized against the internal standard, celecoxib. Celecoxib was used as an internal standard for this study because it gave a retention time (11.51 minutes) distinguishable from the retention times for tamoxifen, C4, and C5 melatonin-tamoxifen drug conjugates.
Pharmacokinetic Analysis of C4 and C5
All original studies using animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health and were approved by the Duquesne University Animal Care and Use Committee or local equivalent (IACUC approval no. 1607-07M1). C57BL/6J female mice (2 months) were used to assess the pharmacokinetic properties of C4 and C5. The drugs were initially dissolved in ethanol [5% (v/v)] and then added to hydroxypropyl methylcellulose (5 mg/ml in ddH20) to obtain a final concentration of 0.5% (w/v). C4 and C5 were each administered separately by two routes of administration—subcutaneous and oral. The outcomes were compared with the subcutaneous administration of tamoxifen, which served as the control group. One group of mice (n = 8) received a subcutaneous dose of 1 mg/kg body weight, whereas another group received an oral dose of 10 mg/kg body weight. Different doses for the two routes were given to achieve similar exposures for the two routes based on experience with tamoxifen (Reid et al., 2014). For both routes of administration, blood was collected at similar time points (0, 30, 60, 120, 180, 360, 480, and 1440 minutes) except that one earlier time of 15 minutes was also taken for the subcutaneous route. One C56BL/6J female mouse aged 2 months was used for each time point. Blood was collected in a K2-EDTA–coated tube by decapitation after CO2 euthanasia. Blood was then centrifuged at 1166g (4°C for 5 minutes) to separate the plasma. The plasma was then stored at −80°C until liquid chromatography (LC)–MS was performed.
LC–Tandem MS Method for Quantification of Tamoxifen, C4, and C5 in Mouse Plasma
Deuterated tamoxifen (TAM-d5) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). Mouse plasma in Li- Heparin was purchased from Lampire Biologic Laboratories. Acetonitrile (HPLC grade), isopropanol, hexane, and formic acid (MS grade) were purchased from Fisher Chemicals. Individual stock solutions (20 μg/ml) of tamoxifen, C4, and C5 were prepared in methanol and stored at −20°C until use. The standards for the calibration curve were prepared by performing serial dilution of working stock solutions (64 ng/ml) of tamoxifen, C4, and C5 in methanol and then dividing them into 125-μl aliquots. TAM-d5 was used as the internal standard. Working stock solutions (50 ng/ml) of TAM-d5 were prepared in methanol, divided into 20 μl aliquots, and stored at −20°C until use.
Calibration standards (0.25, 0.5, 1, 2, 4, 8, 16, and 32 ng/ml) of tamoxifen, C4, or C5 were prepared in 115 μl mouse plasma and then spiked with TAM-d5 (2 ng/ml final concentration) to quantify all of the compounds during the extraction process. Quality control (QC) samples of 0.1 ng/ml (the lower limit of quantification, LLQ), 2 ng/ml (QC low), and 32 ng/ml (QC high) were run in parallel and in triplicate for each experimental run of tamoxifen, C4, and C5. Before the extraction process, 10 μl of 1 M NaOH was added to 200 μl of each sample followed by vortexing for 30 seconds. Next, 1000 μl of extraction solvent [hexane/isopropanol, 95:5 (v/v)] was added to each tube and then placed on a rotary shaker for 5 minutes. Samples were then centrifuged for 5 minutes at 9500g to separate the aqueous phase (bottom layer) from the organic phase (top layer). Next, 900 μl supernatant was transferred to a clean test tube. The remaining aqueous layer in the original tube was subjected to the same extraction process by adding another 1000 μl extraction solvent. The total transferred supernatant (organic phase) was air dried under N2 flux. The dry residue was dissolved in 200 μl methanol and subjected to LC with MS detection.
HPLC followed by tandem MS was conducted using a system model 1200 and triple quadrupole mass spectrometer (model 6460) with a jet stream electrospray ion source (Agilent Technologies, Santa Clara, CA). Data acquisition and chromatographic peak integration were performed using Agilent MassHunter software (versions B02.01 and B02.00). All analyses were performed in positive ion mode and chromatograms were recorded in multiple reaction monitoring scan mode. MS collision parameters for tamoxifen, TAM-d5, C4, and C5 are shown in Supplemental Table 1, whereas MS acquisition and source parameters for the instrument are summarized in Supplemental Table 2.
Chromatographic separation was achieved with a Hypersil GOLD reverse phase C18 column (50 × 4.6 mm, 3 μm packing; Thermo Scientific, Waltham, MA) thermostated at a temperature equal to 25°C. Deionized water and acetonitrile, both acidified with 0.1% formic acid, were used as mobile phase A and B, respectively, at the flow rate of 0.5 ml/min. A linear gradient separation was used where mobile phase B was initially set at 20% for 1 minute, then increased to 80% over 5 minutes, and then further increased to 95% over 0.1 minutes where it remained for 1.9 minutes. Next, mobile phase B was decreased to initial conditions over 0.1 minutes, and the system was re-equilibrated for 3.9 minutes before the following injection. The total run time was 12 minutes with an injection volume of 40 μl. The injection needle was washed in between analyses using a wash solution of acetonitrile containing 0.1% formic acid (v/v) for 20 seconds. The needle draw and ejection speed was set to 100 μl/min.
Calibration samples and QC samples (described above and performed in triplicate) were used for the determination of linearity, accuracy, and precision. Calibration curves were plotted by correlating the peak area ratio of each compound (relative to TAM-d5 as the internal standard) as a function of the concentration of spiked standard solutions with a weight factor of 1/y2 or 1/relative response2. The concentration range (0.25–32 ng/ml with an LLQ of 0.1 ng/ml) for the calibration standards was chosen based on the level of concentration expected in the unknown samples. The concentration of the QC samples was set to 0.1 ng/ml (LLQ), 2 ng/ml (QC low), and 32 ng/ml (QC high) for tamoxifen, C4, and C5 based on the range of calibration concentrations for each compound.
Middle concentrations of the calibration curve were used to determine matrix effects and extraction recovery. The matrix effect was determined by comparing the peak area in postextracted spiked samples versus the peak area in a standard methanol solution. At least six independent plasma samples with different concentrations were tested for the matrix effect for each compound. Extraction recovery was calculated by comparing the peak area of each extracted sample (spiked standard in the blank matrix) with the postextracted spiked sample at the same concentration; this represented 100% recovery. Recovery was calculated for at least four independent plasma samples with different concentrations for each compound. Matrix effects and extraction recoveries for tamoxifen, C4, and C5 are shown in Supplemental Table 3.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software (version 6; GraphPad Software, La Jolla, CA) and determined in advance. Data represent the mean ± S.D. unless mentioned otherwise. Data points that were determined to be outliers by the Grubbs test were excluded. The mean IC50 values of cell viability and cell migration were obtained by nonlinear regression analysis [log (inhibitor) versus response (three parameters)] and fit by least sum of squares. For the saturation binding analyses, Bmax values comparing WT MCF-7 cells to TamR MCF-7 cells used the unpaired t test. For all biphasic curves, two sites (Fit Ki nonlinear regression model in GraphPad Prism) were used. The total cell number obtained via the MTT analysis comparing WT versus TamR MCF-7 cells was analyzed by two-way ANOVA followed by the Bonferroni post hoc test. For other statistical comparisons, one-way ANOVA followed by the Newman–Keuls multiple comparisons test was performed. Mean differences between treatment groups were considered significant at P < 0.05.
Results
Effects of C2, C4, C5, C9, and C15 Conjugates on MCF-7 (ER+/PR+) Cells
Viability.
Cells were exposed to varying concentrations of C2, C4, C5, C9, and C15 and their effect on cell viability was assessed. As controls, MCF-7 cells were exposed to vehicle, melatonin, tamoxifen, 4-OH-tamoxifen, and combinations of melatonin plus tamoxifen or 4-OH-tamoxifen. With respect to viability, C4 trended toward being the most potent (IC50 = 4 nM) versus C2, C5, C9, and C15 (IC50 = 69 mM, 440 mM, 107 mM, and 34 μM, respectively) (Figs. 1 and 2A; Table 1). With respect to efficacy, although all five drug conjugates inhibited MCF-7 cell viability compared with vehicle, C4 and C5 were most efficacious at inhibiting viability by 70%–90% compared with vehicle (Fig. 2B; Table 2). To determine whether the drug conjugates displayed equal or superior potency and/or efficacy against the controls with respect to effects on MCF-7 cell viability, each drug conjugate was compared against melatonin alone, tamoxifen alone, 4-OH-tamoxifen alone, melatonin plus tamoxifen (unlinked), and melatonin plus 4-OH-tamoxifen (unlinked) by one-way ANOVA. The potency of C2, C4, C5, and C15 was similar to all of the controls except for C9, which was least potent (Fig. 2C; Table 3). Regarding efficacy, C5 displayed the greatest efficacy (∼90% compared with vehicle), which was not significantly different from the other controls except for melatonin, which only inhibited viability by ∼10% compared with vehicle (Fig. 2D; Table 4).

Chemical structure of melatonin-tamoxifen drug conjugates.

Comparison of potency and efficacy between C2, C4, C5, C9, and C15 conjugates to inhibit BC cell viability. (A and B) The potency (IC50) (A) or efficacy (B) of C2, C4, C5, C9, and C15 on cell viability in MCF-7, MMC, MDA-MB-231, and BT-549 cells. (C and D) The potency (IC50) (C) or efficacy (D) of melatonin, tamoxifen, 4-OH-tamoxifen, unlinked melatonin plus tamoxifen and unlinked melatonin plus 4-OH-tamoxifen on cell viability in MCF-7, MMC, MDA-MB-231, and BT-549 cells. Each bar represents the mean ± S.D. of three independent experiments for each compound (n = 9 for vehicle, since it was run with each experiment conducted). Data were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was defined as P < 0.05. Letters denote significance between groups as follows: for figure A-a, P < 0.05 vs. C2; b, P < 0.05 vs. C4; c, P < 0.05 vs. C5; d, P < 0.05 vs. C9; for figure B-a, P < 0.05 vs. vehicle; b, P < 0.05 vs. C2; c, P < 0.05 vs. C4; d, P < 0.05 vs. C5, e, P < 0.05 vs. C9; for figure C-a, P < 0.05 vs. melatonin; for figure D-a, P < 0.05 vs. vehicle; b, P < 0.05 vs. melatonin; d, P < 0.05 vs. 4-OH-tamoxifen. Abs, absorbance.
Comparison of potencies between C2, C4, C5, C9, and C15 conjugates to inhibit BC cell viability and migration
Each value represents the mean ± S.D. of potency (IC50) values generated from three to nine individual curves fit by nonlinear regression analysis.
Comparison of efficacies between C2, C4, C5, C9, and C15 conjugates to inhibit BC cell viability and migration
Each value represents the mean ± S.D. of maximum inhibitory effect of each drug conjugate represented as the percentage of vehicle for viability and the percentage of baseline for migration assays.
Comparison of potencies between C2, C4, C5, C9, and C15 conjugates and controls to inhibit BC cell viability and migration
Each value represents the mean ± S.D. of potency (IC50) values generated from three to nine individual curves fit by nonlinear regression analysis.
Comparison of efficacies of C2, C4, C5, C9, and C15 conjugates and controls to inhibit BC cell viability and migration
Each value represents the mean ± S.D. of maximum inhibitory effect of each drug conjugate represented as a percentage of vehicle (n = 9/cell line tested) for viability and a percentage of baseline for migration assays.
Migration.
With respect to migration and when compared with each other, C5 and C15 trended toward being most potent to inhibit MCF-7 cell migration (IC50 = 4 and 1 μM, respectively), whereas C2, C4, and C9 were least potent (IC50 = 100 μM, 9 mM, and 218 mM, respectively) (Fig. 3A; Table 1). Regarding efficacy, all drug conjugates, except for C15 (−13% of baseline), inhibited MCF-7 cell migration equally (+2% to +20% of baseline) compared with vehicle (Fig. 3B; Table 2). All drug conjugates, except for C9, were more potent than melatonin plus 4-OH-tamoxifen to inhibit MCF-7 cell migration but similar to melatonin, tamoxifen, 4-OH-tamoxifen, and melatonin plus tamoxifen controls (Fig. 3C; Table 3). Regarding efficacy, C2, C4, C5, and C9 displayed similar efficacies to inhibit MCF-7 cell migration compared with all controls (+0% to +39% of baseline). Both melatonin (−14% of baseline) and C15 were without effect on MCF-7 cell migration (Fig. 3D; Table 4).

Comparison of potency and efficacy between C2, C4, C5, C9, and C15 conjugates to inhibit BC cell migration. (A and B) The potency (IC50) (A) and efficacy (B) of C2, C4, C5, C9, and C15 on cell migration in MCF-7, MMC, MDA-MB-231, and BT-549 cells. (C and D) The potency (IC50) (C) or efficacy (D) of melatonin, tamoxifen, 4-OH-tamoxifen, unlinked melatonin plus tamoxifen, and unlinked melatonin+4-OH-tamoxifen on cell migration in MCF-7, MMC, MDA-MB-231, and BT-549 cells. Each bar represents the mean ± S.D. of three independent experiments for each compound (n = 9 for vehicle, since it was run with each experiment conducted). Data were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was defined as P < 0.05. Letters denote significance between groups as follows: for figure A-a, P < 0.05 vs. C2; b, P < 0.05 vs. C4; c, P < 0.05 vs. C5; d, P < 0.05 vs. C9; for figure B-a, P < 0.05 vs. vehicle; b, P < 0.05 vs. C2; c, P < 0.05 vs. C4; d, P < 0.05 vs. C5, e, P < 0.05 vs. C9; for figure C-a, P < 0.05 vs. melatonin; b, P < 0.05 vs. tamoxifen; c, P < 0.05 vs. 4-OH-tamoxifen; for figure D-a, P < 0.05 vs. vehicle; b, P < 0.05 vs. melatonin; c, P < 0.05 vs. tamoxifen; d, P < 0.05 vs. 4-OH-tamoxifen, e, P < 0.05 vs. melatonin+tamoxifen.
Effects of C2, C4, C5, C9, and C15 Conjugates on MMC (HER2+) Cells
Viability.
Cells were exposed to varying concentrations of C2, C4, C5, C9, and C15 and their effect on cell viability was assessed. C4 trended toward being most potent (IC50 = 243 nM), followed by C2, C5, and C9 (IC50 = 13, 6, and 6 μM, respectively), and then C15, which was least potent (IC50 = 333 mM) (Fig. 2A; Table 1). Concerning efficacy, C2, C4, C5, and C9 inhibited MMC cell viability to a similar extent (∼20%–50% inhibition compared with vehicle). C15 produced a minimal nonsignificant effect (3% inhibition compared with vehicle) (Fig. 2B; Table 2). All conjugates, except C2, had similar potency to the controls except for C4, which was more potent than melatonin plus 4-OH-tamoxifen (Fig. 2C; Table 3). Regarding efficacy, all drug conjugates, except for C15, inhibited MMC cell viability to a similar extent, which was similar (∼40% inhibition compared with vehicle) to controls (tamoxifen with or without melatonin; 4-OH-tamoxifen with or without melatonin) except for melatonin (9% inhibition compared with vehicle), which did not inhibit MMC viability (Fig. 2D; Table 4).
Migration.
The potencies of C4, C5, C9, and C15 to inhibit MMC cell migration, although variable, were not significantly different from one another; however, they were significantly more potent than that of C2, which had an IC50 of 221 mM (Fig. 3A; Table 1). Regarding efficacy to inhibit MMC cell migration, all drug conjugates, except for C15 (−32% of baseline), inhibited MMC cell migration equally (+1% to +44% of baseline) compared with vehicle (Fig. 3B; Table 2). C2 and C9 were the least potent of all drug conjugates compared with the controls (melatonin alone, tamoxifen alone, 4-OH-tamoxifen alone, and melatonin plus tamoxifen unlinked; Fig. 3C; Table 3). C4, C5, and C15 were similar to all controls except C5, which was more potent than melatonin plus 4-OH-tamoxifen to inhibit MMC cell migration (Fig. 3C; Table 3). Regarding efficacy, C2 and C9 displayed the greatest efficacy (∼+40% of baseline) to inhibit MMC cell migration compared with all controls. C4 and C5 were similar to all controls (between +1% and 7% of baseline) except melatonin, which was without effect versus vehicle. C15 was without effect on MMC cell migration compared with vehicle (Fig. 3D; Table 4).
Effect of C2, C4, C5, C9, and C15 Conjugates on MDA-MB-231 (ER−/PR−/HER−) Cells
Viability.
Cells were exposed to varying concentrations of C2, C4, C5, C9, and C15 and their effect on cell viability was assessed. C9 was the least potent (IC50 = 145 mM), whereas C15 was most potent (IC50 = 477 pM) versus C2, C4, and C5 (IC50 = 246 μM, 37 nM, and 12 μM, respectively) (Fig. 2A; Table 1). Regarding efficacy, C2, C5, and C9 inhibited cell viability to a similar extent (25%–37% inhibition compared with vehicle), whereas C4 and C15 were without effect and similar to vehicle (Fig. 2B; Table 2). The potencies, although variable, were not significantly different from one another except for C5, which was more potent than combination melatonin plus tamoxifen (Fig. 2C; Table 3). Regarding efficacy, C2, C5, and C9 displayed significant inhibition (25%–37%) compared with vehicle, whereas C4 and C15 were without effect. C2 and C9 were also significantly different than melatonin alone, and C2, C5, and C9 were also more efficacious at inhibiting MDA-MB-231 cell viability versus 4-OH-tamoxifen alone (Fig. 2D; Table 4).
Migration.
Melatonin-tamoxifen drug conjugate potency and efficacy to inhibit MDA-MB-231 cell migration revealed no significant differences in potency between C4, C5, and C9; however, they were significantly higher compared with C2 and C15, which displayed micromolar potency (Fig. 3A; Table 1). Regarding efficacy, all drug conjugates, except for C15, inhibited MDA-MB-231 cell migration equally (+10% to +35% of baseline) compared with vehicle; C15 worsened migration of the cells (Fig. 3B; Table 2). Compared with the controls (i.e., melatonin alone, tamoxifen alone, 4-OH-tamoxifen alone, melatonin plus tamoxifen unlinked, and melatonin plus 4-OH-tamoxifen unlinked), C2 was the least potent of all drug conjugates to inhibit MDA-MB-231 cell migration (IC50 = 559 mM). C4, C5, C9, and C15 were similar to all controls, with C5 displaying greater potency (IC50 = 3 μM) than combination melatonin plus tamoxifen (IC50 = 229 mM; Fig. 3C; Table 3). Regarding efficacy, all drug conjugates inhibited MDA-MB-231 cell migration compared with vehicle and melatonin; however, C9 had the highest efficacy (+35% of baseline) and inhibited cell migration greater than melatonin plus tamoxifen (+0% of baseline) and melatonin plus 4-OH-tamoxifen (+2% of baseline). C15 was least efficacious (−43% of baseline), whereas melatonin was without effect (Fig. 3D; Table 4).
Effects of C2, C4, C5, C9, and C15 Conjugates on BT-549 Cells
Viability.
Another TNBC line, BT-549, was screened to assess the potency and efficacy of the drug conjugates on viability and migration. With respect to viability, all drug conjugates had similar potency to inhibit BT-549 cell viability with potency in the low micromolar range (IC50 = 2–34 μM; Fig. 2A; Table 1). Concerning efficacy, only C4 and C5 inhibited BT-549 cell viability (∼35% inhibition compared with vehicle); C2, C9, and C15 were without effect and similar to vehicle (Fig. 2B; Table 2). All drug conjugates had a similar potency to the controls (melatonin, 4-OH-tamoxifen, melatonin plus tamoxifen, and melatonin plus 4-OH-tamoxifen) except for tamoxifen, which was least potent (IC50 = 781 mM; Fig. 2C; Table 3). Regarding efficacy, C2, C4, and C5 inhibited BT-549 cell viability (∼20%–35% inhibition compared with vehicle) and melatonin with C15 were least efficacious (∼10% inhibition compared with vehicle).
Migration.
Exposure of BT-549 cells to C2, C4, C5, C9, and C15 did not result in significant differences in potencies between drug conjugates to inhibit their migration (Fig. 3A; Table 1). With respect to migration, all drug conjugates had similar potency. Regarding efficacy, C2, C4, C5, and C9 inhibited BT-549 cell migration (+0% to +56% of baseline) compared with vehicle. C15 increased BT-549 cell migration (−61% of baseline) versus vehicle (Fig. 3B; Table 2). With respect to control potencies, which ranged between 3 and 116 μM, no significant differences in potencies occurred for any of the drug conjugates except for the C2, which had low potency (IC50 = 579 mM; Fig. 3C; Table 3). Regarding efficacy, C9 inhibited BT-549 cell migration compared with vehicle; C2 and C9 also inhibited cell migration to a greater extent than melatonin, tamoxifen, or 4-OH-tamoxifen—all of which were without effect on BT-549 cell migration. Furthermore, C9 was more efficacious compared with combination melatonin plus tamoxifen (+15% of baseline) and combination melatonin plus 4-OH-tamoxifen (+32% of baseline). C15 made the BT-549 cells more aggressive (−61% of baseline) compared with all controls (Fig. 3D; Table 4).
Binding Affinities of C2, C4, C5, C9, and C15 Conjugates to ERs and MT1Rs
Competition binding analysis was performed to identify potential mechanisms of action for the anticancer actions of the drug conjugates. Specifically, analyses of competition of tamoxifen, 4-OH-tamoxifen, and each of the drug conjugates (C2, C4, C5, C9, and C15) for [3H]-estradiol or [125I]-estradiol binding to ERs or competition of melatonin and each of the drug conjugates for 2-[125I]-iodomelatonin binding to MT1R binding assays were conducted. ER binding affinity assays were conducted in both the mouse uterus (Witt-Enderby et al., 2014) and the MCF-7 cell line (Fig. 4). Tamoxifen and 4-OH-tamoxifen were used as controls for mouse uterus and MCF-7 cells, respectively. As previously published (Witt-Enderby et al., 2014), the affinity of tamoxifen (Ki,high = 3 pM and Ki,low = 6 nM) for ERs expressed in the mouse uterus displayed a biphasic curve (Fig. 4A). In MCF-7 cells, the affinity of 4-OH-tamoxifen for ERs displayed a monophasic curve with an affinity Ki of 46 nM (Fig. 4C). No concentration-dependent inhibition of [3H]-estradiol binding occurred for C2, C9, and C15 (Fig. 4, E–G), whereas C4 had a binding affinity (Ki = 41 nM) similar to 4-OH-tamoxifen (Ki = 46 nM) between concentrations of 1 pM and 10 μM (Fig. 4D). As already reported, C5 showed a similar binding affinity (Ki = 2 nM) as tamoxifen (Witt-Enderby et al., 2014) between concentrations 1 pM and 1 μM (Fig. 4B). Interestingly, all drug conjugates displayed an increase in total ER binding sites (∼225%–800% of control) at higher (10–100 µM) concentrations.

Competition of C2, C4, C5, C9, or C15 for [3H]-or [125I]-estradiol binding to ERs expressed in mouse uterus or MCF-7 cells. (A-G) Binding of [3H]-or [125I]-estradiol to ERs expressed in mouse uterus (A and B) and MCF-7 cells by competition binding using tamoxifen (A), 4-OH-tamoxifen (C), or conjugates C2, C4, C5, C9, or C15 (B, D–G) and G1 effects on total [3H]-estradiol binding to ERs expressed in MCF-7 cells (H). Data represent the mean ± S.D. of three independent experiments performed in duplicate. (A) and (B) were taken from Witt-Enderby et al. (2014). Data in (H) were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was indicated with lowercase letters as follows: a, P < 0.05 vs. vehicle; b, P < 0.05 vs. tamoxifen; c, P < 0.05 vs. G1; d, P < 0.05 vs. C4; and e, P < 0.05 vs. C5.
For human MT1Rs, all drug conjugates demonstrated concentration-dependent inhibition of 2-[125I]-melatonin binding with binding affinity (Ki: C2 = 35 nM, C4 = 12 μM, C5 = Ki,high = 6 pM and Ki,low= 9 nM, C9 = 111 nM, and C15 = 56 nM) similar to that of melatonin (Ki = 2 nM) (Fig. 5). Unlike the increases in ER binding sites observed for all drug conjugates at concentrations greater than 10 μM, no drug conjugates except for C5 produced an increase in 2-[125I]-melatonin binding sites (∼90% of control) at 100 µM (Fig. 5D).

Competition of C2, C4, C5, C9, or C15 for 2-[125I]-iodomelatonin binding to MT1Rs expressed in Chinese hamster ovary (CHO) cells. (A–G) Competition of 2-[125I]-iodomelatonin for binding to MT1Rs expressed in CHO cells in the presence of melatonin (A and B) or conjugates C2, C4, C5, C9, or C15 (C–G). Data represent the mean ± S.D. of three independent experiments performed in duplicate. (B) and (D) were taken from Witt-Enderby et al. (2014).
Binding Affinities of C4, C5, and G1 to ERs
Total binding assays were conducted on MCF-7 cells using [3H]-estradiol to elucidate the role of C4 and C5 on GPR30. As shown in Fig. 4H, all treatments, except for G1, decreased [3H]-estradiol binding compared with vehicle, whereas G1 increased [3H]-estradiol binding. The combination of C5 with G1 demonstrated the lowest [3H]-estradiol binding compared with all groups, including C5 alone (Fig. 4H).
Effects of C4 and C5 Conjugates on TamR MCF-7 Cells
C4 and C5 were then further tested for their anticancer actions in TamR MCF-7 cells. Regarding viability, both C4 (IC50 = 4.27 µM; maximum inhibition = 83% of vehicle) and C5 (IC50 = 6.03 µM; maximum inhibition = 81% of vehicle) displayed similar potency and efficacy to inhibit TamR cell viability (Fig. 6A), whereas the controls were without effect except for tamoxifen, which demonstrated 27% inhibition of TamR cell viability compared with vehicle (Fig. 6A). Regarding migration (Fig. 6B), both C4 and C5 (IC50 = 4.53 and 5.07 µM; maximum inhibition = 126% and 137%, respectively) exhibited similar potency and efficacy to inhibit TamR cell migration similar to melatonin, tamoxifen, or melatonin plus tamoxifen (IC50 = 1.85 µM, 153.4 mM, and 12.19 mM; maximum inhibition = 15%, 129%, and 161%, respectively); 4-OH-tamoxifen was least potent and efficacious (IC50 = 19.11 µM; maximum inhibition = 50%). Maximum inhibition greater than 100% indicates that the cells were detached from the plate or the border width after 24 hours was wider than the 0-hour border width. Further characterization of TamR cells revealed that the binding affinity and density of [3H]-estradiol did not change in TamR MCF-7 cells (KD = 1.1 nM, Bmax = 21.4 pmol/mg protein) versus WT MCF-7 cells (KD = 2.2 nM, Bmax = 4.8 pmol/mg protein) even though ER expression was decreased in TamR cells versus WT cells (Supplemental Fig. 6, A, B, and F). Analysis of growth patterns of TamR cells revealed that over a 7-day period, TamR cells grew at a faster rate and to a greater extent than WT MCF-7 cells in the presence of 4-OH-tamoxifen (Supplemental Fig. 6C) and this was accompanied by a mesenchymal phenotype (Supplemental Fig. 6, D and E) perhaps due to an increase in pERK5 and a decrease in pERK1/2 (Supplemental Fig. 6F).

Effect of C4, C5, melatonin, tamoxifen, 4-OH-tamoxifen, and unlinked melatonin plus tamoxifen on TamR MCF-7 cell viability and cell migration. (A and B) Data in (A) demonstrate the effect of C4, C5, melatonin, tamoxifen, 4-OH-tamoxifen, or unlinked melatonin plus tamoxifen on TamR MCF-7 cell viability and cell migration (B). Data represent the mean ± S.D. of three independent experiments performed in duplicate. Complete characeterization of TamR cells can be found in Supplemental Fig. 6. Abs, absorbance.
Effects of C4 and C5 Conjugates on TU-BcX-4IC Cells and TU-BcX-4QAN Tumor Tissue
C4 and C5 were then tested for their anticancer actions in TU-BcX-4IC cells. Regarding viability, both C4 and C5 (IC50 = 181.5 and 304.2 mM; maximum inhibition = 78% and 65% of vehicle, respectively) displayed similar potency and efficacy to inhibit cell viability (Fig. 7A), whereas melatonin was without effect; melatonin plus tamoxifen demonstrated 80% inhibition of cell viability compared with vehicle (Fig. 7A). Regarding migration (Fig. 7B), both C4 and C5 (IC50 = 211.1 and 80.38 µM; maximum inhibition = 145% and 145%, respectively) exhibited similar and greater potency and efficacy compared with melatonin, tamoxifen, or melatonin plus tamoxifen (IC50 = 168.6, 514.9, and 116.8 mM; maximum inhibition = 21%, 159%, and 155%, respectively). Maximum inhibition greater than 100% indicates that the cells were detached from the plate or the border width after 24 hours was wider than the 0-hour border width. In TU-BcX-4IC cells, Western blot data demonstrated that C4 and C5 decreased pERK1/2 levels, and C5 increased NF-κB with trends toward an increase occurring with C4 (Fig. 7C) similar to what occurred in the other triple-negative cells, MDA-MB-231 and BT-549 (Table 5). However, C4 and C5 had no significant effects on pERK5 levels in TU-BcX-4IC cells. Both conjugates did not change pERK1/2, pERK5, and NF-κB levels in PDX tumor tissue, although C4 and C5 trended toward a reduction in pERK5 levels (Fig. 7C). Further characterization of the TU-BcX-4IC cells for melatonin binding sites or estrogen binding sites revealed total specific binding of 2-[125I]-iodomelatonin (7.6 ± 3.9 fmol/mg protein) or [3H]-estradiol (117 ± 56 fmol/mg protein; Fig. 7D).

Effect of C4, C5, melatonin, tamoxifen, 4-OH-tamoxifen, and unlinked melatonin plus tamoxifen on TU-BcX-4IC cell viability and cell migration and TU-BcX-4QAN tumor tissue. (And B) Data demonstrate the effect of C4, C5, melatonin, tamoxifen, or unlinked melatonin plus tamoxifen on TU-BcX-4IC cell viability (A) and cell migration (B). (C and D) Expression of pERK1/2, pERK5, and NF-κB was analyzed in TU-BcX-4QAN tumor tissue and TU-BcX-4IC cells (C), and total specific binding of 2-[125I]-iodomelatonin or [3H]-estradiol was measured in TU-BcX-4IC cells (D). Data represent the mean ± S.D. of three independent experiments performed in duplicate. Significance was defined as P < 0.05. In (A) and (B), asterisks indicate significance from all treatments except 100 μM treatments for all groups. In (C), “a” indicates significance compared with vehicle and “b” indicates significance compared with 10 μM melatonin. Images of cells in (A) and (B) were exposed to vehicle or 100 μM of the treatment listed within each series of graphs. OD, optical density.
The effect of MEK1/2 (PD98059), MEK5 (Bix02189), and PI3K (pictilisib) on C4/C5-mediated effects on pERK1/2, pERK5, pAKT, NF-κB, RUNX2, and β1-INTEGRIN levels in MCF-7, MMC, MDA-MB-231, and BT-549 cell lines
Data represent the mean ± S.D. of three independent experiments.
Role of MEK1/2, MEK5, and PI3K in Mediating C4 and C5 Effects on BC Cell Viability and Migration
MCF-7.
All inhibitors (PD98059, Bix02189, and pictilisib) attenuated MCF-7 (ER+) cell viability on their own; when combined with C4 or C5, only pictilisib (PI3K inhibitor) enhanced C4- and C5-mediated MCF-7 cell viability, whereas PD98059 and Bix02189 produced subtle (if any) inhibitory effects (Figs. 8, A–D, and 12A). Regarding migration and when added alone, PD98059 and Bix02189 were without effect and pictilisib inhibited MCF-7 cell migration versus vehicle. When combined with C4, all inhibitors (PD98059, Bix02189, and pictilisib) enhanced C4-mediated inhibition of MCF-7 cell migration (Fig. 8, E and G; see Fig. 12B for a schematic). For C5, coadministration with PD98059 was without effect; coadministration with pictilisib enhanced the inhibitory effect of C5, and coadministration with Bix02189 blocked C5’s effects (Figs. 8, F and H, and 12C).

Effect of MEK1/2, MEK5, or PI3K inhibitors on cell viability and migration of C4 or C5 in MCF-7 cells. (A–H) Cell viability (A–D) and migration (E–H) of C4 or C5 in the absence or presence of PD98059, Bix02189, or pictilisib in MCF-7 cells. Each point represents the mean ± S.D. of three independent experiments. Data were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was defined as P < 0.05. Significance of the letters is mentioned in each panel (A-H).
MMC.
Bix02189 and PD98059 alone were without effect, whereas pictilisib alone inhibited MMC viability (Fig. 9). Combination of Bix02189 (Fig. 9A) or pictilisib (Fig. 9C) with C4 enhanced MMC viability. PD98059 added in combination with C4 blocked C4-mediated MMC viability at C4 concentrations (1 pM to 1 μM). C5 inhibited MMC viability, which was enhanced in the presence of Bix02189 (Fig. 9B) or pictilisib (Fig. 9D) but not PD98059 (Fig. 9B). Regarding MMC migration, PD98059, Bix02189 (Fig. 9E), or pictilisib (Fig. 9G) each alone inhibited MMC migration; however, when combined with C4 only PD98059 and pictilisib enhanced C4’s inhibitory actions (Fig. 12F). For C5, PD98059 and Bix02189 enhanced C5’s inhibitory actions on MMC migration at C5 concentrations ranging from 1 pM to 1 μM where no further enhancement occurred at a higher (>1 μM) concentration of C5 (Fig. 9F). Pictilisib enhanced C5-mediated MMC migration at all concentrations tested except the highest concentration of 100 μM (Figs. 9H and 12G).

Effect of MEK1/2, MEK5, or PI3K inhibitors on cell viability and migration of C4 or C5 in MMC cells. (A–H) Cell viability (A–D) and migration (E–H) of C4 or C5 in the absence or presence of PD98059, Bix02189, or pictilisib in MMC cells. Each point represents the mean ± S.D. of three independent experiments. Data were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was defined as P < 0.05. Significance of the letters is mentioned in each panel (A-H).
MDA-MB-231.
No effect of the inhibitors occurred either alone or in combination with C4 and C5 except for pictilisib, which inhibited MDA-MB-231 cell viability when added alone and when compared with vehicle (Fig. 10). Only at one concentration (1 nM C5) did pictilisib block its effect (Fig. 10, C and D). Regarding migration, PD98059 and Bix02189 alone were without effect, whereas pictilisib inhibited cell migration versus vehicle. When combined with C4, all inhibitors enhanced C4-mediated inhibition of cell migration (Fig. 10, E and G). For C5, pictilisib added in combination with C5 enhanced cell migration, whereas PD98059 and Bix02189 blocked C5’s inhibitory effects (Fig. 10, F and H).

Effect of MEK1/2, MEK5, or PI3K inhibitors on cell viability and migration of C4 or C5 in MDA-MB-231 cells. (A–H) Cell viability (A–D) and migration (E–H) of C4 or C5 in the absence or presence of PD98059, Bix02189, or pictilisib in MDA-MB-231 cells. Each point represents the mean ± S.D. of three independent experiments. Data were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was defined as P < 0.05. Significance of the letters is mentioned in each panel (A-H).
BT-549.
PD98059 or Bix02189 alone was without effect on BT-549 cell viability, whereas pictilisib alone produced some inhibitory effects. When added in combination with C4, all inhibitors slightly, albeit significantly, inhibited C4-mediated inhibition of BT-549 viability at C4 concentrations higher than 1 μM and the same occurred for C5 in the presence of Bix02189 or pictilisib but not PD98059 (Figs. 11, A–D, and 12J). For migration, PD98059 and Bix02189 alone were without effect, whereas pictilisib inhibited BT-549 cell migration (vs. vehicle). When coadministered with Bix02189 or pictilisib but not PD98059, C4-mediated effects on BT-549 cell migration were enhanced (Figs. 11, E–H, and 12K). Pictilisib added in combination with C4 or C5 enhanced their inhibitory effects on BT-549 cell migration, whereas PD98059 was without effect (Fig. 11, G and H). The same findings occurred for C5 except that Bix02189 blocked, but did not enhance, the inhibitory effect of C5 on BT-549 cell migration (Fig. 11, E and F).

Effect of MEK1/2, MEK5, or PI3K inhibitors on cell viability and migration of C4 or C5 in BT-549 cells. (A–H) Cell viability (A–D) and migration (E–H) of C4 or C5 in the absence or presence of PD98059, Bix02189, or pictilisib in BT-549 cells. Each point represents the mean ± S.D. of three independent experiments. Data were analyzed by one-way ANOVA followed by the Newman–Keuls post hoc t test where significance was defined as P < 0.05. Significance of the letters is mentioned in each panel (A-H).

Summary of mechanisms of action underlying C4 or C5 in BC cells. Depicted is a summary of the mechanisms of action underlying C4 (A, B, D, F, H, J, and K) or C5 (C, E, G, I, J, and L) in MCF-7 (A–C), MMC (D–G), MDA-MB-231 (H and I), or BT-549 (J–L) BC cells. The lines connecting the proteins indicated in black were the relationships deduced from the viability and migration assays, and the lines indicated in gray were derived from the Western blot analyses.
Effect of C4 and C5 Melatonin-Tamoxifen Drug Conjugates on pERK5, pERK1/2, pAKT, NF-κB, RUNX2, and β1-INTEGRIN Protein Levels
To identify the signaling proteins/cascades involved in C4- or C5-mediated inhibition of BC, Western blot analysis was performed in each cell line under basal (unstimulated) conditions or after 15-minute exposure to each conjugate (C4 or C5) alone or in combination with PD98059 (MEK1/2 inhibitor), Bix02189 (MEK5 inhibitor), or pictilisib (PI3K inhibitor). Treatment effects on pERK1/2, pERK5, pAKT, RUNX2, NF-κB, and β1-INTEGRIN were analyzed. Due to the enormity and complexity of the analyses, all data are presented in Supplemental Tables 4–7 and schematics are shown in Fig. 12 (indicated in blue) for clarity, whereas significant changes in protein levels are mentioned in Table 5.
In general, the major findings of the Western blot analyses for MCF-7 cells demonstrate that all three pathways (MEK1/2, MEK5, and PI3K), when inhibited by PD98059, Bix02189, or pictilisib, respectively, enhance the inhibitory effects of C4 on MCF-7 viability and migration and crosstalk (mainly inhibitory actions) between MEK/pERK1/2 and MEK5/pERK5 occurs. For C5, although both MEK1/2 and PI3K pathways are parallel to and enhance the inhibitory effects of C5 when inhibited by PD98059 and pictilisib, respectively, MEK5 was critical in mediating C5’s actions on MCF-7 viability and migration. RUNX2 expression was inhibited in the presence of C4 and C4 plus pictilisib. β1-INTEGRIN expression levels were not significantly changed in the presence of C4 or C5 (Fig. 12, A–C; Supplemental Tables 4–7).
For MMC (HER2+) BC cells, the Western blot analysis revealed that the MEK1/2/pERK1/2 pathway appeared to play a more central role possibly through comodulation of NF-κB, pERK5, and β1-integrin. Western blot analyses revealed that C5 activated (1 nM) or inhibited (10 μM) NF-κB or β1-integrin levels that were concentration dependent (see the dotted lines; Fig. 12, E and G). For C5, the inhibitor analysis revealed that crosstalk between MEK1/2/pERK1/2 and MEK5/pERK5 occurred and that inhibition of pERK5 may be through a MEK1/2/pERK1/2-mediated inhibition of MEK5 (Fig. 12G). No effect of C4 or C5 on Runx2 expression was observed (Supplemental Tables 4–7).
For MDA-MB-231 TNBC cells, relationships between PI3K/pERK1/2 and MEK5/pERK5 on NF-κB expression were observed where pictilisib inhibited pERK1/2 levels and Bix02189 increased NF-κB expression (Fig. 12H). C5 increased NF-κB and β1-INTEGRIN levels, whereas MEK1/2-mediated crosstalk with MEK5/pERK5 occurred and PI3K-mediated modulation of NF-κB, RUNX2, and β1-INTEGRIN was observed (Fig. 12I).
For BT-549 TNBC cells, the MEK5/pERK5 and PI3K signaling pathways were parallel to and facilitated the inhibitory actions of C4 in the presence of Bix02189 and pictilisib, respectively, possibly by modulating pERK1/2 as revealed through Western blot analysis (Fig. 12K; Table 5). PD98059 alone inhibited BT-549 cell migration and these effects may be mediated through NF-κB (Fig. 12K). For C5, Western blot analysis revealed that C5 modulates NF-κB; however, how NF-κB fits into these signaling cascades remains unclear and needs further study (Fig. 12L). MEK1/2 inhibition by PD98059 inhibited BT-549 migration and these effects are likely mediated through an inhibition of pERK1/2 revealed by Western blot analysis. No effect of C4 or C5 on β1-INTEGRIN or RUNX2 expression occurred in these cells (Fig. 12L; Supplemental Tables 4–7).
As shown in Supplemental Fig. 7, basal expression of all proteins was assessed between cell lines and significant differences in some of these proteins existed between the TNBC lines MDA-MB-231 and BT-549 and the ER+-expressing BC line MCF-7. Specifically, pERK1/2 levels were highest in MDA-MB-231 cells compared with MCF-7 and BT-549 cells; in BT-549 cells, levels of NF-κB were greatest compared with MCF-7 cells and β1-INTEGRIN levels were highest compared with MCF-7 or MDA-MB-231 cells (Supplemental Fig. 7).
Assessment of C4 and C5 Drug Conjugate Metabolism Both In Vitro and In Vivo
Microsomal Incubation of Tamoxifen, C4, and C5.
Tamoxifen was incubated for up to 20 minutes in the absence of microsomes and levels were assessed by HPLC-UV. No change in drug levels occurred, suggesting that tamoxifen was stable over time during the incubation period (data not shown). To assess tamoxifen oxidative metabolism by liver microsomes, incubations were conducted in the presence of NADPH for either 10 or 20 minutes in mouse or human microsomes, respectively. As shown in Table 6, there was an ∼40% loss in 10 minutes in mouse microsomes and in 20 minutes in human microsomes. The observed loss in the human experiment is consistent with the preponderance of cytochrome P450–mediated metabolism of tamoxifen (Dahmane et al., 2014). The same assay in both species was conducted as described for tamoxifen to assess the susceptibility of C4 and C5 conjugates to liver microsomal metabolism. Loss of either compound was similar to that observed for tamoxifen in the respective species (Table 6).
In vitro metabolism and in vivo pharmacokinetic parameters of tamoxifen, C4, or C5 by MLM/human liver microsomal incubation and by subcutaneous/oral administration in female C57BL/6J mice, respectively
Data represent the mean ± S.D. percentage of drug conjugate remaining from microsomal incubations (n = 3) unless indicated otherwise.
Pharmacokinetics of Tamoxifen, C4, and C5 in Mice.
Tamoxifen was administered by the subcutaneous route, whereas C4 and C5 were administered through subcutaneous and oral routes. As summarized in Table 6, exposure to C5 after oral administration was substantially lower compared with C4. In contrast, exposure parameters (Cmax and Area Under Curve, AUC0-24 hours) were similar for the two compounds after subcutaneous administration, and they were similar to those calculated after the same dose of tamoxifen by this route (Table 6). Based on the differences between C4 and C5 after oral dosing relative to the similar exposures obtained after subcutaneous dosing, the oral bioavailability relative to subcutaneous administration of C4 was much higher than C5.
Discussion
Our data demonstrate that C4 and C5 drug conjugates have the greatest potential as novel BC drugs, supported by their binding profiles at ERs and MT1Rs, and in their efficacy to inhibit an array of diverse BC cells, including TamR, TNBC, and PDX-TNBC cells. C4 and C5 demonstrated low nano- or micromolar potency to inhibit BC cell viability (65%–80% inhibition compared with vehicle), making them attractive anticancer drug candidates in ER+ BC, HER2+ BC, and TNBCs that include PDX-TNBC. Both C4 and C5 also decreased TamR MCF-7 cell viability and migration, making these compounds potentially useful for treating recurrent or TamR BC. The involvement of MEK1/2, MEK5, and PI3K in C4- or C5-mediated anticancer actions was chosen because of their involvement in MT1R or ESR1-mediated BC signaling (Grant et al., 2009; Temraz et al., 2015; Simoes et al., 2016; Araki and Miyoshi, 2018; Kastrati et al., 2017; Martinelli et al., 2017; Kumar et al., 2018). In this study, it was observed that the anticancer actions of C4 and C5 were “context specific” dependent on the BC phenotype (i.e., ER+, HER2+, or triple negative), the linkers connecting melatonin to tamoxifen, the endpoints measured (i.e., viability or migration), and the inhibitors used in the analysis (i.e., PD98059, Bix02189, or pictilisib). C4 (for viability and migration; Fig. 12, A and B, respectively) and C5 (for viability; Fig. 12A) inhibited MCF-7 cell viability in parallel with MEK1/2, MEK5, and PI3K but not through these pathways. For C5, the inhibitory effects were shown to be mediated through MEK5 and not through MEK1/2, whereas the PI3K pathway (when inhibited) worked in parallel with C5 to enhance its inhibitory action (Fig. 12C). These data suggest that C4 and C5 work similarly with respect to viability but not migration and that the number of carbons linking melatonin with tamoxifen influenced the outcome with respect to MCF-7 cell migration and the signaling cascade to which the drug conjugate modulated. For MMC cells, the findings suggest that lower concentrations of C4 may be working through the MEK1/2 pathway to influence MMC viability (Fig. 12D) and that C5-mediated inhibition of MMC viability works in concert with but not through PI3K and possibly MEK5 but not MEK1/2 (Fig. 12E). For migration, the PI3K and MEK1/2 pathways enhanced C4’s inhibitory action (Fig. 12F), whereas combination of PD98059, Bix02189, and pictilisib with C5 enhanced its inhibitory actions (Fig. 12G). For MDA-MB-231 cells, the data suggest that the MEK1/2, MEK5, and PI3K pathways (when inhibited) work in concert with C4 to inhibit MDA-MB-231 migration; for viability, only the PI3K pathway cooperated with C4 to inhibit MDA-MB-231 (Fig. 12H). Pictilisib enhanced C5-mediated inhibition of viability and migration like C4; however, C5 inhibited MDA-MB-231 cell migration through MEK1/2 and MEK5 (Fig. 12I). For BT-549 cells, C4-mediated inhibition of viability occurred through the MEK1/2, MEK5, and PI3K signaling pathways, whereas C5 inhibited viability through MEK5 and PI3K and not MEK1/2 (Fig. 12J). For C4, the MEK5 and PI3K pathways (when inhibited) worked in concert with C4 to enhance its inhibitory actions on BT-549 migration (Fig. 12K). For C5, Bix02189 blocked C5’s inhibitory effects on BT-549 cell migration, suggesting that the C5-mediated inhibition of BT-549 cell migration occurs through MEK5; however, the PI3K pathway worked in concert with C5 to enhance its antimigratory effects but only when PI3K was inhibited by pictilisib (Fig. 12L).
These inhibitor studies demonstrate that C4 and C5 are not acting “typically” at ERs and MT1Rs and suggest that novel pharmacophores are being created in a cancer cell–specific manner to produce their diverse anticancer actions. These unique pharmacophores created by C4 and C5 may be attributed to the type of linker connecting melatonin to tamoxifen (i.e., cleavable or noncleavable) to influence the pharmacological characteristics of anticancer drugs as reviewed (Hasan et al., 2018). This idea is supported by the findings that: 1) C4 and C5 inhibited BC lines devoid of ERs (MMC, MDA-MB-231, BT-549); 2) that C4 and C5 displayed unique binding characteristics at ERs and MT1Rs; and 3) uterine protection occurred for C5, which was not observed when melatonin and tamoxifen were coadministered but unlinked. Regarding the third point, it was demonstrated that a 3-day administration of C5 prevented uterine stimulation compared with 17β-estradiol (E2) alone, tamoxifen alone, and melatonin plus tamoxifen coadministered but unlinked in FVB/n ovariectomized mice (Witt-Enderby et al., 2014).
These data demonstrate that unique pharmacophores are being produced in the presence of C4 or C5 that explain, in part, their anticancer actions and their uterine-protective actions not observed with the coadministered but unlinked controls (i.e., melatonin plus tamoxifen or melatonin plus 4-OH-tamoxifen). The data demonstrate enhanced efficacy of C4 and C5 compared with the unlinked controls likely due to unique ER/MT1R and/or intracellular protein interactions. For example, in MCF-7 (ER+) BC cells, pERK1/2, pERK5, and NF-κB were significantly modulated by C4 or C5 (Fig. 12, A–C). For MMC (HER2+) BC cells, pERK1/2 appeared to play a more central role possibly through comodulation of NF-κB, pERK5, and β1-integrin (Fig. 12, D–G). For the TNBC line, MDA-MB-231, pERK1/2- and pERK5-mediated inhibition of NFκB was modulated by C4, whereas C5 effects on NF-κB and β1-INTEGRIN expression levels, or pERK1/2 crossmodulation of pERK5 and NF-κB, or PI3K-dependent inhibition of NF-κB, RUNX2, and β1-INTEGRIN may underlie C4’s or C5’s anticancer effects, respectively (Fig. 12, H–I). For BT-549 TNBC cells, C4- and C5-mediated effects on NFκB or PI3K-dependent regulation of pERK1/2 may underlie its anticancer actions in this TNBC line (Fig. 12, J–L).
Basal expression of pERK1/2, NF-κB, and β1-INTEGRIN could also account for the differential actions of C4 and C5 on BC cell viability, migration, and modulation of signaling cascades in MCF-7, MMC, MDA-MB-231, and BT-549 cells. Phospho-ERK1/2 levels were highest in MDA-MB-231 cells versus MCF-7 and BT-549 cells, NF-κB levels were highest in BT-549 cells versus MCF-7 cells, and β1-INTEGRIN levels were highest in BT-549 cells compared with MCF-7 or MDA-MB-231 cells. The preference of C4 and C5 for the MEK1/2 pathway in MDA-MB-231 cells may be attributed, in part, to its high basal levels of pERK1/2, and the strong PI3K inhibitory effect in BT-549 cells when combined with C4 or C5 may be attributed to its high NF-κB and β1-INTERGIN levels. All of the proteins analyzed have been shown to either lie downstream of MEK1/2, MEK5, and/or PI3K and play significant roles in BC development, growth, and progression (Temraz et al., 2015; Simoes et al., 2016; Araki and Miyoshi, 2018; Kastrati et al., 2017; Martinelli et al., 2017; Kumar et al., 2018). Their inhibition by C4 or C5 alone or in combination with the MEK1/2, MEK5, or PI3K inhibitors opens a novel and rich area for BC drug development.
Our study also demonstrated that C4 and C5 inhibited cell viability and migration in TamR MCF-7 cells with excellent efficacy and high potency. As described in the Supplemental Methods, these TamR MCF-7 cells have a mesenchymal morphology, indicating that the cells have undergone an epithelial to mesenchymal transition and they also display an accelerated growth rate in the presence of 4-OH-tamoxifen compared with WT MCF-7 cells perhaps due to increased pERK5 (prosurvival kinase) activity and lower pERK1/2 (kinase involved in differentiation and antiproliferative when located in cytoplasm) activity (Supplemental Fig. 6, C–F) (Mebratu and Tesfaigzi, 2009; Sethi et al., 2010; Eapen et al., 2011). Although ER-α expression was lower in TamR MCF-7 cells compared with WT MCF-7 cells and low ERα is associated with a poor prognosis in patients with tamoxifen failure (Vendrell et al., 2008), the findings that [3H]-estradiol binding characteristics (affinity and Bmax) were not different between cells suggest that other ERs (i.e., nongenomic ERs like GPR30) may be playing more of a role in C4- and C5-mediated anticancer actions. This idea is supported by the findings that tamoxifen could activate GPR30 in endometrial (Ishikawa) cells (Lin et al., 2009). The detection of [3H]-estradiol binding sites in TU-BcX-4IC (PDX-TNBC) cells also supports this idea. Future studies should explore downstream effectors like ERK1/2, ERK5, or PI3K pathways (see schematics in Fig. 12).
Using a mouse and human microsomal system, C4 and C5 followed similar metabolic loss as tamoxifen, demonstrating oxidative, presumably cytochrome P450–dependent metabolism. In vivo pharmacokinetic analysis in the mouse demonstrated that both C4 and C5 had exposures similar to tamoxifen after subcutaneous administration, suggesting similar pharmacokinetic characteristics to tamoxifen. After oral administration to mice, rats, and humans, tamoxifen is extensively metabolized (Fromson et al., 1973; Kisanga et al., 2004). In mice, the oral bioavailability of tamoxifen compared with the subcutaneous route was <10% after doses of either 4 or 10 mg/kg for each route (Reid et al., 2014). A similar estimate of oral relative to subcutaneous bioavailability was observed for the C4 conjugate (Table 6). The lower bioavailability of C5 compared with C4 after oral administration suggests higher first-pass elimination of C5 and that other modes of delivery (e.g., subcutaneous) should be considered for C5. This idea is supported in Witt-Enderby et al. (2014) in which C5, given subcutaneously at a dose of 200 μg/kg body weight, was sufficient to produce uterine protection and modulate estrogen/ER-dependent progesterone receptor mRNA expression in the mammary gland (Witt-Enderby et al., 2014). These data suggest that C5 is bioavailable and has the potential to protect against the adverse effects on the uterus associated with chronic tamoxifen usage.
Overall, this study identified novel melatonin-tamoxifen drug conjugates exhibiting anticancer actions against BC, including TNBC and TamR BC, that are associated with poor prognosis. Future, more comprehensive preclinical and clinical BC studies are warranted.
Acknowledgments
The authors thank Drs. David Lapinsky and Vicki Davis for their coinvention with Dr. Witt-Enderby regarding these drug conjugates (U.S. Patent 8,785,501). They also thank Alex Alfortish and the Krewe de Pink for support in this project (to M.E.B.).
Authorship Contributions
Participated in research design: Hasan, Zlotos, Stratford, Witt-Enderby.
Conducted experiments: Hasan, Adhikari, Matossian, Elliott, M. Wright, Alzoubi, Collins-Burow, Burow, Zlotos, Stratford, Witt-Enderby.
Contributed new reagents or analytic tools: Marzouk, T. D. Wright, Matossian, Elliott, M. Wright, Alzoubi, Collins-Burow, Burow, Holzgrabe, Zlotos.
Performed data analysis: Hasan, Miller, Zlotos, Stratford, Witt-Enderby.
Wrote or contributed to the writing of the manuscript: Hasan, Matossian, Zlotos, Stratford, Witt-Enderby.
Footnotes
- Received February 11, 2019.
- Accepted June 7, 2019.
This work was supported by Duquesne University [Marie-Clement Rodier, C.S.Sp. Endowed Chair (to P.A.W.-E.) and Duquesne University Aging Research and Teaching Consortium funding (to R.E.S. and P.A.W.-E.)], the National Institutes of Health National Cancer Institute [Grant R01 CA174785 (to B.M.C.-B.)], and the Louisiana Cancer Research Consortium Biospecimen Core Laboratory.
This work was previously presented as a poster presentation at the following meeting: Hasan M, Marzouk MA, Adhikari S, Wright T, Miller B, Peckich B, Yingling S, Stratford R, Zlotos D, Cavanaugh J, et al. Melatonin-tamoxifen hybrid ligands and their effects on breast cancer (Abstract 3915). American Association for Cancer Research 2018 Annual Meeting; 2018 April 14–18; Chicago.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- BC
- breast cancer
- C2
- 3-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]propionamide
- C4
- 5-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]pentanamide
- C5
- 6-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]hexanamide
- C9
- 10-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]decanamide
- C15
- 16-[[4-[4-[(1Z)-1,2-diphenyl-1-buten-1-yl]phenoxy]ethylmethylamino]-N-[2-(5-methoxy-1H-indole-3-yl)ethyl]hexadecanamide
- DMEM
- Dulbecco’s modified Eagle’s medium
- ER
- estrogen receptor
- ESR1
- estrogen receptor 1
- FBS
- fetal bovine serum
- GPR30
- G protein–coupled receptor for estrogen
- HER2
- human epidermal growth factor receptor 2
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- LLQ
- lower limit of quantification
- MEK
- mitogen-activated protein kinase kinase
- MMC
- mouse mammary carcinoma
- MS
- mass spectrometry
- MT1R
- melatonin receptor 1
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB
- nuclear factor κB
- PDX
- patient-derived xenograft
- PI3K
- phosphoinositide 3-kinase
- QC
- quality control
- PR
- progesterone receptor
- TAM-d5
- deuterated tamoxifen
- TamR
- tamoxifen resistant
- TNBC
- triple-negative breast cancer
- WT
- wild type
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
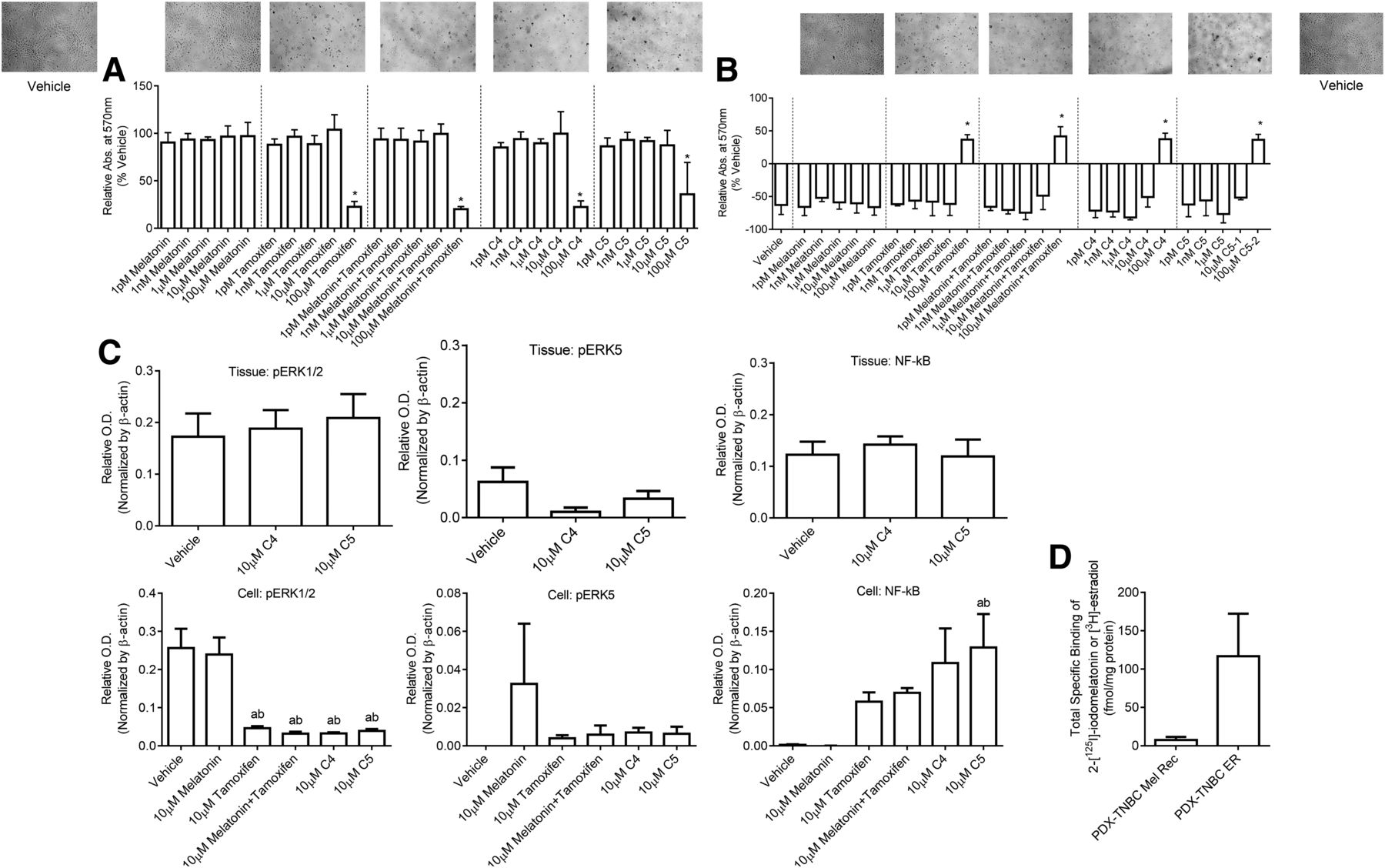{kind=link}
{kind=link}
{kind=link}
{kind=link}
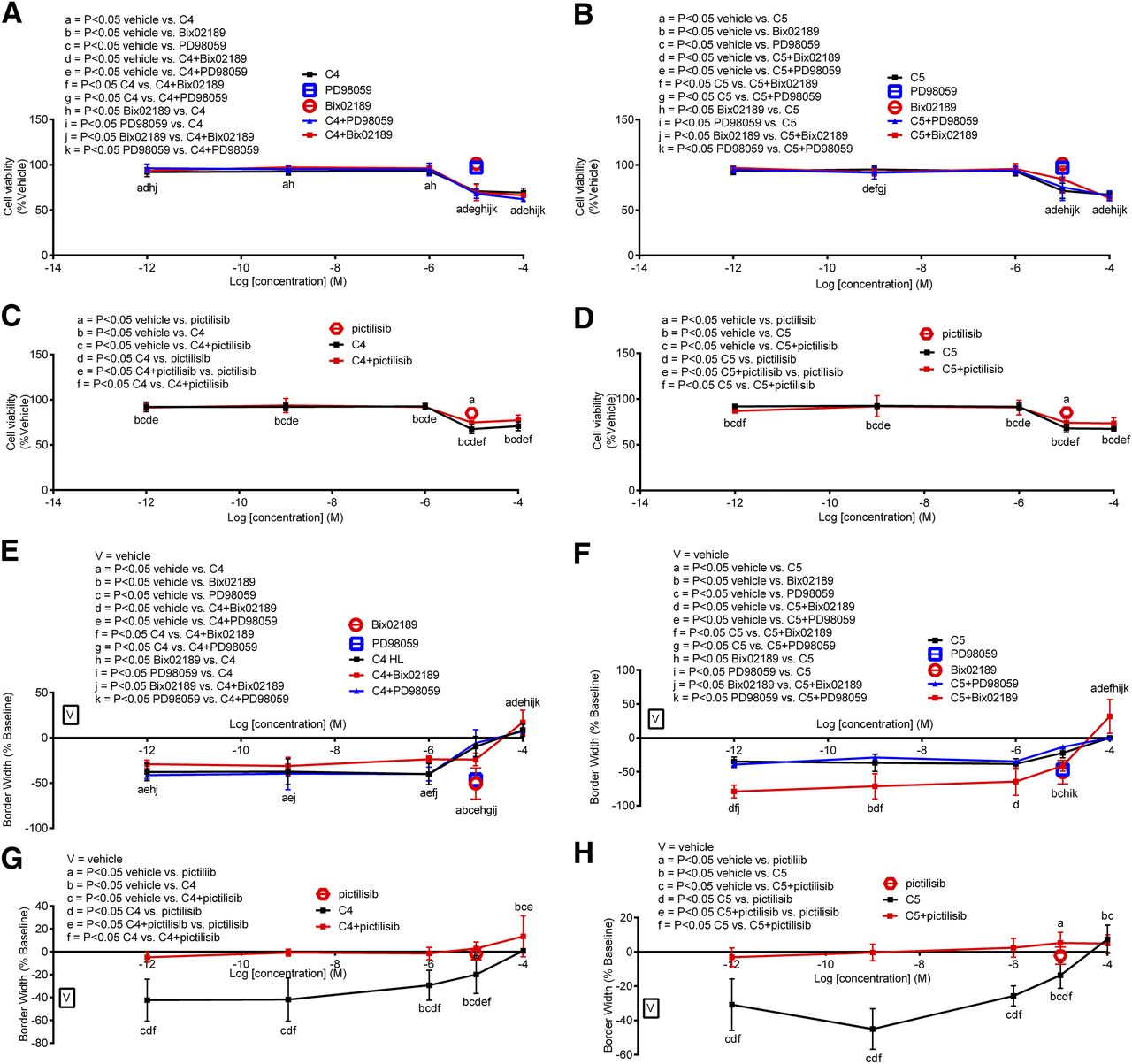{kind=link}
{kind=link}